
Die europäische In vitro Diagnostic Medical Device Regulation (IVDR) müssen Hersteller beachten, die in der EU In-vitro-Diagnostika in den Verkehr bringen wollen.
Die Verordnung (EU) 2017/746 In vitro Diagnostic Medical Device Regulation (IVDR) regelt im europäischen Markt den gesamten Lebenszyklus von in vitro diagnostischen Medizinprodukten (IVD). Die IVDR ist zeitgleich mit der Verordnung (EU) 2017/745 Medical Device Regulation (MDR) am 25. Mai 2017 in Kraft getreten, welche alle übrigen Medizinprodukte und deren Zubehör reguliert. Gültig ist die IVDR seit dem 26. Mai 2022. Einen Zeitstrahl und Erläuterungen zu den Übergangsfristen finden Sie in diesem Artikel des Johner Instituts.
In diesem Fachartikel wollen wir Ihnen einen Überblick über die IVDR geben: die relevanten Adressaten, ihr Aufbau, die Anforderungen sowie Hintergrundinformationen zum Rechtsakt. Zu den jeweiligen Themen haben wir Ihnen Fachartikel des Johner Instituts verlinkt, um eine Einarbeitung in das teilweise schwer zu bändigende Thema zu erleichtern.
1. Wen betrifft die IVDR?
1.1 Anwendungsbereich
Die IVDR ist für den gesamten Markt der IVD in der EU zuständig: von der Entwicklung über die Marktüberwachung bis zur Anwendung. Insbesondere regelt die IVDR die Voraussetzungen, die IVD erfüllen müssen, um im europäischen Markt in Verkehr gebracht und betrieben werden zu dürfen.
Die IVDR definiert In-vitro-Diagnostikum als ein Medizinprodukt, das vom Hersteller dazu bestimmt ist, aus dem menschlichen Körper stammende Proben zu untersuchen und relevante Informationen zu liefern. Diese Informationen können unter anderem physiologische oder pathologische Zustände, Prädispositionen für Krankheiten, voraussichtliche Wirkung von Behandlungen und die Therapieüberwachung betreffen.
Weitere Produkte wie Reagenzien, Kalibratoren, Kontrollmaterialien, Instrumente und Probengefäße gelten in der IVDR ebenfalls als In-vitro-Diagnostika.
1.2 Betroffene Organisationen
- Somit sind die Hauptadressaten dieser Verordnung die Hersteller von IVD.
- Weitere Wirtschaftsakteure, die die Anforderungen der IVDR beachten müssen, sind Händler, Importeure, Hersteller aus Drittländern und deren Bevollmächtige.
- Neben den Wirtschaftsakteuren stellt die IVDR auch Anforderungen an die verantwortlichen nationalen Behörden und die Benannten Stellen, welche in die Konformitätsprüfung einer großen Spannweite an Produkten einbezogen werden.
- Zur dritten Gruppe, die von der IVDR in Verantwortung genommen wird, gehören Gesundheitseinrichtungen, z. B. Krankenhäuser und Kliniken, die IVD betreiben, oder Labore, die als Hersteller von eigengenutzten Inhouse-IVD agieren.
1.3 Weiterführende Tipps
… für Anfänger
Falls das Thema für Sie neu ist, dann laden Sie sich das kostenlose Starter-Kit herunter. Es verschafft Ihnen einen Überblick über die Regularien, zeigt Ihnen die Schritte zur Konformität Ihres Medizinprodukts und enthält die IVDR-Checkliste zum Download. Nutzen Sie die Fachartikel unter dem Schlagwort Einsteiger.
… für Fortgeschrittene
Arbeiten Sie mit der konsolidierten Version der IVDR in Deutsch bzw. in Englisch. Diese fasst alle Änderungen an der IVDR zusammen, auch die verlängerten Übergangsfristen. Interne Links erleichtern Ihnen die Navigation in der über 170 Seiten langen Verordnung.
Steigen Sie mit den Fachartikeln, die weiter unten verlinkt sind, tiefer in die Details ein. Hilfreich sind auch die Fachartikel unter dem Schlagwort IVD.
… für Hersteller von IVD gemäß Richtlinie 98/79/EG (IVDD)
Hersteller, die IVD bereits gemäß der IVDD in Verkehr gebracht haben, sollten den Fachartikel zu den Unterschieden zwischen der IVDR und IVDD lesen. Die Kapitel der IVDR sind im Vergleich zur alten In vitro Diagnostic Directive (IVDD) völlig neu strukturiert. In dem verlinkten Fachartikel finden Sie Informationen zu den Veränderungen der IVDR im Vergleich zur IVDD.
… für Hersteller von Medizinprodukten
Hersteller von Medizinprodukte sollten den Fachartikel zur MDR lesen.
… für Importeure, Händler, Krankenhäuser und andere Betreiber sowie Labore und RUO-Hersteller
Lesen Sie die in Kapitel 3 b) verlinkten betreffenden Fachartikel.
2. Aufbau und Struktur der IVDR (EU 2017/746)
Die IVDR umfasst 113 Artikel, die sich in zehn Kapitel gliedern. Abbildung 1 zeigt die Struktur der IVDR mit ihren Kapitelthemen. Die Inhalte der zehn Kapitel haben wir für Sie in Tabelle 1 zusammengefasst.
Die 15 Anhänge der IVDR sind nicht zu vernachlässigen. Sie konkretisieren viele der Anforderungen, die in den Artikeln der IVDR beschrieben sind. Zum Beispiel enthält Anhang I die Grundlegenden Sicherheits- und Leistungsanforderungen, die für alle IVD gelten und daher erfüllt werden müssen. Die Inhalte der 15 Anhänge haben wir für Sie in Tabelle 2 zusammengefasst.
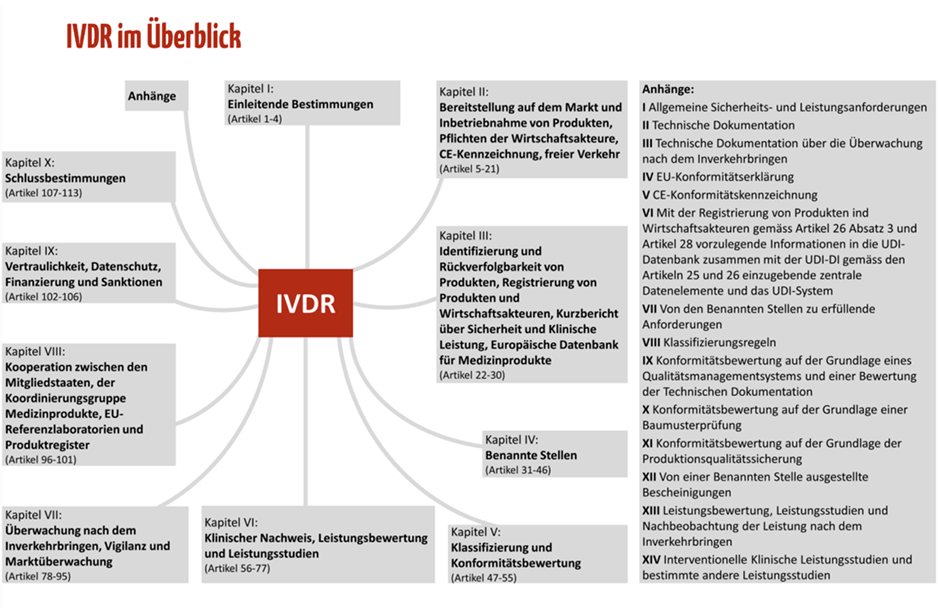
Die Mindmap mit Anhängen ist Teil des kostenfreien Starter-Kits, das Ihnen einen schnellen Überblick über die mehr als 170 Seiten umfassende IVDR gibt.
