
Wahrscheinlich gibt es wenige Dinge, die Hersteller von Medizinprodukten mehr fürchten, als einen Warning Letter durch die FDA.
Wenn Sie einen FDA Warning Letter erhalten, darf Ihnen kein Fehler mehr unterlaufen.
Erfahren Sie hier,
- wann Sie einen FDA Warning Letter bekommen,
- auf was Sie dann achten müssen und
- wie Sie die Kuh wieder vom Eis bekommen.
1. FDA Warning Letter versus Formular 483
FDA Warning Letters sind meist das Ergebnis einer vorausgegangenen Inspektion, bei der der Auditor Abweichungen bzw. Gesetzesverstöße entdeckt hat, beispielsweise beim QM-System (Quality System Regulations gemäß 21 CFR 820). Diese Abweichungen dokumentiert er mithilfe eines Formulars, das die Nummer 483 trägt.
Wohlgemerkt, die Forms 483 und ein FDA Warning Letters sind zwei verschiedene Dinge. Der Warning Letter erfolgt jedoch oft als nächste Stufe.
a) FDA Warning Letters
FDA Warning Letters sind als solche leicht zu erkennen, denn sie enthalten im Betreff die Worte Warning Letter:

Die FDA Warning Letters sind öffentlich einsehbar. Sie enthalten üblicherweise folgende Abschnitte:
- Ergebnisse der Inspektion (wenn eine solche voranging)
- Beobachtungen und Verweise auf verletzte Regularien, z. B. auf 21 CFR part 820
- Erwartete Reaktion. Das ist typischerweise eine Antwort innerhalb von 15 Tagen, in der steht, wie man diese Abweichungen behoben hat oder beheben will und wie man plant, künftig diese Vorkommnisse zu vermeiden.
- (Mögliche) Konsequenzen
In Europa wird die Behördenkommunikation üblicherweise nicht veröffentlicht. In den USA schon, und die Konsequenzen sind groß: Die Hersteller laufen nicht nur Gefahr, mit einem Produktions- oder Importverbot und strafrechtlichen Maßnahmen konfrontiert zu werden, sondern sie sind auch öffentlich bloßgestellt.
b) Das Formular 483
Das „Form 483“ sieht wenig spektakulär aus. Es enthält einen Kopf mit den Daten zur Behörde, dem Datum der Inspektion (Onsite Inspection) und der Firma:

Dann folgen die eigentlichen Beobachtungen und schließlich eine Fußzeile, die neben der Seitenzahl auch die Nummer dieses Formulars beinhaltet:

Die 483er-Formulare der FDA sind zumindest teilweise öffentlich zugänglich.
2. Ablauf
a) Behörde entdeckt Abweichung
Wenn die FDA eine signifikante Verletzung der Regularien entdeckt, informiert sie den Hersteller. Das geschieht bei schwerwiegenden Verstößen in Form eines Warning Letters, der die Verletzung beschreibt.
Wenn die FDA diese Abweichung bei einer Inspektion entdeckt, erhält der Hersteller am Ende der Inspektion den „483er“ ausgestellt und einige Wochen später einen finalen Bericht, den Establishment Inspection Report (EIR). Hersteller sollten darauf innerhalb von 15 Werktagen mit einer geeigneten Ursachenanalyse und einem Maßnahmeplan reagieren.
Die Hersteller müssen auf einen 483er nicht antworten, aber es ist empfehlenswert – auch, um ggf. die nächste Stufe abzuwenden, den FDA Warning Letter.
b) Hersteller hat Warning Letter erhalten
Der Warning Letter lässt keinen Zweifel daran, dass ein großes Problem vorliegt. Die FDA bringt darin zum Ausdruck,
- welches Problem aus ihrer Sicht vorliegt,
- wie das Unternehmen das Problem aus Sicht der Behörde lösen kann oder soll,
- wie lange das Unternehmen Zeit hat, um das Problem zu lösen.
c) Hersteller löst das Problem (hoffentlich)
Jetzt ist es ernst: Die Firma sollte alle Energie darauf verwenden, um die notwendigen Korrekturen und Korrekturmaßnahmen zu ergreifen. Beispiele dafür sind:
- Prozesse grundlegend überarbeiten
- Mitarbeiterinnen und Mitarbeiter ausbilden lassen
- Ressourcen bereitstellen, z. B. Qualitätsabteilung aufstocken
- Lieferanten wechseln
- Produktionsanlagen überholen
- Dokumentation neu erstellen
- Produkte vom Markt holen und neu entwickeln
Oft empfiehlt es sich oft, für diese Remediation externe Hilfe in Anspruch zu nehmen. Sie können uns in solch einem Fall gerne kontaktieren.
Ob die Korrekturen und Korrekturmaßnahmen angemessen und wirkungsvoll waren, prüft die FDA.
Viele Firmen ändern zu wenig bzw. antworten nicht angemessen auf die gefundenen Abweichungen. Andere schießen bei diesen Maßnahmen über das Ziel hinaus. Aber knapp darunter zu zielen, wäre fatal.
d) Wie es weiter geht
Wenn die FDA mit der Fehlerbeseitigung zufrieden ist, verschickt sie ggf. einen Close-out Letter. Diesen stellt die Behörde aber nur aus, wenn sie sich von der Wirksamkeit der Maßnahmen überzeugt hat.
Wenn die Verletzungen aufgrund ihrer Art nicht behebbar sind, verschickt die FDA diesen Close-out Letter nicht.
In jedem Fall bleibt die Firma auf dem Radar der Behörde und muss mit verstärkten Überwachungsaktivitäten rechnen.
Wenn die FDA erneut Abweichungen feststellt, muss der Hersteller mit sofortigen und drastischen Maßnahmen rechnen. Diese reichen von Vermarktungsverboten über Schließung von Betriebsstätten bis hin zum Einsammeln aller Produkte durch die Behörde auf Kosten des Herstellers. Strafrechtliche Konsequenzen bis hin zu Verhaftungen können dazukommen.
3. Auswertung der FDA Warning Letters
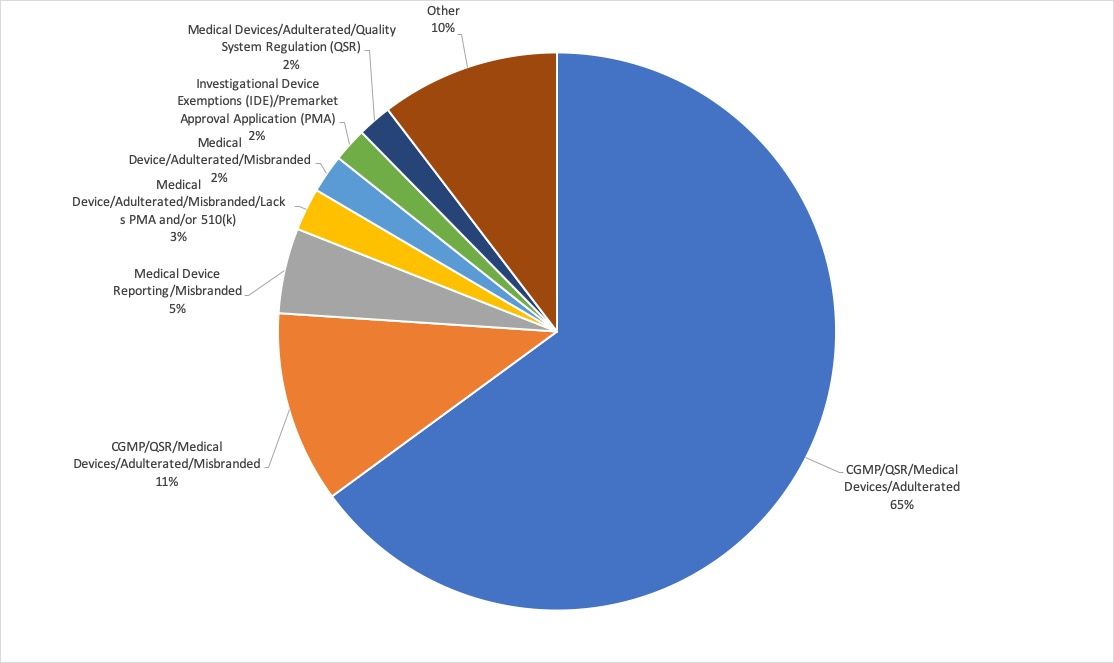
Die FDA publiziert die Warning Letters, was eine Auswertung ermöglicht. Die häufigsten Ursache für diese Briefe sind:
- Nichtkonformes QM-System (Verstoß gegen 21 CFR part 820)
- Irreführende oder falsche Angaben, z. B. den Nutzen des Produkts betreffend
- Fehlende Zulassung
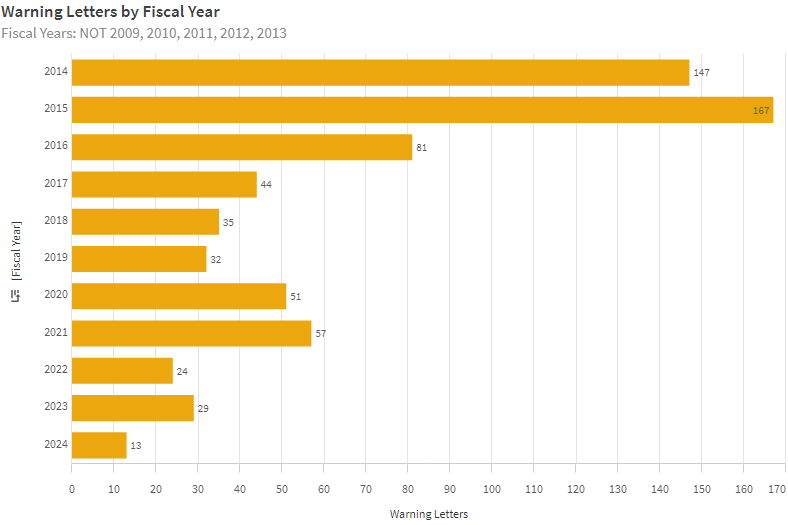
Interessanterweise hat die Anzahl der Warning Letters in den letzten Jahren abgenommen. Ob das an der zurückgehenden Anzahl von Inspektionen liegt, die wiederum durch kleinere Budgets verursacht ist, ist unklar.
4. Fazit
Wer einen FDA Warning Letter erhält, muss wissen, dass allerhöchste Not herrscht. Jetzt heißt es, alle Energien aufzuwenden, um das Problem an der Wurzel zu packen.
Wenn die Geschäftsführung nicht aufrichtig hinter dieser „Remediation“ steht und die notwendigen Ressourcen freigibt und der Behebung der Missstände nicht die erste Priorität einräumt, ist auf die Zukunft der Firma nicht mehr zu wetten.
Änderungshistorie
- 2024-02-12: Aktualisierung der Statistiken und kleine Korrekturen


