
Das Medizinprodukterecht-Durchführungsgesetz (MPDG) löst in Deutschland das Vorgängergesetz, das Medizinproduktegesetz (MPG) ab. Sie finden hier die Version des MPDG vom 28.04.2020.
- Für Medizinprodukte ist ab dem 26.05.2021 das MPDG für Hersteller, Betreiber und weitere Akteure verbindlich.
- Für IVD ist ab dem 26.05.2022 das MPDG für Hersteller, Betreiber und weitere Akteure verbindlich.
Im Gegensatz zum MPG ist das MPDG kein eigenständiges Werk, sondern ergänzt nur die EU-Medizinprodukteverordnungen (MDR, IVDR) um nationale Vorgaben.
Dieser Artikel verschafft Ihnen einen ersten Einblick und verrät, auf was Sie als Medizinproduktehersteller achten müssen, um den erweiterten Straf- und Bußgeldvorschriften zu entgehen. Er enthält auch ein PDF zum Download, das das MPDG und das MPG gegenüberstellt, um die Änderungen einfacher zu identifizieren und darauf zu reagieren.
Am 15.04.2021 haben der Deutsche Bundestag und am 07.05.2021 der Deutsche Bundesrat eine weitere Änderung des MPDGs beschlossen. Am 20.05.2022 kamen weitere Änderungen, u. a. bezügl. Freiverkaufszertifikate, Versicherungsschutz und klinische Prüfungen hinzu.
1. Medizinprodukterecht-Durchführungsgesetz (MPDG)
a) Hintergrund: Wie es zum MPDG kam
Das bisherige Medizinproduktegesetz überführt(e) die Anforderungen der EU-Richtlinien wie der MDD und der IVD in nationales Recht. Hingegen benötigen die MDR und die IVDR wie alle EU-Verordnungen keine solche Überführung. Sie haben bereits gesetzlichen Charakter.
Dennoch bedarf es nationaler Vorgaben, die beispielsweise regeln:
- Welche Behörden, die im jeweiligen Land zuständig sind
- Strafen wie Freiheitsstrafen und Bußgelder (das ist Sache der Nationalstaaten)
- Besondere Anforderungen im jeweiligen Land, die allerdings nicht im Widerspruch zu den EU-Verordnungen stehen dürfen und auch nicht den Wettbewerb behindern dürfen
Diese Festlegungen nimmt das Medizinprodukterecht-Durchführungsgesetz MPDG sowie (künftige) nationale Verordnungen vor. Das MPDG wurde als Teil des MPAnpG-EU eingeführt.
b) Zusammenspiel der Gesetze
Das Medizinprodukte-EU-Anpassungsgesetz – MPEUAnpG (zeitweise auch als Medizinprodukte-Anpassungsgesetz-EU MPAnpG-EU bezeichnet) enthält nicht nur das MPDG, welches das MPG ablösten soll. Das MPEUAnpG beschreibt auch Änderungen an acht weiteren Gesetzen wie am
- Sozialgesetzbuch, Fünftes Buch,
- Gesetz über die Werbung auf dem Gebiet des Heilwesens sowie am
- Gesetz über den Verkehr mit Arzneimitteln.
Zudem ergänzt es Verordnungen,
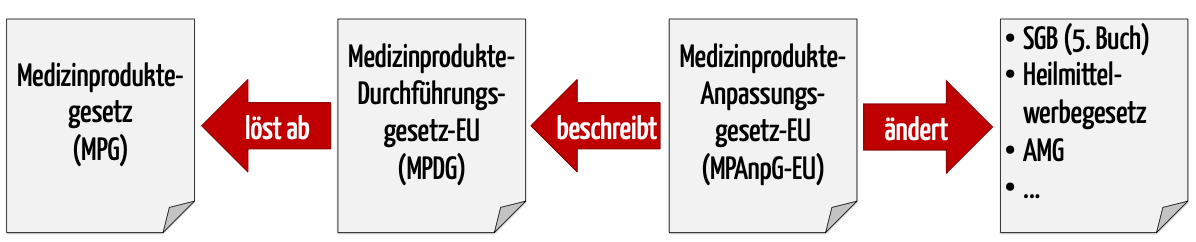
Diese Änderungen sind allerdings weitgehend marginal und passen ungültig gewordene Referenzen insbesondere auf die EU-Richtlinien an.
Fast die Hälfte des 163-seitigen Referentenentwurfs umfasst Begründungen.
Beachten Sie, dass die Mindmap noch nicht die im Mai 2021 beschlossene erneute Änderung des MPDG berücksichtigt. Sobald eine konsolidierte Version verfügbar ist, aktualisieren wir diese Mindmap.
c) Gesetze und Verordnungen von und nach dem MPEUAnpG
Nach der Umsetzung des Anpassungsgesetzes gibt es neue Gesetze und Verordnungen:
| Bis 25.05.2021 | Ab 26.05.2021 | Kommentar |
| MPG | MPDG | Für IVD bleibt das MPG bis 25.05.22 gültig. |
| MPBetreibV | MPBetreibV | Verordnung wurde leicht geändert |
| MPSV | MPAMIV | |
| MPKPV | — | Wird im Wesentlichen durch MDR geregelt. Ergänzungen im MPDG. |
| DIMDIV | — | Tw. im MPDG geregelt. Weitere Verordnung geplant. |
| MPV | — |
2. Was das MPDG ändert und was es beibehält
a) Der Umfang wächst
Die MDR und die IVDR geben den gesetzlichen Rahmen bereits vor. Daher enthält das MPDG im Gegensatz zum MPG viele Festlegung wie die Konformitätsbewertungsverfahren nicht mehr. Dennoch ist das MPDG mit ca. 21.000 Wörtern deutlich umfangreicher als das MPG (knapp 16.000 Wörter). Davon zeugen auch die Anzahl der Paragraphen, die von 44 auf 69 nennenswert gestiegen ist.
b) Die Struktur bleibt ähnlich
Das Medizinprodukterecht-Durchführungsgesetz MPDG ist wie das MPG in neun „Kapitel“ untergliedert, die das MPG „Abschnitte“ nennt. Die direkte Gegenüberstellung der Artikel zeigt, dass v.a. die Kapitel zu den klinischen Prüfungen, zur Vigilanz und zu den Übergangsbestimmungen deutlich mehr Kapitel enthalten.
c) Es gibt höhere Strafen und mehr Strafgründe
Die Freiheitsstrafen ändert das MPDG auf bis zu 10 Jahren. Zudem droht das Gesetz auch denjenigen Freiheitsstrafen an, die gefälschte Produkte in den Verkehr bringen. Neu ist auch, dass gewerbsmäßiger oder bandenmäßiger Verstoß explizit genannt wird und mit bis zu 10 Jahren Freiheitsentzug geahndet werden kann.