2.1 Die zehn Kapitel der IVDR (EU 2017/746)
In Tabelle 1 ist die Kapitelstruktur mit den jeweiligen Inhalten übersichtlich dargestellt.
| Kapitelnummer (Mit Links auf IVDR) | Titel / Inhalt / Anforderungen (Mit Links zu Fachartikeln) |
| I | Anwendungsbereich und Definitionen |
| II | Wichtigste Anforderungen an Hersteller, Händler, Importeure und Mitgliedsstaaten Viele Verweise auf Artikel in anderen Kapiteln und auf die Anhänge |
| III | Nachverfolgbarkeit von Produkten, v. a. Anforderungen zur UDI |
| IV | Anforderungen an die Benannten Stellen |
| V | Klassifizierung und Zulassung bzw. Konformitätsbewertungsverfahren |
| VI | Leistungsbewertungen und Leistungsstudien |
| VII | Post-Market Surveillance, Marktüberwachung, Meldewesen |
| VIII | Zusammenarbeit von Mitgliedsstaaten, MDCG und anderen Experten |
| IX | Vertraulichkeit, Datenschutz, Strafen |
| X | Übergangsfristen und mehr |
2.2 Die Anhänge der IVDR
Die IVDR hat 15 Anhänge, welche verschiedene wichtige Themen in Bezug auf die Konformität von IVD-Produkten behandeln. Die Inhalte der Anhänge sind Tabelle 2 zu entnehmen.
| Nummer des Anhangs (Mit Links zur IVDR) | Titel / Inhalt / Anforderungen (Mit Links zu weiterführenden Informationen) |
| I | Allgemeine Sicherheits- und Leistungsanforderungen |
| II | Technische Dokumentation |
| III | Technische Dokumentation für die Post-Market Surveillance |
| IV | EU-Konformitätserklärung |
| V | CE-Kennzeichnung |
| VI | UDI, EUDAMED |
| VII | Anforderungen an die Benannten Stellen |
| VIII | Klassifizierungsregeln |
| IX bis XI | Konformitätsbewertungsverfahren: – Vollständiges QM-System – Baumusterprüfung – Produktkonformitätsprüfung |
| XII | Zertifikate durch Benannte Stellen |
| XIII | Leistungsbewertung, Leistungsstudien und Nachbeobachtung der Leistung |
| XIV | Interventionelle klinische Leistungsstudien |
| XV | Entsprechungstabelle |
Es ist zu erkennen, dass Informationen zur Technischen Dokumentation, mit der die Hersteller die Konformität ihrer Produkte beweisen, gleich in den ersten Anhängen zu finden sind. Außerdem sind Themen wie die Registrierung in der EUDAMED, die UDI, die Klassifizierungsregeln und die Konformitätserklärung in den Anhängen der IVDR erklärt. Auch die Leistungsbewertung und klinische Leistungsstudien werden in den Anhängen der IVDR behandelt.
3. Überblick über die Anforderungen der IVDR
Nachdem der Aufbau und die Struktur der IVDR (EU 2017/746) nun bekannt sind, gehen wir im Folgenden auf die Anforderungen der Hersteller (3 a)) und auf die Anforderungen der anderen Wirtschaftsakteure wie Händler, Importeure und Labore (3 b)) ein.
3.1 Anforderungen an die Hersteller selbst und deren IVD
Tabelle 3 zeigt die produktübergreifenden Anforderungen an die Hersteller von IVD. Strebt der Hersteller für eines seiner IVD eine Konformität nach IVDR an, muss er ein Qualitätsmanagementsystem etablieren und über den gesamten Lebenszyklus der Produkte aufrechterhalten. Hier gilt die ISO 13485 als harmonisierte Norm zu beachten.
3.1.1 Anforderungen an die Hersteller
| Anforderung der IVDR (Mit Links zu Fachartikeln) | Erläuterung |
| Qualitätsmanagementsystem | Alle Hersteller benötigen ein QM-System, u. a. für die Entwicklung, Produktion und Überwachung der Produkte im Markt. Mit Ausnahme von Produkten der Klasse A wird das QM-System von den Benannten Stellen im Rahmen des Konformitätsbewerbungsverfahren geprüft. Bei sterilen Produkten der Klasse A beschränkt sich die Involvierung einer Benannten Stelle auf „die Aspekte der Herstellung, die mit der Erreichung und dem Erhalt des sterilen Zustands zusammenhängen“ (gemäß IVDR, Art. 48 (10) und Anhang XII, Kapitel I, Nr. 7.)“. |
| Risikomanagementsystem | Über ein Risikomanagementsystem stellen Hersteller sicher, dass die unternehmensspezifische Risikopolitik über den Lebenszyklus der individuellen Produkte gelebt und eingehalten wird. Die Risikomanagementakte wiederum stellt produktspezifisch die Nutzen-Risiko-Bewertung dar. |
| Für die Einhaltung der Regulierungsvorschriften verantwortliche Person | Die Hersteller sind verpflichtet, eine Person zu beschäftigen, die für die regulatorische Konformität verantwortlich ist (gemäß IVDR, Artikel 15; auch PRRC genannt) und z. B. sicherstellt, dass die Technische Dokumentation konform mit dem eigenen QM-System erstellt wird. Auch EU-Bevollmächtigte müssen Zugriff auf einen PRRC haben. |
3.1.2 Anforderungen an die Produkte
Zu den produktübergreifenden Anforderungen kommen Anforderungen an jedes einzelne Produkt hinzu. Diese produktspezifischen Anforderungen ergeben sich aus der Zweckbestimmung des jeweiligen IVD. Aus dieser ergibt sich wiederum die Qualifizierung als IVD, die Klassifizierung in eine der vier Risikoklassen, die Anforderungen an die Leistungsbewertung, das produkt-spezifische Risikomanagement und das Labeling. Weitere Verifikations- und Validierungstätigkeiten wie z. B. die Usability oder Stabilitätsnachweise, sowie die Entwicklungsakte gehören ebenso zu einer Technischen Dokumentation. Auch Post-Market Surveillance und Vigilanz sind produktspezifische Aufgaben, die nach der IVDR von den Herstellern erfüllt werden müssen.
Nutzen Sie unseren IVDR-Klassifikator für eine korrekte Klassifizierung Ihres IVD.
Eine weitere neue Pflicht für Hersteller ist die Vergabe einer UDI für jedes IVD und dessen Registrierung in der EUDAMED. Dies soll die Rückverfolgbarkeit und das Überwachen nach dem Inverkehrbringen für alle Stakeholder erleichtern. Zusammengefasst dient die Technische Dokumentation eines IVD nach Anhang II und III dem Nachweis, dass alle gültigen Grundlegenden Sicherheits- und Leistungsanforderungen (Anhang I, IVDR) erfüllt sind. Tabelle 4 zeigt eine Übersicht dieser produktspezifischen Anforderungen.
3.1.3 Weitere regulatorische Anforderungen an die Produkte, deren Entwicklung, „Zulassung“ und Überwachung
| Anforderung der IVDR (Mit Links zu Fachartikeln) | Erläuterung |
| Klassifizierung der IVD und Klassifizierung von Software-IVD | Für jedes IVD muss der Hersteller die Risikoklasse festlegen, welche maßgeblich aus der Zweckbestimmung hervorgeht. |
| Grundlegende Sicherheits- und Leistungsanforderungen und Technische Dokumentation | Die TD muss den Anforderungen des Anhangs II genügen und alle Nachweise erbringen, dass die grundlegenden Anforderungen des Anhangs I erfüllt sind. Für diese Nachweise sind die Hersteller angehalten, harmonisierte Normen und die gemeinsamen Spezifikationen (Common Specifications) anzuwenden. |
| Risikomanagement | Für jedes IVD muss der Hersteller produktspezifische Risiken identifizieren, bewerten und beherrschen. In einem kontinuierlichen Prozess muss der Hersteller stetig das Nutzen-Risiko-Verhältnis beurteilen. |
| Leistungsbewertung | Der Hersteller muss kontinuierlich prüfen, ob Sicherheit, Leistungsfähigkeit und Nutzen der IVD gegeben sind. Reichen die klinischen Daten nicht aus, ist eine Leistungsstudie notwendig. |
| Unique Device Identification (UDI) | Alle IVD müssen eine eindeutige Identifizierung erhalten, die UDI. Damit müssen die Produkte in der EUDAMED registriert werden. |
| Labeling | Die IVDR legt Anforderungen an die Gebrauchsanweisung, weitere Begleitmaterialien und das sonstige Labeling wie Aufdrucke und Verpackungen genau fest. |
| Konformitätsbewertung und Zulassung | Abhängig von der Risikoklasse kann der Hersteller ein Konformitätsbewertungsverfahren wählen. Dabei muss er – außer bei Produkten der Klasse A – eine Benannte Stelle einbeziehen. Im Erfolgsfall muss er eine Konformitätserklärung ausfüllen und das CE-Kennzeichen anbringen. |
| Vigilanz und Überwachung nach dem Inverkehrbringen | Hersteller sind verpflichtet, ihre IVD über deren komplette Lebensdauer im Markt zu überwachen, kontinuierlich Daten dazu zu sammeln und ggf. darauf zu reagieren. Erkennen sie Risiken, müssen sie die Behörden informieren. |
3.1.4 Hinweis zum Erfüllen der Anforderungen
Nicht nur die IVDR stellt Anforderungen an IVD und deren Hersteller. Viele weitere Gesetze und Normen sind zu beachten. In diesem Artikel zur Zulassung von IVD werden sie vorgestellt. Während die IVDR rechtlich bindend ist, sind Normen und Richtliniendokumente dies nicht. Trotzdem wird ein Hersteller in der Praxis Schwierigkeiten haben die Konformität seiner Prozesse und Produkte zu belegen, wenn er sich nicht an die für sein Produkt gültigen harmonisierten Normen und Richtlinien hält.