In den Katalog der mit Freiheitsstrafen bewehrten Handlungen neu oder präziser aufgenommen wurden:
- Beginnen und Fortführen klinischer Prüfungen und Leistungsbewertungen entgegen mehrerer Artikel der MDR bzw. IVDR
- Inverkehrbringung von Produkten, ohne diese bzw. ohne den Hersteller registriert zu haben
- Irreführende Angaben machen z.B. auf dem Produkt, der Verpackung, der Gebrauchsanweisung oder in Werbematerialien
Den Bußgeldkatalog hat das MPDG ebenfalls erweitert, die Geldbußen belässt es aber bei maximal 30.000 EUR.
d) Die Sicherheitsbeauftragten sind Vergangenheit
Beachtenswert ist, dass das Medizinprodukterecht-Durchführungsgesetz keinen Sicherheitsbeauftragten mehr kennt. Entsprechend lautet beispielsweise die Forderung „nur“, dass der Hersteller „Sicherheitskorrekturmaßnahmen“ mitteilen muss, nicht aber, dass dies die Pflicht einer spezifischen Rolle, nämlich des Sicherheitsbeauftragten ist.
Es findet sich auch kein Hinweis darauf, dass die „qualifizierte Person“ nach Artikel 15 MDR die Pflichten des Sicherheitsbeauftragten übernehmen müsse. Entsprechend enthält das MPDG Referenzen auf den Artikel 15 nur bei den Bußgeldvorschriften.
e) Die Medizinprodukteberater bleiben
Die Anforderungen an die Medizinprodukteberater belässt das Medizinprodukterecht-Durchführungsgesetz MPDG. Das betrifft auch die vorausgesetzte Sachkenntnis.

Lernen Sie alles Wichtige zum Medizinprodukteberater in unserem E-Learning-Kurs!
Lernen Sie in unserem E-Learning-Kurs zum Medizinprodukteberater alle rechtlichen Grundlagen, die Sie für die Ausübung dieser Rolle benötigen. Nach dem Kurs wissen Sie genau, welche Anforderungen an die Rolle gestellt werden, welche Pflichten die Rolle hat und welche Konsequenzen aus Missachtung der Pflichten resultieren können.
f) Mehr Anforderungen an klinische Prüfungen und Leistungsbewertungen
Das Kapitel im MPDG, dass den Herstellern vorschreibt, wie sie klinische Prüfungen zu beantragen, zu beginnen, durchzuführen und zu überwachen haben, hat sich im Umfang vervielfacht und geht teilweise über die Anforderungen der MDR hinaus, wie Sie weiter unten noch lesen.
g) Erweiterte Zuständigkeiten der Behörden
Genau wie beim MPG legen viele Kapitel des MPDGs die Zuständigkeiten und Rechte der einzelnen Behörden fest. Das betrifft konkret das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und das Paul-Ehrlich-Institut (PEI).
Nicht mehr die Länderbehörden, sondern (auch) diese Bundesbehörden, sollen künftig:
- Maßnahmen zum Schutz vor unvertretbaren Risiken ergreifen, die von einem Medizinprodukt ausgehen.
- Produkte zurückrufen oder vom Markt nehmen
- Die Bereitstellung, das Betreiben oder Anwenden untersagen oder einzuschränken
- Die Öffentlichkeit zu unterrichten
Diese Rechte räumt der §45 des MPDG den Bundesoberbehörden ein. Er verlangt aber zuvor eine Risikobewertung nach §38.
Des Weiteren können Sie künftig, das Produkt zurückzurufen oder vom Markt zu nehmen. Bislang sind allein die Behörden der Länder dafür zuständig.
Das MPDG soll auch für die überfällige Umsetzung der sog. „Medicrime“-Konvention des Europarates in Bezug auf die Fälschung von Medizinprodukten umsetzen. Diese Umsetzung in nationales Recht ist notwendig, damit Deutschland dieses Übereinkommen ratifizieren kann.
3. Was das MPDG zusätzlich zur MDR und IVDR fordert
Das Medizinprodukterecht-Durchführungsgesetz MPDG regelt – anders als der Name es vermuten lässt – nicht nur die Durchführung der MDR bzw. IVDR, sondern stellt teilweise zusätzliche Anforderungen wie beispielsweise die folgenden:
a) Schwerpunkt klinische Prüfungen
Die Anforderungen an klinische Prüfungen müssen auch dann eingehalten werden, wenn diese Studien, nicht dem Nachweis des Nutzens sowie der Sicherheit und Leistungsfähigkeit der Produkte dienen, sondern neuen wissenschaftlichen Erkenntnissen. Das regelt das MPDG genauer als die MDR. Beachten Sie sie dazu unseren Artikel zu klinischen Prüfungen.
Das MPDG verpflichtet Hersteller, auch bei weniger kritischen Produkten erst nach Zustimmung durch die Bundesoberbehörde klinische Prüfungen zu beginnen.
Zudem legt es die Latte für klinische Prüfungen an besonderen Probanden (Minderjährige, nicht in der Lage zu erkennen, um was es geht, Gefangene usw.) höher als die MDR.
Es beschreibt auch die Interaktion mit der Ethik-Kommission und deren Vorgehen genauer.
Personenbezogene Daten, die bei klinischen Prüfungen erhoben werden, müssen durch die Prüfer vor der Übermittlung pseudonymisiert werden.
b) Erweiterter Anwendungsbereich
Auch Betriebe, die Medizinprodukte sterilisieren oder keimarm für andere aufbereiten, müssen sich bei den Behörden registrieren. Das gilt auch für Firmen, die implantierbare Sonderanfertigen der Klasse III herstellen.