3.2 Anforderungen an andere (Wirtschafts-)Akteure
Neben den IVD-Hersteller als Adressaten der IVDR, sind weitere Akteure betroffen. Tabelle 5 zeigt eine Aufstellung von weiteren (Wirtschafts-)Akteuren, welche im Gültigkeitsbereich der IVDR handeln.
| Akteure (Mit Links zu Fachartikeln) | Erläuterung | |
| Händler von IVD | Die Händler von IVD müssen sich nicht in der EUDAMED registrieren. Allerdings sind sie nicht befreit von Pflichten, wie zum Beispiel das Weiterleiten und Melden von Vorkommnissen und Rückmeldungen der Anwender. | |
| Importeure von IVD aus Drittstaaten | Importeure, die Produkte aus einem Drittland auf dem Unionsmarkt in Verkehr bringt, stellt die IVDR zusätzliche Pflichten. Im Gegensatz zu den Händlern müssen die Importeure sich in der EUDAMED registrieren. | |
| EU-Bevollmächtigte | Hersteller außerhalb der EU‑Mitgliedsstaaten müssen einen EU‑Bevollmächtigten ernennen, der den nationalen Behörden als Kontaktstelle dient. Die mindestens auszuführenden Aufgaben der EU-Bevollmächtigten sind in Artikel 11 der IVDR beschrieben. Neben der Registrierung in der EUDAMED müssen EU‑Bevollmächtigte Zugriff auf eine in Artikel 15 beschriebene Verantwortliche Person (PRRC) haben. | |
| Krankenhäuser und andere Betreiber | An Gesundheitseinrichten richten sich manche Artikel der IVDR ebenfalls. Hinzukommen nationale Gesetzte wie das Medizinproduktedurchführungsgesetz (MPDG) und die Medizinprodukte-Betreiberverordnung (MPBetreibV). | |
| Labore, die als Hersteller von Inhouse IVD agieren | Labore die eigenentwickelte IVDs anbieten stellen sogenannte Inhouse IVD her. Hierfür müssen wesentliche Anforderungen der IVDR erfüllt sein, wie zum Beispiel die Anforderungen aus Anhang I, IVDR. | |
| Lieferanten von RUO-Produkten | „For Research Use Only“ (RUO) Produkte sind nicht für die Diagnostik am Menschen entwickelt und fallen somit nicht in den Geltungsbereich der IVDR. Labore, die diese Produkte nutzen, um diagnostische Tests anzubieten, befinden sich im Bereich der Inhouse IVD und werden somit durch die IVDR reguliert. Herstellern, Händler und Betreibern, die das RUO- Label falsch anbringen, um damit regulatorische Hürden zu umgehen, gefährden die Gesundheit von Patienten und müssen mit Strafen rechnen. | |
| Pharmahersteller im Kontext von Companion Diagnostics (CDx). | Hersteller von therapiebegleitenden Diagnostika für ihre Arzneimittel werden ebenfalls von der IVDR reguliert. Die Anforderungen für CDx sind höher als für IVD derselben Risikoklasse. |
4. Hintergrundinformationen zum Rechtsakt
4.1 Wie es zu den EU-Verordnungen EU 2017/745 MDR und EU 2017/746 IVDR kam
Es ist bisher unklar, weshalb 2017 die Neuregulierung des Medizinprodukte- und IVD-Marktes von der EU angegangen worden ist. Der Brustimplantate-Skandal wird in Fachkreisen als Auslöser diskutiert, was jedoch von den handelnden Akteuren bestritten wird. Zielsetzung der Verordnungen ist es aber definitiv die Sicherheit und Gesundheit von Patienten, sowie ein barrierefreier EU-Markt für Medizinprodukte zu gewährleisten.
4.2 Unterschied von EU-Verordnungen und EU-Richtlinien
Die „alten“ EU-Medizinprodukte-Richtlinien mussten wie alle EU-Richtlinien in nationale Gesetze und nationale Verordnungen überführt werden, um Gesetzeskraft zu erlangen. Das waren in Deutschland das Medizinproduktegesetz (MPG) und Verordnungen wie die Medizinprodukte-Betreiberverordnung (weiterhin gültig) und die Medizinprodukte-Sicherheitsplanverordnung (inzwischen ungültig).
Die EU-Verordnungen (hier: MDR, IVDR) haben direkt gesetzlichen Charakter. Nationale Gesetze ergänzen diese Verordnungen nur noch um u. a. Strafvorschriften und die Festlegung der zuständigen Behörden. In Deutschland ist das das Medizinproduktedurchführungsgesetz MPDG. Dieses legt neben den oben genannten Vorschriften auch Anforderungen an sogenannte Medizinprodukteberater fest, welche Fachkreise über die Produkte ihres Herstellers beraten.
4.3 Änderungen der IVDR im Vergleich zur IVDD
Dieser Fachartikel beleuchtet die Änderungen, die die IVDR im Vergleich zur IVDD, der In-vitro Diagnostic Device Directive, eingeführt hat.
5. Fazit und Zusammenfassung
Die In vitro Diagnostic Device Regulation (IVDR) ist ein sehr umfangreiches Gesetzeswerk, das alle Akteure (Hersteller, Benannte Stellen, Importeure, Händler, Labore) vor große Herausforderungen stellt.
Im Gegensatz zur IVDD hat die IVDR nicht nur die regulatorischen Anforderungen deutlich erhöht, sondern auch den Gültigkeitsbereich ausgeweitet. Produkte wie Inhouse IVD (Lab‑developed Tests, LDT) unterliegen jetzt der IVDR.
Die Akteure haben keine andere Wahl, als sich mit diesem Gesetzeswerk intensiv auseinanderzusetzen und dessen Anforderungen zu erfüllen. Diese erhöhten Anforderungen betreffen sowohl die „Zulassung“ (Pre-Market-Phase) als auch explizit die Post-Market-Phase (Post-Market Surveillance, Vigilanz).
6. Weitere Fachartikel und Links
6.1 Klassifizierung
Die Risikoklasse eines IVD ist maßgeblich für den regulatorischen Aufwand den Hersteller aufbringen müssen, um die Konformität unter Beweis zu stellen. In diesem Fachartikel können Sie sich einen Überblick auf die Klassifizierung von IVDs nach EU‑Verordnung 2017/746 verschaffen und werden angeleitet wie sich Produkte geschickt und rechtssicher klassifizieren lassen.
6.2 Übergangsfristen
Die EU hat die Übergangsfristen für den Umstieg auf die IVDR mehrfach verschoben. Diese Regeln sind so komplex, dass ein Fachartikel zu den Übergangsfristen helfen wird.
6.3 Besondere IVDs
In einem weiteren Fachartikel des Johner Instituts werden sogenannte Probenahme-Sets im Detail besprochen. Diese Sets liegen regulatorisch den Behandlungseinheiten der MDR nahe. Zusammengefasst können mehrere Produkte zu einem Probenahme-Set zusammengestellt werden, wenn ihre Zweckbestimmung nicht verändert wird.