Zudem verbietet das MPDG, gefälschte Produkte, gefälschtes Zubehör und gefälschte Teile und Komponenten herzustellen, auf Lager zu halten, in den Verkehr zu bringen und in Betrieb zu nehmen.
c) Kennzeichnungen
Die Begleitmaterialien müssen in deutscher Sprache abgefasst sein. Ausnahmen bei professionellen Anwendern sind gestattet (Englisch).
Das MPDG besteht darauf, dass Vorführgeräte explizit gekennzeichnet sind, zumindest wenn sie den Anforderungen der Verordnungen nicht genügen.
d) Und noch vieles mehr
Im Fall eines Streites zwischen Hersteller und Benannter Stelle über die Klassifizierung entscheidet die Bundesoberbehörde.
4. Konsequenzen für Hersteller und Betreiber
a) Mehrere Quellen studieren
Firmen, die in Deutschland Medizinprodukte herstellen oder in den Verkehr bringen, Firmen, die klinische Prüfungen durchführen oder auch nur als Sterilisationsdienstleister auftreten, müssen das MPDG studieren. Das ist mühsam, weil sie die Anforderungen aus drei Quellen – den EU-Verordnungen, dem MPDG und den nationalen Verordnungen – zusammentragen müssen. Es gibt keinen konsolidierten Anforderungskatalog.
Besonders bei den klinischen Prüfungen inklusive den Ethik-Anträgen heißt das, das Gesetz sehr genau zu studieren und nicht dem Irrglauben zu erliegen, dass MDR-Konformität ausreichen würde.
b) Verfahrensanweisungen überarbeiten
Hersteller tun gut daran, ihre Verfahrensanweisungen zu überprüfen bzw. zu überarbeiten, um Konformität mit dem Medizinprodukterecht-Durchführungsgesetz zu erreichen. Das gilt insbesondere für die Verfahrensanweisungen für
- Klinische Prüfungen
- Registrierung von Herstellern und Produkten
- Vigilanz
c) Rollen anpassen
Die Hersteller müssen die „Person Responsible for Regulatory Compliance“ gemäß Artikel 15 etablieren. Beachten Sie dabei, dass deren Aufgabenspektrum und deren Kompetenzen sich von denen eines Sicherheitsbeauftragten unterscheiden.
Der bloße Austausch der Rollenbezeichnung droht, zu einer Nicht-Konformität mit den gesetzlichen Anforderungen zu führen.
5. Aktuelles
Mai 2021
Der Bundestag und Bundestag haben nochmals Änderungen am Medizinproduktedurchführungsgesetz beschlossen, die u.a. die Benannten Stellen und klinischen Prüfungen betreffen. Diese Änderungen sind hier zu finden. Eine konsolidierte Version des MPDG liegt Mitte Mai 2021 noch nicht verfügbar, sollte aber hier veröffentlicht werden.
April 2020
In einem Gesetz, genauer gesagt im Entwurf eines Zweiten Gesetzes zum Schutz der Bevölkerung bei einer epidemischen Lage von nationaler Tragweite wird das neue Medizinprodukterecht-Durchführungsgesetzes um ein Jahr verschoben. Sie finden die relevante Stelle im Artikel 19, die Begründung auf Seite 99 unten.
März 2020
Gegen die Stimmen der AfD hat der Deutsche Bundestag am 5. März 2020 einen Gesetzentwurf der Bundesregierung angenommen, der das Ziel verfolgt, das deutsche Medizinprodukterecht an EU-Vorgaben anzupassen.
Über 217 Seiten umfasst die entsprechende Drucksache 19/15620. Dieser Entwurf unterscheidet sich an einigen Stellen von dem weiter oben verlinkten Referentenentwurf.
Ende des DIMDIs
Am auffälligsten ist die Änderung, dass die Bezüge zum DIMDI entfernt wurden und das BfArM nun als die wesentliche Behörde aufgeführt wird. Das betrifft insbesondere das „Deutsche Medizinprodukteinformations- und Datenbankbanksystem“ nach §86.
Korrekturen, Verschärfungen und Konkretisierungen
Weiter finden sich einige Korrekturen, Verschärfungen und Konkretisierungen:
- Bei Änderungen von klinischen Prüfungen muss die Ethik-Kommission doch umfangreicher involviert werden (§39).
- Einige Verantwortlichkeiten wurden von den Bundes- an die Länderbehörden übergeben (z.B. §76). An anderer Stelle (§74) wird die Bundesbehörde verpflichtet, die Länderbehörden zu informieren.
- Umgekehrt erhält die Bundesoberbehörde das Recht, bei Gefahr im Verzug selbst aktiv zu werden (§74 (4)).
- Bei Korrekturmaßnahmen darf die Bundesoberbehörde vom Sponsor oder Prüfer alle für die Sachverhaltsaufklärung oder Risikobewertung erforderlichen Auskünfte und Unterlagen verlangen (§69).
- Auch die Betreiber unterliegen der Überwachung durch die Behörden (§77).
- Wer eine Anzeige z.B. vor Inverkehrbringen seines Produkts (§4) nicht, nicht richtig, nicht vollständig oder nicht rechtzeitig erstattet, begeht nun auch eine Ordnungswiedrigkeit.
Streitpotenzial
§74 verpflichtet die Behörden, bei Untätigkeit eines Wirtschaftsakteurs zu handeln. Es fehlt allerdings die Definition, wie schnell ein Wirtschaftsakteur handeln muss. Dies wäre wichtig, da die Ursachenanalyse Zeit in Anspruch nimmt. Erst dann kann ein Hersteller entscheiden,
- ob sein Produkt ursächlich für das Problem ist,
- ob Korrekturmaßnahmen notwendig sind und
- welche Korrekturmaßnahmen er ergreifen sollte.
Da sowohl der Hersteller als auch die Behörde eine Risikobewertung durchführen müssen, stellt sich die Frage, wer letztlich entscheidet. Die ISO 14971 verpflichtet beispielsweise in Kapitel 4.2 das Management zu dieser Festlegung.