Nicht erst seit Corona sind IVD zur Eigenanwendung (Selbsttest) und patientennahe Tests ein wichtiger Teil des Gesundheitssystems in Europa. In diesem Fachartikel haben wir Ihnen die regulatorischen Besonderheiten für diese IVD Produkte zusammengefasst.
6.4 Weitere regulatorische Anforderungen
Viele weitere Gesetze und Normen sind zu beachten, die der Artikel zur Zulassung von IVD vorstellt.
Zudem hat die Medical Device Coordination Group (MDCG) Erklärungen und Anforderungen publiziert.
Hilfe bei der Umsetzung der IVDR
Kostenfreie Angebote
Haben Sie noch Fragen zur IVDR und zu ihrer Umsetzung? Antworten erhalten Sie in unserem kostenlosen Micro-Consulting.
Laden Sie sich das kostenlose Starter-Kit herunter, das Ihnen einen Überblick über die regulatorische Landschaft verschafft und die IVDR-Checkliste als PDF und im DOCX-Format enthält.
Videos und E-Learning
Die Videotrainings im Auditgarant zeigen Ihnen Schritt für Schritt, wie Sie Ihre Technische Dokumentation und Ihr QM-System schlank, schnell und IVDR-konform erstellen. Dazu stehen über 100 Templates und Musterdokumente zum Download bereit. So schaffen Sie die Voraussetzungen dafür, Ihre Produkte schnell und sicher zuzulassen und in den Markt zu bringen.
Produktprüfung
Die Experten des Johner Instituts helfen Ihnen beim Prüfen Ihrer Produkte durch:
- Usability Tests
- Penetrations-Tests
- Test der elektrischen Sicherheit und EMV (z.B. IEC 61010-1)
- Prüfung der Biokompatibilität
- Leistungsbewertungen und Leistungsstudien
Beratung
Nutzen Sie die Expertise der Regulatory-Affairs-Experten des Johner Instituts, um Ihre
- regulatorische Strategie zu bestimmen und Produkte zu klassifizieren,
- Technische Dokumentation (TD) zu erstellen,
- QM-Systeme (QMS) aufzubauen,
- TD und QMS zu prüfen und Sie auf Audits und Reviews vorzubereiten sowie in diesen zu begleiten, und die
- Post-Market Surveillance zu übernehmen.
Mit dem IVDR-Readiness-Check erlangen Sie schnell Klarheit darüber, wie konform Ihr QM-System und Ihre Technische Dokumentation sind. Das hilft, unnötige Schwierigkeiten und Verzögerungen bei Audits und Reviews der Technischen Dokumentation und damit der Vermarktung der Produkte zu vermeiden.
Nehmen Sie gleich Kontakt auf, damit wir gemeinsam klären können, wie Sie die regulatorischen Anforderungen der IVDR schnell und einfach erfüllen und Ihre Produkte sicher in den Markt bringen können.
Änderungshistorie (ab Mai 2021; Rest gelöscht)
- 2024-02-25: Kapitelnummerierung überarbeitet, Zwischenüberschriften ergänzt
- 2024-02-06: Artikel überarbeitet
- 2023-05-03: Artikel komplett neu geschrieben



Hallo Herr Prof. Johner,
sie schreiben oben, dass die Übergangsfrist 3 Jahre beträgt und die IVDR 2020 verpflichtend wird, gilt es aber nicht nur für die Medizinprodukte? Für IVD soll 5 Jahre Übergangsfrist gelten oder?
Viele Grüße
Anastasia Wolf
Sie haben Recht, liebe Frau Wolf! Ist korrigiert. Danke!!
Hallo Herr Johner,
ich frage mich ob die Übergangsfrist auch für Produkte gilt, die erst nach Verabschiedung der Verordnung erstmalig in Verkehr gebracht werden. Ich habe die Übergangsfrist auch so interpretiert gelesen: …Alle Hersteller sind betroffen, da auch existierende Produkte (mit entsprechender Übergangsregelung) nach MDR/IVDR zertifiziert werden müssen… (Quelle: Zühlke-blog). Was ist hier Ihr Wissenstand?
Vielen Dank
Udo Klinger
Sehr geehrter Herr Klinger,
ich denke, es gilt die einzelnen Aktivitäten zu unterscheiden. Ein Produkt das vor der Frist legal in Verkehr gebracht wurde, ist legal im Verkehr. Hier hat die MDR keine unmittelbare Auswirkung.
Produkte, die bereits vor der Übergangsfrist „zugelassen“ aber auch noch danach weiter produziert werden, werden die Anforderungen der UDI erfüllen müssen.
Die Marktüberwachung und andere Post-Market-Aktivitäten müssen den Anforderungen der MDR nach der Übergangsfrist genügen. Auch für „alte Produkte“.
Hallo Herr Johner, vielen Dank für diesen Artikel!
vor allem die Abbildung zu den Konformitätsbewerungsverfahren gibt eine gute Übersicht.
Allerdings bin ich der Meinung, dass die Klasse C hier noch nicht vollständig in der Konformitätsbewertung über Annex VIII abgebildet ist. Hier spricht die Verordnung von „in the case of devices classified as class C, the quality management system assessment shall be accompanied by the assessment of the technical documentation for devices selected on a representative basis in accordance with provisions in Sections 5.3a to 5.3e of Chapter II of this Annex.“ (Annex VIII, 3.3 c und äquaivalent 4.5) Man will also meines Erachtens bei Klasse C eine abgeschwächte Form der Bewertung der Techn. Dokumentation, die gemäß den Abschnitten 5.3a und 5.3e hauptsächlich auf den Review der Leistungsbewertung abzielt und im Rahmen der Überwachungsaudits stattfinden soll.
Wie sehen Sie das?
Viele Grüße, Sebastian Grömminger
Hallo,
ich bin noch ganz neu in dem Feld (Laie) und hätte eine eher banale Frage. Oben ist bei Klasse C Produkten die Rede von Selbsttests (geht man hier von Schwangerschafts-, Drogentests etc. aus?) und weiter unten kommt dann “ Die IVDR unterscheidet sogar noch weitere Typen von In-vitro-Diagnostika: … Produkte zur Eigenanwendung“ was wäre das denn für ein IVD das nicht unter Liste C aber in diesen weiteren Typus fallen würde?
Und wie werden diese weitere Typen eingestuft? Ich müsste ja eine Klasse D-A angeben oder nicht?
Vielen Dank und liebe Grüße!
Danke für Ihre sehr gute Frage! Die Frage, in welche Klasse ein Produkt fällt, hängt auch von der Frage ab, ob es für die Eigenanwendung vorgesehen ist. Hierzu besagt die Regel 4 im Anhang VIII:
Die Schwangerschaftstests fallen nicht darunter. Denn hier greift die Regel 6:
Hallo,
ich bin gerade die Klassifizierungsregeln durchgegangen. Liege ich richtig in der Annahme, dass Blutentnahmeröhrchen auf Basis von Regel 5 (Probenbehältnisse) der Klasse A zugeordnet werden?
Ist es für die Klassifizierung von Bedeutung, ob es sich um patientennahe Tests handelt?
Vielen Dank und liebe Grüße!
Ich teile Ihre Vermutung bezüglich der Klassifizierung.
Die Patientennähe ist bei der Frage wichtig, ob es ein IVD oder ein MDD ist. Die Behörden schreiben: „“Produkte, die zu diagnostischen Untersuchungszwecken im oder am menschlichen Körper angewendet werden, sind somit grundsätzlich keine In-vitro-Diagnostika.“
Hallo Herr Prof. Dr. Johner,
ich bin gerade dabei mir die IVDR zu erarbeiten und Ihre Seiten zu dem Thema sind schon sehr hilfreich. Es war mir bislang nur nicht ganz eindeutig, welche Art von Konformitätsbewertungsverfahren auf Klasse C Produkte zu treffen und durchgeführt werden müssen. Habe ich das richtig verstanden, dass diese Produkte alle drei Konformitätsbewertungsverfahren (Anhang IX-XI) durchlaufen bzw. den genannten Anforderungen nachgehen müssen?
Außerdem habe ich noch nicht so ganz durchschaut, wo die gemeinsamen Spezifikationen nachzulesen sind. Ich habe verstanden, was sie sind und wer sie erlässt, aber gibt es denn tatsächlich schon welche? Sind gemeinsame Spezifikationen das gleiche wie gemeinsame technische Spezifikationen?
Ich danke bereits für die Antwort(en).
Mit freundlichem Gruß
F. Dresche
Sehr geehrter Herr Dresche,
danke für Ihre wichtigen Fragen!
Sie müssen nur einen der beiden Wege durchlaufen, also IX XODER X und XI.
Die gemeinsamen Spezifikationen sind derzeit nicht veröffentlicht. Damit hat sich die Kommission quasi in die Lage versetzt, zu gegebenen Anlässen eigene „Normen“ zu veröffentlichen. Die gemeinsamen Spezifikationen können technische Spezifikationen entsprechen, können aber auch Anforderungen z.B. an die klinische Leistungsbewertung oder die Dokumentation stellen.
Beste Grüße, Christian Johner
Ich bin Chirurg und benutze für Operationen Instrumente,welche teilweise kein CE Zeichen aufweisen aber von nahmenhaften Firmen ( Leibinger,Äskulap) hergestellt wurden. Für die Fremdvergabe der Wiederaufbereitung wird von mir verlangt nur Instrumente mit CE Kennzeichen oder Instrumente mit Herstellergarantie der CE Zertifizierung benutzen zu dürfen.
Leider wollen die obigen Firmen und Händler über die ich die Instrumente bezogen habe diese nicht ausstellen.
Habe ich ein Anrecht auf Bestätigung der nachträglichen CE Zertifizierung von Firmen und Händlern für meine Instrumente(Scheren,Klemmen Pinzetten Wundhaken)?
Sehr geehrter Herr Dr. Rapke,
danke für Ihre spannende Frage! Die Firmen müssen Ihnen eine Konformitätserklärung zur Verfügung stellen. Dass namhafte Firmen das nicht tun, wundert mich sehr. Da würde ich erst einmal meine Hand ins Feuer legen.
Haben Sie die Produkte von den Herstellern selbst oder von Dritten bezogen?
Da wir hier möglicherweise in den strafrechtlichen Bereich rutschen, würde ich gerne die Diskussion in den nicht-öffentlichen Teil verschieben. Unsere E-Mail-Adresse finden Sie oben auf der Webseite.
Viele Grüße, Christian Johner
Wenn es ein Medizinprodukt ist, muss es das CE-Zeichen und eine Konformitätserklärung haben.
Könnte es sein, dass es sich bei No .12 um Instrumente handelt, bei denen das CE-Zeichen lediglich auf der Verpackung ist, weil auf dem Instrument selbst zu wenig Platz zur Verfügung steht?
Sehr geehrter Herr Brauns,
die MDR und IVDR verpflichten Hersteller, die CE-Kennzeichnung anzubringen und eine Konformitätserklärung bereitzustellen.
Es gibt Ausnahmen bei der Pflicht, das CE-Kennzeichen aufzubringen.
Was Sie mit No. 12 meinen, ist mir nicht ganz klar. Beziehen Sie sich auf die MDR oder IVDR? Ich frage auch, weil Sie den Beitrag zu IVDR kommentieren, der Begriff „Instrumente“ aber auch in den MDR-Kontext fallen könnte.
Beste Grüße, Christian Johner
Sehr Prof. Johner
Vielen Dank für diese sehr gute Übersicht. Ich habe eine kleine Unsicherheit in Bezug auf die Einteilung: sind diagnostische Kits für den Nachweis von Bakterien im Bereich Food und Umwelt (Salmonellen, Listeten, Enterobakterien, E. Coli etc.) Gegenstand der IVDR oder nicht? Es handelt sich dabei zwar nicht um Probenmaterial vom Menschen, die Erreger selbst können aber vom Mensch sein? Wie findet die Einteilung in sehr ansteckend und ansteckend statt: ist der Nachweis von Staphylokokken resp. die Resistenzbestimmung dieser oder anderer Keime eher der Klasse D oder C zuzuschreiben? Herzlichen Dank und freundliche Grüsse Christoph
Sehr geehrter Christoph,
Die Untersuchung von Lebensmitteln auf Keime fällt in den Anwendungsbereich der Regulation (EC) No 178/2002 und nicht in die IVDR. Das ist vor allem drin begründet, dass die Definition eines In-Vitro-Diagnostikums festlegt, dass die Untersuchung mit menschlichem Probenmaterial erfolgen muss. Daher ist die IVDR auch nur auf Produkte anwendbar, die menschliches Probenmaterial verwenden um Information für medizinische Zwecke zu gewinnen. Ob die Keime, die in Lebensmitteln nachgewiesen wurden zuvor schon einmal den menschlichen Körper besiedelt hatten ist dabei nicht relevant.
Ein solches Produkt kann daher auch nicht in Klasse C oder D fallen.
Beste Grüße, Sebastian Grömminger
Sehr geehrter Prof. Johner,
wie sieht es mit vom Labor hergestellten Verdünnungen aus? In der Pathologie werden zum Teil IVD Antikörperkonzentrate eingesetzt und nach Anweisung des Pathologen verdünnt. Die Hersteller bieten aber auch „Ready to Use“, vorverdünnte Antikörper an. Wenn das Labor nun seine eigene „Verdünnung“ ansetzt, ist es dann Hersteller? Wie wird das bewertet? Ich habe da unterschiedliche Aussagen gehört und bin verunsichert.
Liebe Svenja,
vielen Dank für Ihre Frage!
Fall 1: Wenn diese Antikörper-Konzentrate CE-markierte IVD-Produkte sind, sollte die Gebrauchsanweisung den Verdünnungsvorgang beschreiben. Sofern diese Verdünnungen jedoch beim Pathologen individuell auf das Verfahren abgestimmt sein müssen, müssten im Rahmen der Akkreditierten Laborverfahren auch entsprechende Validierungsunterlagen zum Verdünnungsverfahren vorliegen. Fall 2: Die „ready to use“ Verdünnungen sind hingegen Konzentrationen, die bereits etabliert sind. Hierbei müsste man in der Gebrauchsanweisung des Herstellers Angaben zu Grenzen der Untersuchung finden sowie die Bedingungen, in denen eine sichere Anwendung gewährleistet ist. Dann sollte das Produkt korrekte Ergebnisse entsprechend den Leistungsdaten liefern. In beiden Fällen befinden Sie sich also im jeweils korrekten Anwendungsszenario des Herstellers. Sollten Sie von den Hersteller-Vorgaben abweichen, befinden Sie sich im „Off-Label-Use“ und müssten als Pathologisches Labor selbst die Konformität des Untersuchungsverfahrens gemäß IVD-Richtlinie bzw. IVD-Verordnung sicherstellen. Zu letzterem Fall finden Sie mehr Information hier: https://www.johner-institut.de/blog/regulatory-affairs/laboratory-developed-test-ldt/
Bemerkung zu „Was die IVDR nicht geändert hat“
Es entsteht beim Lesen des letzten Abschnittes der Eindruck, dass hinsichtlich Konformitätsverfahren mit / ohne NB alles beim alten bleibt:
„Die IVDR hat auch nichts daran geändert, dass – außer bei unkritischen Produkten – benannte Stellen bei der Konformitätsbewertung beteiligt werden müssen und die Konformität durch ein CE-Zeichen ausgedrückt werden muss.“
Das Verhältnis von unkritischen Produkten (Klasse A) mit Eigendeklaration zu kritischen Produkten (Klasse As, B, C, D) wird sich – je nach Quelle – von heute 7- 20% auf 80 – 85% aller Produkte verschieben. Das ist eine massive Änderung und ein Engpass bei den NB zeichnet sich jetzt schon ab.
Laboratory Developed Tests sind heute gang und gäbe, unter der IVDR ist das nur noch erlaubt, wenn es keine kommerziell erhältliche Kits gibt.
Lieber Herr Pianegonda, vielen Dank für Ihren Hinweis! Da gebe ich Ihnen vollkommen recht. Ich habe vor geraumer Zeit zu den von Ihnen genannten Zahlen bereits versucht herauszufinden, welche Quellen mir diese Zahlen evidenzbasiert nennen können. Leider habe ich bisher nur Aussagen erhalten, die auf groben Schätzungen beruhen. Wenn Sie mir eine Quelle nennen können, die eine belastbare Aussage auf Basis von Fakten nennt ergänze ich das gerne im Artikel. Die aktuelle Formulierung im Artikel werde ich allerdings direkt abändern, denn auch im IVDR-Seminar ziehen wir ein vergleichbares Fazit.