Auch die Entscheidung über die Angemessenheit behördlich angeordneter Maßnahmen birgt Streitpotenzial.
Leichte Entspannung bei CAPAs
Zu begrüßen ist hingegen die Klarstellung im §85 (2). Dort heißt es nun:
Das Bundesinstitut für Arzneimittel und Medizinprodukte ist […] zuständig für die Bewertung von Meldungen über sicherheitsrelevante Präventiv- und Korrekturmaßnahmen nach Artikel 83 Absatz 4 der Verordnung (EU) 2017/745,
MPDG §85 (2)
Der entsprechende Artikel der MDR liest sich so, als müssten alle „CAPAs“ gemeldet werden.
Klarstellung des Scopes
Besonders IVD-Hersteller sollten diesen Satz lesen:
Microsoft Word – 17589.docx
Mit dem Entwurf eines Medizinprodukterecht-Durchführungsgesetzes (MPDG) soll das nationale Medizinprodukterecht an die neuen unionsrechtlichen Vorschriften angepasst werden. Das MPDG solle ab 26. Mai 2020 das Medizinproduktegesetz (MPG) ersetzen und zunächst nur für die der Verordnung (EU) 2017/745 unterfallenden Produkte gelten. Nicht erfasst seien zunächst die In-vitro-Diagnostika im Sinne der Verordnung (EU) 2017/746. Für diese gelte das MPG in der bis einschließlich 25. Mai 2020 geltenden Fassung bis zum Geltungsbeginn der Verordnung (EU) 2017/746 am 26. Mai 2022 fort.
Drucksache 19/17589, S. 189 oben
Danke an Bernhard Reszel für die Informationen und Analysen!
6. Fazit
Das Bundesgesundheitsministerium hatte wenige Monate vor Inkrafttreten der MDR einen Referentenentwurf für das Medizinprodukterecht-Durchführungsgesetz MPDG vorgelegt. Damit erhielten die Hersteller und Betreiber zumindest eine Vorstellung davon, wie das endgültige Gesetz aussehen wird, und können sich darauf einstellen.
Die Tatsache, dass dieser Referentenentwurf sehr spät kam und dafür mit einem erstaunlichen Umfang aufwartet, wird die Einschätzung vieler bestärken: Deutschland (und Europa) schafft den Herstellern keine guten Rahmenbedingungen, um im Markt wettbewerbsfähig zu bleiben. Ressourcen, die man für die Bewältigung von Bürokratie benötigt, fehlen für die Innovation.
„Zum Glück“ wurde sowohl die MDR als auch das Inkrafttreten des MPDGs um ein Jahr verschoben.
Änderungshistorie
- 2022-06-30: Hinweis auf Änderungen des MPDG ergänzt.
- 2021-05-17: Einleitenden Text komplett geändert. Historie mit MPAnpG-EU gekürzt. Historie aktualisiert. Hinweise zu erneuter Änderung am Mai 2021 ergänzt. Tabelle eingefügt.
- 2021-04-14: Vergleich MPDG und MPG aktualisiert. Die ursprüngliche Version basierte noch auf dem Referentenentwurf. Zudem Mindmap als PDF zum Download eingefügt.



Liebes Team vom Johner Institut,
vielen Dank für die Zusammenfassung, wie immer sehr hilfreich!
Ich möchte kurz darauf hinweisen, dass es im Abschnitt 2 f) und 4 b) nicht um die „klinische Bewertung“ geht, sondern um die „klinische Prüfung“ (Studie).
Das MPAnpG-EU bzw. MDG enhält keine zusätzlichen Anforderungen an die klinische Bewertung, welche über die Anforderungen von MDR Artikel 61 und Anhang XIV hinausgehen.
Beste Grüße,
Sarah Panten
Du hast absolut Recht, liebe Sarah! Danke!
Ich hatte in der Überschrift von „Bewertungen“ und im Text von „Prüfungen“ geschrieben.
Danke Deiner Hilfe, konnte ich das gleich fixen. Danke!!
Herzliche Grüße, Christian
Hallo,
vielen Dank für den aufschlussreichen Artikel.
Ich habe den Entwurf des MDG ebenfalls gelesen und stelle mir bei § 53 „Deutsches Informations- und Datenbanksystem über Medizinprodukte“ die Frage, ob mit dem Geltungsbeginn der MDR alle relevanten Daten für die EUDAMED dem DIMDI gemeldet werden müssen, so dass der Austausch der Daten dann zwischen DMIDS und der EUDAMED stattfindet.
Ich frage das, da wir als Hersteller von Medizinprodukten die technische Infrastruktur zur Übermittlung von Daten zur EUDAMED, z.B. über XML-Export, schaffen müssen.
Sollte der Weg des Datentransfers aber über das DIMDI laufen, scheint es mir zumindest technisch einfacher zu sein.
Vielleicht können sie dazu etwas sagen.
Mit bestem Dank,
Victoria Krüger
Sehr geehrte Frau Krüger,
Sie stellen eine ebenso spannende wie schwierige Frage. Noch haben wir keine endgültigen Informationen dazu, weil noch immer nicht offiziell feststeht, wann die EUDAMED bereitsteht. Die entsprechenden nationalen Regelungen, sprich nationalen Verordnungen, fehlen ebenfalls noch. Derzeit kämpft man noch am MDG.
Sobald wir etwas erfahren, kommunizieren wir das übers Instituts-Journal. Ich lege aber das Ohr aufs Gleis, um möglichst schnell Antworten für Sie zu finden.
Beste Grüße, Christian Johner
Liebes Team vom Johner Institut,
vielen Dank für die gute Übersicht zum MPDG.
Leider hat sich an der ein oder anderen Stelle der Fehlerteufel eingeschlichen. Die Links führen zu anderen pdf.-Dokumenten als angekündigt.
MPDG-2020-03-05 führt zur Drucksache 19/15620 des deutschen Bundestages vom 02.12.2019 statt zur Drucksache 19/17589 vom 4.03.2020, die auch am 5.03.2020 vom Bundestag verabschiedet wurde.
Der Link zum Referentenentwurf führt zum Kabinettsentwurf vom 6.11.2019.
Herzliche Grüße
Sonja Neuhofen
Vielen Dank, liebe Frau Neuhofen!