Klassifizierung C oder D?
Sehr geehrter Herr Prof. Johner,
laut IVDR Anhang VIII Nr. 2.1 Spiegelstrich 2 fallen IVDs, die „…lebensbedrohende Krankheiten mit einem hohen oder mutmaßlich hohen Vebreitungsrisiko…“ nachweisen in die Klasse D.
Jetzt stellt sich die Frage nach der Definition von lebensbedrohenden Krankheiten und eines hohen Verbreitungsrisikos.
Laut Kiehl, W. (2015). Infektionsschutz und Infektionsepidemiologie Fachwörter – Definitionen – Interpretationen Seite 72 sind: „Hochkontagiöse und lebensbedrohliche Krankheiten: Krankheiten, die neben der hohen Ansteckungsgefahr einen sehr schweren klinischen Verlauf aufweisen, sodass von ihnen eine besondere Gefahr für die Allgemeinheit ausgeht (sog. gemeingefährliche Krankheiten). Im IfSG werden sie auch als »bedrohliche Krankheiten« (threatening diseases) bezeichnet.“
Das IfsG hat diese „bedrohlichen Krankheiten“ unter §6 und §7 aufgelistet. Es gibt aber keine Definition der IVDR und keine Information ob die Erkrankung behandelbar sein muss oder nicht.
Insofern würde ein IVD für die Diagnostik von Tuberkulose, die behandelbar ist, von Legionellen oder von Ebola als Klasse D eingestuft, während es damals unter IVDD noch unter sonstiges IVD lief.
Ist das gewollt, da ich den Sprung doch sehr extrem finde. Zumal wir Ebolaviren, Influenzaviren und z.B. Röteln auf die gleiche Ebene stellen würden.
Oh Entschuldigung,
Mit freundlichen Grüßen
Dr. Breß
Lieber Herr Dr. Breß,
Ja, das sehe ich genauso. am Beispiel von SARS-CoV-2 haben wir diese Hoch-Klassifizierung von „sonstiges IVD“ in Klasse D bereits im März diskutiert: https://www.johner-institut.de/blog/regulatory-affairs/corona-virus-ivdr/
Die Positiv-Listen der IVDD sollten mit der IVDR auf ein aus der MDD bewährtes und mit der GHTF bzw. IMDRF global harmonisiertes Klassifizierungs-System umgestellt werden. Dass dies auch die ein oder andere Nebenerscheinung hat, die zur Hoch-Klassifizierung führt ist vorhersehbar. Die Vorteile sind, dass das System lückenlos ist und es keine Schlupflöcher mehr gibt. Eine Aussage, ob die Hoch-Klassifizierung billigend in Kauf genommen wurde oder sogar klar gewollt war wäre an dieser Stelle reine Mutmaßung. Die Corona-Krise zeigt jedoch deutlich, dass Sonderzulassungen im Gesundheitsnotstand mehr denn je klare Vorgaben wie in den USA brauchen. Dass nun auch bekannte und therapierbare Krankheitserreger hoch-klassifiziert werden, liegt daran, dass die Autoren lediglich den Schweregrad (Tod möglich oder nicht) bei einer Lebensbedrohlichen Krankheit heranziehen. Bei der IVDR hat man es versäumt die Wahrscheinlichkeit des Auftretens des Todes mit einzubeziehen. ggf. kann man bei der ein oder anderen Erkrankung, die nicht schnell in die tödliche Richtung verläuft noch in Klasse C über das Patienten-Management argumentieren. Das hängt jedoch stark von de konkreten Fomulierung der Zweckbestimmung ab. Es beleibt abzuwarten ob und die MDCG eine brauchbare Leitlinie zur Klassifizierung von IVDs bereitstellt.
Beste Grüße, Sebastian Grömminger
Sehr geehrter Herr Johner,
wie würden Sie ein Röhrchen mit einer Stabilisierungslösung, welches für den Transport und Lagerung von Abstrichen aus Nase und Rachen bestimmt ist, gemäß IVDR klassifizieren? Die Abstriche werden für Coronatests verwendet. Aus meiner Sicht ist es der Klasse A zuzuordnen, da der Test an sich nicht vom Röhrchen gemacht wird und dieses nur als Probenbehältnis dient. Liege ich mit meiner Einschätzung richtig?
Vielen Dank und beste Grüße,
Maximilian
Lieber Maximilian, prinzipiell würde ich Ihnen gemäß Ihrer formulierten Zweckbestimmung Recht geben, da es sich um ein Probenbehältnis handelt und dann in Klasse A über Regel 5 des Anhang VIII der IVDR fällt. Es kommt nun aber sehr darauf an, wie die Zweckbestimmung im Detail lautet, welche Charakteristika die Stabilisierungslösung hat, ob das Röhrchen steril hergestellt wird und ob die Probennahme von Laien durchgeführt wird. Wenn das Röhrchen speziell für die Detektion von SARS-CoV-2 entwickelt wurde, z.B. weil die Stabilisierungslösung unmittelbar die Sensitivität dieser speziellen Untersuchung sicherstellt, wären wir im schlimmsten Fall sogar in Klasse D über Regel 1. Die Zweckbestimmung muss daher so allgemein wie möglich sein. Wenn die Charakteristika der Stabilisierungslösung dies jedoch nicht zulassen, ist eine Klassifizierung in Klasse A nicht möglich. Beachten Sie bitte, dass für den Swab zur Abnahme der Probe im Rachenraum die MDR gilt.
Beste Grüße, Sebastian Grömminger
Sehr geehrter Herr Professor Johner,
wir sind Händler für Invitrodiagnostika für das medizinische Labor. Unsere Diagnostik Teste laufen mit entsprechenden Testprotokollen (sogenannten Applikationen) auf den Laborgeräten. Wenn jetzt z.B. ein medizinisches Labor, mit einem Analysegerät der Firma Roche arbeitet und den rocheigenen ProteinC Test gegen einen ProteinC Test von uns ersetzen möchte und wir unseren Test mit dem entsprechenden Testprotokoll auf das Roche Gerät applizieren können, wäre in einem solchen Fall IVDR spezifisch etwas zu berücksichtigen ?. Selbstverständlich ist das Produkt selbst, also das Reagenz des Testes entsprechend CE markiert und erfüllt selbst die Kriterien der IVDR. Das erforderliche Testprotokoll um das Reagenz an dem Roche System laufen lassen zu können, wird von uns zusammen mit dem Kunden erstellt und dokumentiert etc.. Was sagt die IVDR zu einer solchen Konstellation ?
In der gesamten neuen Richtlinie finden sich dazu keine Hinweise, auch im Web habe ich keine Hinweise gefunden.
Vielen Dank für eine kurze Rückmeldung dazu und herzliche Grüße
Bernd Kahrmann
Lieber Herr Kahrmann, einen Teil der Antwort auf Ihre Frage finden sie in der IVD-Definition der IVDR (Artikel 2, Abs. 2): „In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das […] — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung […] bestimmt ist“. Damit sagt die IVDR, dass das prinzipiell erlaubt ist. Nun sollten Sie noch klären ob die Verbindung des Analysegerätes und Ihres Tests als Applikation auf dem Analysegerät in Einklang mit der Zweckbestimmung und der Gebrauchsanweisung des Analysegerätes steht. Wenn darin eine andere Kombination als die vom Hersteller des Analysegerätes vorgesehene ausgeschlossen wird, müssen entweder Sie oder das Labor die Konformität der Kombination gewährleisten. Das bedeutet, dass Sie auch für das von Ihnen ggf. mit dem Kunden zusammen erstellte Testprotokoll die Konformität belegen müssen (Erfüllung der Grundl. Sicherheits und Leistungs-Anforderungen sowie Verifizierung und Validierung der neuen Kombination). Die entsprechende Anforderung dazu finden Sie in Anhang I, Abs. 13.1.: Wenn ein Produkt zur Verwendung in Kombination mit anderen Produkten oder Ausrüstungen bestimmt ist, muss die Kombination einschließlich der Verbindungen sicher sein und darf die vorgesehene Leistung der Produkte nicht beeinträchtigen. Beachten Sie bitte: Es treffen mit Sicherheit noch weitere Anforderungen auf Ihren beschriebenen Fall zu.
Viele Grüße,
Ihr IVD-Experte Sebastian Grömminger
Sehr geehrter Herr Johner,
welche Anforderungen der IVDR muss ein steriles sonstiges IVD (gem. IVDD), welches nach IVDR Klasse A steril sein wird, ab Mai 2022 erfüllen? Und wie lange darf das IVD gemäß IVDD produziert und verkauft werden?
Besten Dank!
Karl Heinz
Sehr geehrter Herr Prof. Johner,
wir sind Hersteller eines Blutzuckersystems für das Labor mit der Bedienung durch Fachpersonal. Nach der Richtlinie 98/79/EG gehört es zu den sonstigen IVD Produkten und wurde durch ein eigenständiges Konformitätsverfahren bewertet und zertifiziert.
Nach Regel 5 der neuen IVDR handelt es sich um Spezifische IVD Reagenzien, Instrumente bzw. Probenverhältnisse. Somit wird meines Erachtens diese Produktgruppe zukünftig als Klasse A klassifiziert. Ist dieses richtig ?
Lieber Johannes,
gemäß den Implementierungsregeln in Anhang VIII benötigen Sie die Zweckbestimmung für das Produkt und erst auf dieser Basis können Sie (oder auch gerne wir für Sie) eine finale Klassifizierung vornehmen. Da Diabetes (zumindest bei einer Hypoglykämie) eine lebensbedrohliche Situation darstellen kann, ist es ggf. schwierig aus Regel 3 k) herauszukommen. Das hängt sehr stark davon ab, in welchem Kontext Ihr Produkt eingesetzt wird. Wenn es zum Beispiel für Notfallsituationen zum Zwecke des Patientenmanagements vorgesehen ist, wäre Regel 3 k) sicher anwendbar. Im Labor-Kontext wie Sie es angeben wohl eher nicht. Wenn es ein reines Laborgerät ist, das photometrisch den Glukosewert bestimmt, ist Regel 5 recht sicher zutreffend.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Herr Johner,
wir sind Hersteller eines LIS (Laborinformationssystem = stand alone software). Dieses ist bisher nicht als MD bzw. IVD zertifiziert. Beim letzten ISO 13485 Audit bemerkte der Auditor, dass man in Betracht ziehen sollte, das LIS nach IVDR zertifizieren zu lassen, da es sich hier um Software handelt, die die Benutzung der Laborgeräte beeinflusst. Wir sind gerne bereit, uns dieser Argumentation anzuschließen. Allerdings würde das bedeuten, dass die Software in dieselbe Klasse fällt, wie die Laborgeräte, die angeschlossen werden können. Das würde schlimmstenfalls die Klasse D sein. Die andere Variante wäre aus meiner Sicht, das LIS als Zubehör zu Laborautomaten zu deklarieren. Damit könnten wir es unabhängig klassifizieren. Sehen Sie letztere Möglichkeit als eine gangbare Alternative an?
Viele Grüße
Jochem Stähler
Lieber Herr Strähler, vielen Dank für Ihre spannende Frage. Als Zubehör können Sie Software leider nur in wenigen Einzelfällen definieren, wenn die Software selbst keinen (!) medizinischen Zweck erfüllt. Werden jedoch Auswertealgorithmen in der Software angewandt und damit Ergebnisse für die Diagnose berechnet, handelt es sich um eigenständige Software, die selbst ein MP bzw. IVD ist. Vielleicht hilft Ihnen dieser Beitrag weiter: https://www.johner-institut.de/blog/regulatory-affairs/ivd-software/ oder melden sie sich bei uns, wir helfen gerne.
Herzliche Grüße, Sebastian Grömminger
Sehr geehrter Herr Johner und Team,
Ich weiss diesen Blog_Artikel ist schon ein bisschen älter, aber ich verstehe nicht ganz wie Ihr Berechner auf Basis von Art, 110 funktionniert.
Nach diesem Berrechner könnte ich ein Sonstige IVD noch weiter nach Mai 2022 auf dem Markt bringen….aber nach dem „Joint implementation and preparedness plan for Regulation (EU) 2017/746 on in vitrodiagnostic medical devices (IVDR)“ wäre es nicht möglich. Es steht dort folgendes:
„In addition, devices that were lawfully placed on the market under Directive 98/79/EC by virtue of a certificate may to continue to be made available on the marketor put into servicealso after the date of application of the IVDR and until May 2025at the latestunder certain conditions. “ Somit wären für mich diese Sonstige IVDS die keine Bescheinigung haben nicht von dem Aufschub bis 2024 zw 2025 betroffen. Viele Dank im Voraus für Ihre Antwort Mit freundlichen Grüßen
Liebe Frau Salin, Ihre Schlussfolgerung ist korrekt. Bitte unterscheiden Sie jedoch stets die Begriffe „Inverkehrbringen“ von der „Bereitstellung und Inbetriebnahme“. Gemäß Art. 110 (4) dürfen Sie ein sonstiges IVD nur noch bis 25. Mai 2022 in Verkehr bringen aber noch bis 27. Mai 2025 bereitstellen und in Betrieb nehmen, sofern das Produkt bereits rechtmäßig in Verkehr gebracht wurde. So gibt es auch der Berechner an. Bei den Begrifflichkeiten gibt es jedoch oft Missverständnisse. „Inverkehrbringen“ bezeichnet die erstmalige Bereitstellung eines Produkts, mit Ausnahme von Produkten für Leistungsstudien, auf dem Unionsmarkt. Jedes einzelne Produkt wird demnach in Verkehr gebracht, indem es zu einem Besitzer mit Sitz in der EU wechselt. Dieser neue Besitzer darf das in Verkehr gebrachte Produkt dann wiederum bereitstellen (weiterverkaufen) und der Anwender darf es in Betrieb nehmen, für weitere drei Jahre nach dem Gültigkeitsdatum der IVDR. Dies wird oft als „Abverkaufsregelung“ bezeichnet. Diese Zusammenhänge erklärt die IVDR alleine leider nicht. Mehr dazu finden Sie im „Blue Guide“ der EU: https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:52016XC0726(02)&from=DE; Nun sollte auch das Ergebnis der Berechnung Sinn ergeben: Inverkehrbringen bis 25. Mai 2022 und Bereitstellung und Inbetriebnahme anschließend bis 27. Mai 2025 erlaubt. Haken Sie gerne nach, falls Sie noch etwas unklar ist.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Herr Grömminger
Ich habe Fragen, die mit der Zweckbestimmung verbunden sind.
-Insbesondere NGS Labors sind sich nicht sicher, welches Gerät sie in Zukunft für die Sequenzierung verwenden dürfen. NGS Geräte welche die EMV und Niederspannungsrichtlinien erfüllen, und kein RUO Label haben können auch für die Diagnostik verwendet werden. Kann man ein NGS System nicht besser als allgemeinen Laborbedarf bezeichnen?
-Von einem bekannten Hersteller gibt es eine sogenannte DX Version, welches jedoch nach meinem Verständnis ein bei Labors weithin etabliertes RUO Gerät ist, mit einer Zusatzfestplatte mit entsprechender Software für bestimmte FDA und CE-IVD Applikationen. Folglich darf der RUO Modus nicht für die Diagnostik verwendet werden, obwohl er Teil eines Gerätes mit CE-IVD Zertifikat ist?
-Für den Fall das das Labor über ein anderes kommerzielles NGS System verfügt, wäre es ein „off label use“ wenn der kommerzielle Test X nicht explizit darauf validiert ist. Das Labor müsste folglich den Test X auf seinem System selber zum Homebrew Test Y validieren, für den es aber ein kommerzielles Äquivalent, nämlich Test X validiert auf einem anderen System gibt. Wie müsste das akkreditierte Labor hier entscheiden?
Beste Grüße
Andreas
Lieber Andreas Eckelt,
vielen Dank für die spannenden Fragen!