Sie haben absolut Recht! Dank Ihrer Hilfe konnte ich die aktuellen Dokumente verlinken. Danke für Ihren wichtigen Hinweis!
Viele Grüße, Christian Johner
Liebes Johner-Institut-Team,
mich beschäftigt seit einiger Zeit die Frage, wie die bisherige Ausnahmeregelung bzgl. PMCF (post-market-clinical follow up)-Studiendurchführung und Studiengenehmigung nach MPG 23b (Ausnahmeregelung) im neuen MDR bzw. MPDG abgebildet wird.
In Ihrer sehr übersichtlichen Gegenüberstellung des MPG zu MDG schreiben Sie, dass dies unter dem Aspekt „sonstige klinischen Prüfungen“ (Art 82 MDR / §24 MPDG) abgebildet wird. Im eigentlichen Gesetzestext scheint mir das nicht 100%-ig klar zu werden…
Mir scheint, dass hierbei der im MDR genannt Begriff der „klinischen Nachbeobachtung nach dem Inverkehrbringen“ am ehesten den PMCF-Studien entsprechen, die bisher unter MPG 23b lediglich einer berufsrechtlichen Beratung der EK (nach §15 BO) und eben nicht einer Ethikbewertung und BfArM-Meldung als klinische Prüfung bedurften.
Art 74,1 des MDR besagt, dass folgende Artikel auch für die klinische Nachbeobachtungen nach dem Inverkehrbringen gelten:
„Für klinische Prüfungen nach dem Inverkehrbringen gelten Artikel 62 Absatz 4 Buchstaben b bis k und m, die Artikel 75 bis 77 und Artikel 80 Absatz 5 sowie die einschlägigen Bestimmungen des Anhangs XV.“ In diesen genannten Artikeln wird dann aber wiederum nicht genauer darauf eingegangen wie die „klinischen Nachbeobachtung nach dem Inverkehrbringen“ bei der Ethikkommission gemeldet/vorgelegt wird, und ob auch (wenn keine invasiven Untersuchungen geplant sind) unter vereinfachten Regularien (z.B. keine BfArM Meldung) genehmigt werden kann.
Haben Sie hierzu eine Einschätzung?
Denken Sie, dass eine direkte Anfrage bei der zuständigen Ethikkommission sinnvoll wäre um abzuschätzen wie diese solch Art Studie (PMCF-Studie/Anwendungsbeobachtung von CE-gekennzeichneten MP im Rahmen der Zweckbestimmung) ab Mai 2021 bewerten werden?
Herzliche Grüße,
Dr. Yvonne Mödinger
Sehr geehrte Frau Dr. Mödinger,
vielen Dank für Ihre spannenden Fragen.
Bitte beachten Sie die Unterscheidung und Abgrenzung von Klinischen Prüfungen zum Nachweis der Konformität des Produktes mit den aufgeführten Zweck (MDR, Artikel 62), Klinischen Prüfungen zum Nachweis der Konformität des Produktes, die nicht zu einem der in Artikel 62 Absatz 1 genannten Zwecke durchgeführt werden (Sonstige Klinische Prüfungen (MDR, Artikel 82)) und Klinischen Prüfungen mit CE-gekennzeichneten Produkten mit zusätzlichen Verfahren invasiv oder belastend (MDR, Artikel 74).
Die MDR definiert im Anhang XIV die „klinischen Nachbeobachtung nach dem Inverkehrbringen“ (PMCF) als einen fortlaufenden Prozess zur Aktualisierung der klinischen Bewertung. Bei der klinischen Nachbeobachtung nach dem Inverkehrbringen sammelt und bewertet der Hersteller auf proaktive Weise klinische Daten. PMCF-Aktivitäten beschränken sich nicht nur auf PMCF-Studien, siehe hierzu das MDCG-2020-7, welche verschiedene PMCF-Aktivtäten als Beispiele aufführt.
Dieses MDCG-202-07 Dokument (Section C) ebenso wie die MEDDEV 2.12/2 rev. 2 (Kapitel 6) listen jedoch auch PMCF-Studien als Beispiele auf, die nicht unter die MDR-Artikel 62, 74 oder 82 fallen. Dementsprechend unterscheidet sich, ob und welche zuständigen Landesbehörden und Ethikkommissionen hinzugezogen werden.
Viele Grüße,
N. Jurrmann.
Liebes Johner Institut Team,
danke für den guten, zusammenfassenden Beitrag.
Eine Sache zu den Medizinprodukteberatern ist für mich noch nicht ganz klar, darum möchte ich an dieser Stelle nachfragen. Sie schreiben, dass das neue MPDG die Anforderungen an die Medizinprodukteberater belässt und dies auch die vorausgesetzte Sachkenntnis betrifft. Im Gesetzesentwurf der verlinkt ist, steht bei §83 zur Sachkenntnis jedoch zusätzlich zu der naturwissenschaftlichen, medizinischen und technischen Ausbildung auch die IT-kaufmännische Ausbildung.
Ist dieser Teil der IT-kaufmännischen Ausbildung nun wirklich ergänzt worden, oder habe ich das falsch verstanden?
Vielen Dank für Ihre Hilfe und viele Grüße,
Melanie Bergmann
Sehr geehrte Frau Bergmann,
Sie haben das MDCG perfekt gelesen. Die IT-kaufmännische Ausbildung ist ergänzt worden. Sie sind also noch präziser als ich im Beitrag.
Danke für diesen wichtigen Hinweis!
Beste Grüße, Christian Johner
Sehr geehrtes Johner Team,
ich habe eine Frage zum Thema Vigilanz. Im alten MPG findet sich noch die Verpflichtungen für die Benannten Stellen, im Rahmen von „Gefahr im Verzug“ in Zusammenhang mit Nichtkonformitäten, unverzüglich die BOB bzw. die zuständigen Behörden zu informieren.
Eine solche Anforderung habe ich weder im neuen MPDG noch in der MDR finden können. Bedeutet dies, dass die Benannten Stellen damit komplett aus dem Vigilanzsystem verabschieden können?
Für eine schnelle Antwort bedanke ich mich im Voraus
MfG
Andreas Lauf
Sehr geehrter Herr Lauf,
danke für Ihre sehr relevante Frage!
Die Informationspflicht findet sich genauer im MPSV als im MPG spezifiziert. Diese Meldepflichten an die Hersteller verpflichten in erster Linie, die BOB zu informieren.
Dieses Konzept hat man in der MDR fortgeschrieben. Die Benannten Stellen sind damit aber keinesfalls verabschiedet: Die Artikel 88 und 89 verpflichten die Hersteller zu einer Zusammenarbeit auch mit den Benannten Stellen.
Zudem greifen hier fast immer zivilrechtliche Informationspflichten zwischen Hersteller und Benannter Stelle.
Weiter ist die Benannte Stelle bei der „Überwachungsbewertung“ z.B. gemäß Anhang IX beteiligt. Auch die unangekündigten Audits erfolgen nicht unkorreliert zu den gemeldeten Zwischenfällen.
Fazit: Der erste Ansprechpartner bei Zwischenfällen sind weiterhin die Behörden. Die Benannten Stellen sind aber keinesfalls aus dem Vigilanzsystem verabschiedet worden.
Mit den besten Grüßen, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre schnelle Antwort. Wenn ich sie richtig verstehe, dann ist meine Interpretation, dass weder die MDR noch das MPDG die benannten Stellen als aktives Mitglied in der Meldekette sehen, richtig. Das war ja in der MDD (Artikel 16, Absatz 6) und im MPG (§ 18, Absatz 3) anders. Die MPSV hilftan dieser Stelleleidernicht weiter, dasie nicht die benannten Stellen adressiert, sondern nur die Akteure innerhalb der Lieferkette und die Behörden.
Ich finde es allerdings durchaus erstaunlich, dass die MDR, die es sich- insbesondere als Konsequenz aus dem PIP-Skandal- auf die Fahne geschrieben hat, durch eine effektivere, schnellere und transparentere Vigilanz für sichere Medizinprodukte sorgen zu wollen, die benannten Stellen „in Fällen, in denen sich ein Eingreifen der zuständigen Behörde als erforderlich erweisen könnte“ (Zitat MPG, §18, Abs. 3) aus der Verantwortung nimmt.
Mit freundlichen Grüßen
Andreas Lauf
Gute Tag Herr Prof. Johner,
leider gibt es einen systematischen Fehler im pdf zum Vergleich zwischen MPG und MPDG.
1. wird dort vom MDG gesprochen und
2. sind einige Paragraphen des MDG falsch zugeordnet worden. So ist z.B. die Klassifizierung nicht unter §9 zu finden sondern unter §6 MPDG.
Auch ist § 8 MPDG nicht: “ Abgabe von Prüfprodukten, Produkten für Leis- tungsstudien und Sonderanfertigungen, Ausstellen“ sondern, „Sprachenregelung für die EU-Konformitätserklärung und für Produktinformationen“.
Mit freundlichen Grüßen
Dr. Andreas Breß
Sehr geehrter Herr Dr. Breß,
danke für Ihren Hinweis. Sie haben Recht, wir müssen den Artikel aktualisieren.
Das MPDG wurde während des ersten Referentenentwurfs noch mit MDG abgekürzt. Auch die Nummerierung und Inhalte haben sich geändert. Daher war das Geschriebene zwar damals korrekt. Es entspricht aber, wie Sie richtig feststellen, nicht mehr dem aktuellen Stand.
Danke, dass Sie mich aufmerksam machen. Mir wäre das sonst entgangen.
Viele Grüße, Christian Johner
Guten Tag Herr Prof. Johner,
sie haben in einer Antwort zu einem anderen Kommentar geschrieben, dass genauere Informationen sich in in der MPSV befinden. Mit Erlangung der Gültigkeit der MPDG und Ende der Übergangsfristen kann dann aber die MPSV nicht mehr gelten, da diese auf das MPG referenziert und nicht auf das MPDG.
Es gibt im MPDG keine Referenzierung auf die MPSV.
Für mich stellen sich damit einige Fragen:
1. Wo wird man diese Informationen, die ehemals in der MPSV vorhanden waren finden oder wird man nur noch mit den Informationen der IVDR arbeiten.
2. Es wäre wichtig für die Vorbereitung der IVDR, z.B. zu wissen wie man mit folgenden Begriffen/Verfahren umgehen sollte:
Verantwortliche nach § 5 MPG (Sicherheitsbeauftragter)
Rückruf-Begriff (§2 Satz 2 MPSV)
Vorkommnis (§2 Satz 1, MPSV)
Es scheinen viele Definitionen wegzufallen bzw. nicht mehr relevant zu sein.
Vielen Dank schonmal für Ihre Antwort.
Mit freundlichen Grüßen
Dr. Andreas Breß
Sehr geehrter Herr Dr. Bress,
die Informationen der MPSV finden Sie in der MPAMIV.
Diese Verordnung definiert auch neue Begriffe. Es fallen aber auch Definitionen weg, weil diese in der MDR bzw. IVDR stehen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
danke für den ausführlichen und sehr informativen Artikel. In der PDF-Datei Vergleich-MPG-MPDG stimmen die aus der MPDG referenzierten Paragraphen nicht überein. Zum Beispiel auf Seite 4: Durchführung der Vigilanzaufgaben. Es müsste § 71 heißen. Es sind noch weitere Paragraphen falsch.
Mit freundlichen Grüßen
C. Rockstroh
Sehr geehrte Frau, sehr geehrter Herr Rockstroh,
Sie haben absolut Recht: Der Beitrag bedarf einer Überarbeitung. Viele der Paragrafen beziehen sich noch auf den Referentenentwurf und stimme nicht mehr.
Dank Ihres Hinweises haben wir das auf dem Schrim und werden den Artikel überarbeiten. Vielen Dank dafür!
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner
Vielen Dank für den aufschlussreichen Artikel und die vielen Links.
Ich verstehe nicht, was aus der Allgemeinen Anzeigepflicht (MPG § 25) geworden ist. In Ihrer Vergleichstabelle MPG – MPDG steht bei § 25 MPG rechts bei MPDG nichts. Heisst das, dass mit dem neuen MPDG die Medizinprodukte nicht mehr beim BfArM gemeldet werden müssen? Ich weiss, dass Medizinprodukt irgendeinmal in die EUDAMED gemeldet werden müssen.
Danke für Ihre Erläuterungen.
Beste Grüsse
Oscar Banz
Sehr geehrter Herr Banz,
es ist genau wie Sie sagen: Bis die EUDAMED funktionsfähig ist, gelten die „alten“ Anforderungen, die die Registrierung der Produkte beim BfArM (früher DIMDI) fordert.
Nach aktueller Planung soll es nächstes Jahr soweit sein. Das Modul zur Registrierung der Akteure ist bereits funktionsfähig.
Danke für Ihre wichtigen Nachfrage!
Viele Grüße, Christian Johner
Sehr geehrtes Johner Team, sehr geehrter Herr Johner,
als ehem. langjähriger Leiter eines MP Risikomanagement einer großen Universitätsklinik, irritiert mich im „neuen“ MPDG und MPAMIV die Tatsache, das bei einem „vorsätzlichen Verstoß gegen die Meldeverpflichtung nach § 3 MPAMI i.V. mit dem Wort „mutmaßlich“ weder Bußgeld- noch Strafbewehrt noch anderweitig sanktioniert ist. (siehe Urteil von H. Dänzer, von der Uniklinik Mannheim wegen Verstoßes gegen § 14 MPG)
Gerade in Verbindung mit dem § 13 MPDG sehe ich hier sehr grosse Probleme auf einen Anwender zukommen, wenn durch einen fehlerhaften Zubehöreinkauf seitens eines Einkaufsabteilung als auch einer Medizintechnikabteilung diese Produkte zu einem Patientenschaden kommt? Selbst die §§ 71 und 72 MPDG helfen da nicht weiter. Ich dachte durch die MDR i.V. mit dem MPDG sollte die Patientensicherheit verbessert werden? Das sehe ich hier nicht.
Auch in der MDR ist nicht viel zu diesem brisanten Thema zu finden.
Sehe ich das richtig?
Danke für eine Antwort
Ihr Achim Storm
Sehr geehrter Herr Storm,
danke für die spannende Frage!
Wenn ich Sie richtig verstehe, stört Sie, dass Verstöße gegen die Meldepflichten nicht bzw. nicht ausreichend geahndet werden? Meinten Sie das?
Ich sehe im MPDG §88 2. (9) den Bezug zu den Meldepflichten und damit eine bußgeldbewehrte Ordnungswidrigkeit aber keinen Straftatbestand. Möglicherweise habe ich Sie aber missverstanden. Schreiben Sie mir einfach, dann tauche ich noch tiefer in diesen Teil des Gesetzes ab.
Viele Grüße, Christian Johner
Lieber Herr Johner,
danke für Ihre schnelle Antwort.
Ja, genau darum geht es.
es war schon jahrelang schwer genug in einer Gesundheitseinrichtung, die Anwender unter MPSV Bedingungen von der Wichtigkeit einer „Vorkommnis Meldung“ (Präventiv – Charakter) zu überzeugen. (umso mehr Meldungen an die BfArM erfolgte umso mehr Daten und damit Wissen über die Qualität eines Medizinproduktes) Obwohl es immer schon eine Mitwirkungspflicht und eine Meldepflicht gab. (es hatte aber nie eine Konsequenz, wenn man das nicht tat).
In sehr vielen CIRS Plattformen werden Vorkommnisse publiziert, die dort eigentlich nicht hingehören. Da diese sehr oft „meldepflichtige Vorkommnisse“ sind. Seit Jahren gibt es nicht mal eine aktuelle Vigilanz und Wissenschaftliche Auswertung mehr auf der BfArM Seite nach den Ursachen: was ist wann, mit welchem MP, von welchem Hersteller, wie passiert (Produkt, falsche Wartung oder falsche Instandhaltung oder Anwenderfehler?)
Woher soll denn ein Anwender überhaupt sein Wissen beziehen, um das Produkt sicher am Patienten anzuwenden??
Jetzt soll in Zukunft „der Anwender“ die §§ 11, 12 und 13, MPDG anwenden, dies dann mit dem § 3 MPAMI verknüpfen und melden wenn er der Meldepflicht korrekt nachkommen möchte, um ggf. daraus seine Lehren zu ziehen? um in Zukunft richtig zu handeln. (er hat ja unter MPSV schon selten ein Feedback von der BfArM bekommen) nun braucht er aber noch zusätzlich die MDR um alles richtig zu interpretieren. Also lässt er in Zukunft das ganz…….da ja auch weder die MT, noch die Beschaffungsabteilung Ihn unterstützen……..! Wie oft kommt es denn auf Grund von Vorkomnismeldung zu einem Produktrückruf? oder wenn auch nur eine schlecht lesbare GA geändert werden sollte, weil die Gebrauchsfähigkeit eines MP einfach nicht richtig möglich ist. Und wie soll ein Anwender denn durch § 13 MPDG darauf Einfluss nehmen? Wenn der Anwender davon betroffen wird, ist es für Ihn und ggf. für den Patienten zu spät. (falsche Akkus, falsche Anschlüsse, falsche Leistungsdaten, falsche PIN Belegung etc. da kein Original Zubehör) Die „Nichtanwendung“ durch den Anwender dieser Produkte unter § 13 MPDG hätte eigentlich mit aufgeführt werden müssen. Da der Anwender die „letzte“ Instanz einer Patientenanwendung ist um ggf. den Zwischenfall vor der eigentlichen Anwendung zu verhindern. Und das löst weder der § 71 noch § 72 noch die MDR, wenn ich der Meldepflicht aus welchem Grunde auch immer, nicht nachkomme. Wo bleibt da die „Marktbeobachtungspflicht“ Und auf welcher Datenlage bauen wir in Zukunft das „klinische Risikomanagement“ oder das „Fehlermanagement“ auf?……durch Kaffeesatz lesen, war das doch oft die letzten Jahre der Fall.
Danke
Ihr Achim Storm
Sehr geehrtes Johner Team,
in der MDR liest man nicht selten das Wort „Feld“
so z.B. unter „Vigilanz“ im Artikel 87
„Meldung von schwerwiegenden Vorkommnissen und Sicherheitskorrekturmaßnahmen im Feld“
Was bedeutet jetzt genau das Wort „Feld“ in diesem Zusammenhang? und was ist dabei zu beachten?
Danke
Ihr Achim Storm
Sehr geehrter Herr Storm,
mit „Feld“ ist „Markt“ gemeint. Also die Orten, an denen die Produkte eingesetzt werden wir Krankenhäuser, Arztpraxen, Wohnungen usw.
Viele Grüße, Christian Johner
Sehr geehrtes Johner Team,
da jetzt ab 26.05.2021 das MPDG mit seiner MPAMIV gilt, hätte ich gerne erfahren: mit welchen Formularen und wie denn jetzt u.a. „mutmaßliche“ Vorkommnisse gemeldet werden sollen? wenn die BfArM Seite bisher weder aktualisiert noch andere Formulare, die der aktuellen Gesetzeslage entsprechen, eingestellt wurden. Wie soll oder muss jetzt vorgegangen werden? (Zeit in der BfArM war eigentlich genug, um alles gesetzeskonform vorzubereiten)
Danke für eine Antwort
Ihr Achim Storm
Sehr geehrter Herr Storm,
das BfArM hat nach eigener Aussage auf seiner Webseite die Formulare am 01.01.2020 erneuert. Solange dort nichts Neues veröffentlicht ist, sollten Sie diese Formulare verwenden. Diese Formulare sind bereits für die MDR vorgesehen und nennen z.B. bereits die EUDAMED und UDI.
Dass ausreichend Zeit gewesen wäre, um vieles (besser) vorzubereiten, ist sehr wahr und kommentiert sich leider selbst.
Beste Grüße, Christian Johner
Viele Grüße, Christian Johner
Guten Tag,
ich hätte eine Frage zu der Forderung in §72. Darin steht, dass vor Untersuchung des Produktes eine Einwilligung der Patienten einzuholen ist.
Das MPDG ist deutsches Gesetz und damit müssen deutsche Hersteller es anwenden. Die Forderung gilt aber für alle Produkte die europaweit oder sogar weltweit in Verkehr gebracht wurden? Oder nur für die in Deutschland in Verkehr gebrachten Produkte?
Vielen Dank im Voraus!
Beste Grüße
Nadine Langguth
Sehr geehrte Frau Langguth,
die Forderung gemäß §72 gelten für Produkte, welche in Deutschland in Verkehr gebracht wurden. In weiteren Ländern müssen die Produkte gemäß der Gesetzgebung der jeweiligen Länder in Verkehr gebracht werden.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
ich wollte kurz fragen, ob die IVDR-Hersteller auch in DMIDS sich anmelden müssen.
Vielen Dank im Voraus1
MfG
Younis Skaik
Sehr geehrter Herr Skaik,
IVD’s sind Medizinprodukte, weshalb diesbezüglich die gleichen Anforderungen gelten und eine Registrierung/Anmeldung in DMIDS (Deutsches Medizinprodukte-Informations- und Datenbanksystem) erforderlich ist.
Herzliche Grüße
Sehr geehrte Damen und Herren,
eine banale Frage, aber ich komme nicht dahinter.
Bspw. in §11 Betreiben und Anwenden von Produkten heisst es:
…Produkte und Produkte nach §2 Absatz 2 dürfen nur nach Maßgabe der Rechtsverordnung nach §88 Absatz 1 Satz 1 Nummer 6 betrieben und angewendet werden.
Welche Rechtsverordnung ist hier gemeint? Die Medzinprodukteverordnung kann es m. E. nicht sein, da geht es in Artikel 88 um die Meldung von Trends.
Die MPBetreibV hat keine 88 Paragraphen sondern nur 19.
Wo finde ich die Maßgabe, nach der Produkte betrieben und angewendet werden dürfen?
Sehr geehrter Herr Lichtenthal,
es ist die Rechtsverordnung „Medizinprodukterecht-Durchführungsgesetz (MPDG)“ gemeint. Diese definiert in §88 Absatz 1 Satz 1 Nummer 6, welche Anforderungen an das Errichten, Betreiben, Anwenden und Instandhalten von Produkten zu erfüllen sind.
Herzliche Grüße
Sehr geehrter Hr. Prof. Johner,
das MDPG ist deutsches Gesetz und deutsche Hersteller müssen es anwenden. Was gilt denn in den einzelnen Ländern der EU equivalent für die Ergänzung der MDR mit nationalen Vorgaben? Gibt es eine Übersicht der entsprechenden Gesetze (oder der Behörden)?
Freundliche Grüsse
Dagmar Biegon
Sehr geehrte Frau Biegon,
eine derartige Übersicht ist mir leider nicht bekannt.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
mir wird leider nicht deutlich, wann man einen Betrieber ist bzw. was weitere Akteure sind ?
Wir sind kein Hersteller, betreiben die MP aber auch nicht und wenden sie selbst auch nicht an, wir bereiten sie auf (Desinfizieren sie) und vermieten sie in Gesundheitseinrichtungen.
Für Medizinprodukte ist ab dem 26.05.2021 das MPDG für Hersteller, Betreiber und weitere Akteure verbindlich.
Erbitte Rückinfo.
Vielen Dank
Sehr geehrter Herr T.,
bitte verzeihen Sie die langsame Antwort.
Wenn Sie Medizinprodukte aufbereiten, dann sind sie ggf. ein Hersteller. Das ergibt sich einmal aus der Definition des Begriffs Hersteller:
Zum anderen ergibt es sich ggf. aus dem 17 der MDR:
Falls Sie nicht unter die beiden o.g. Fälle fallen, dann sind Sie ein Dienstleister im Auftrag eines Betreibers. Dann legt Ihnen dieser Betreiber die Pflichten aus, die sich wiederum aus dessen Pflichten sowie aus dessen QM-Vorgaben erbeben.
Zusätzlich sollten Sie prüfen, ob Sie unter den §4 des MPDG fallen (was ich vermute):
Dieser Paragraph wendet sich auch an Organisationen, die weder Hersteller noch Betreiber sind.
Beste Grüße, Christian Johner