Im NGS-Bereich wird das aktuell aufgrund der Technologieführerschaft durch einen Hersteller dominiert. Ein Hersteller kann, wenn auch nicht notwendig, seine Produkte in der IFU aneinander koppeln und keine andere Kombination erlauben. Gleichzeitig kann er die Research Use Only (RUO) -Funktionalität für die nicht-medizinische Anwendung frei schalten. Sofern die Kennzeichnungsvorgaben eingehalten werden und die Konformität darstellbar ist, sehe ich hier keinen Verstoß gegen geltende Regularien. Die Verwendung eines RUO-Produktes, bzw. RUO-Funktionalitäten erfordern, dass das medizinische Labor die regulatorische Verantwortung übernimmt und die Konformität gewährleistet. Eine Kennzeichnung „for general laboratory use“ hält auch die EU-Kommission gemäß MEDDEV 2.14/2 für sinnvoller, daher würde ich auch dafür plädieren.
Inwiefern jedoch in beiden Fällen Artikel 5 (5) d) durch den Hersteller einklagbar ist und das Labor gezwungen werden kann, das CE-IVD-Markierte Produkt (DX) des IVD-Herstellers zu verwenden ist jedoch noch ungeklärt.
für den letzten Fall in dem das Labor auf ein alternatives NGS-System umsteigt, halte ich es für sinnvoll, wenn medizinische Labore mit Herstellern intensiv zusammenarbeitet um Daten zu Liefern, damit Klasse A IVD-Instrumente konform in Verkehr gebracht werden. So bleibt eine ausreichende Flexibilität für Drittanbieter und für Labore Panel Kits und Lab developed Tests zu entwickeln.
Herzliche Grüße, Sebastian Grömminger
Hallo,
verstehe ich es richtig, dass für sonstige IVDs (gemäß IVDD), die Klasse A – nicht steril nach IVDR sind, gemäß dem Vorschlag zur Änderung der Übergangsfristen weiterhin ab 27.05.2022 die IVDR Anforderungen erfüllen müssen?
Beste Grüße,
Martina
Liebe Frau Marker,
Ja das ist korrekt. Der Vorschlag bietet nur für Produkte, die unter der IVDR von einer Benannten Stelle bewertet werden müssen eine erweiterte Übergangsfrist. Da Klasse-A-Produkte (mit Ausnahme von sterilen Produkten) in reiner Selbsterklärung konformitätsbewertet werden können, sieht die EU offenbar keinen Handlungsbedarf.
Herzliche Grüße,
Sebastian Grömminger
Hallo,
verstehe ich es richtig, dass die neue Produkte, die davor nicht auf dem Markt waren und keine CE-Markierung hatten? Was ist mit den Produkten, die vorher keine Bennate Stellen brauchten, und die jetzt unter Klasse C klassfiziert wurden, gemäß IVDR? Sind die jetzt neue Produkten?
Vielen Dank im Voraus!
MfG
Ron
Lieber Ron,
ja, korrekt. „neue Produkte“ sind diejenigen, die noch nicht vor dem 26. Mai 2022 unter IVDD In Verkehr gebracht wurden (bzw. noch werden).
Diese werden nicht von den vorgeschlagenen verlängerten Übergangsfristen profitieren können. Das Produkt das von der längeren Übergangsfrist profitieren soll, muss zwei Kriterien erfüllen: Es muss noch vor dem 26. Mai 2022 CE-Konformitätserklärt sein und muss unter IVDR in Klasse B, C, D oder A-steril fallen. Je nach klasse gelten dann die unterschiedlichen Fristen.
Beste Grüße,
Sebastian Grömminger
Guten Tag,
„für IVD, für die Hersteller unter IVDD ohne Einbeziehen einer Benannten Stelle die Konformitätserklärung selbst vor dem 26. Mai 2022 ausgestellt haben„. Was genau wurde hier gemeint bei selbst Konformitätserklärung?
Dankeschön!
Schöne Grüße
Mcmillian
Liebe Frau Mcmillian,
die meisten IVDs unter der IVD-Richtline 98/79/EG werden als sogenannte sonstige IVDs gemäß Anhang III in der Verantwortung des Herstellers selbst konformitätsbewertet und konformitätserklärt durch Austellen einer CE-Konformitätserklärung und der CE-Markierung des Produktes. Das ist mit „selbst konformitätserklärt“ gemeint.
Herzliche Grüße,
Sebastian Grömminger
Guten Tag,
wir werden unser Produkt dieses Jahr unter sonstige IVDs (gemäß IVDD), Klasse A – Selbsterklärung Konformitätsbewertung – CE kennzeichnen. Danach fällt unser Produkt laut IVDR in die Klasse C.
Meine Frage;
– wenn wir vor dem 26 Mai 2022 die Zweckbestimmung unseres Produktes ändern, können wir dann wieder eine Konformitätserklärung ohne benannte Stelle durchführen?
– was ist nach dem 26 Mai 2022 – gelten dann die neuen Übergangsfristen – je nach Klasse – in unserem Fall Klasse C?
Vielen dank und beste Grüße
Vorschlag der EU-Kommission zur Anpassung der Übergangsfristen
Liebe Frau Schwarz,
ich schätze, Sie meinen, Sie wollen Ihr „sonstiges“ IVD gemäß IVDD vor dem 26. Mai 2022 mit geänderter Zweckbestimmung konformitätserklären und dieses wird ein Klasse C-Produkt unter der IVDR sein.
Das heißt ja, das können Sie noch ohne Benannte Stelle machen.
Dann können Sie, sofern Sie die Konformität für das geänderte Produkt unter IVDD belegen können UND der Vorschlag der Kommission auch so angenommen wird, die verlängerte Übergangsfrist nutzen.
Ergänzend möchte ich aber betonen: Das geht nur OHNE wesentliche Änderung am Produkt! Unter Einhaltung dieser Voraussetzungen können Sie Ihre (zukünftigen) Klasse-C-Produkte noch bis 26. Mai 2026 unter IVDD-Konformität in Verkehr bringen und dann noch ein Jahr länger bis 26. Mai 2027 „abverkaufen“ (Bereitstellung und Inbetriebnahme). Es ist ein bisschen wie bei „Zurück in die Zukunft“: Sie müssen jetzt wissen, welche Klasse ihr Produkt haben wird, um nach Gültigkeitsbeginn der IVDR noch die geringeren Anforderungen der alten IVDD nutzen zu können.
Herzliche Grüße,
Sebastian Grömminger
Guten Tag Herr Grömminger,
unter 6 b) Spiegelstrich 2 und 3 heißt es: Bescheinigungen … verlieren spätestens am 27. Mai 2025 ihre Gültigkeit. Bedeutet das eine Gültigkeit inklusive 27. Mai (Ablauf 27. Mai 23:59 Uhr) oder exklusive 27.Mai (Ablauf 26.Mai 23:59 Uhr)?
Vielen Dank und beste Grüße
Lieber Herr Graß, wenn die Änderung so umgesetzt wird, lautet die IVDR in Art. 110 abs. 2 in beiden Unterabsätzen zu den ausgestellten IVDD-Zertifikaten wie folgt: „[…] shall become void at the latest on 27 May 2025“. Daraus schließe ich, dass es kein Zertifikat mit einem späteren Datum als der 26. Mai 2025 geben darf. Und falls doch entsprechende Zertifikate ausgestellt werden, verlieren diese die Gültigkeit, sobald das Kalenderdatum 27. Mai 2025 eintritt. Also um 0:00 Uhr. Entsprechend wäre die Gültigkeit exklusive dem 27. Mai 2025.
Herzliche Grüße,
Sebastian Grömminger
Dear Dr. Sebastian,
The Progressive roll-out of the In Vitro Diagnostic Medical Devices Regulation just published (www.https://ec.europa.eu/commission/presscorner/detail/en/ip_21_6965). I just wanted to ask you about this sentence (No change is proposed for CE-marked devices that do not require notified body involvement under the IVD Regulation, or for devices that are ‘new‘, i.e. devices that have neither a notified body certificate nor a declaration of conformity under the current Directive 98/79/EC. For those types of devices, the IVD Regulation will therefore apply from 26 May 2022 as planned.) if it contradicts with what you have already mentioned hier in this discussion.
Thank you in advance!
Regards, Rony
Dear Rony,
Thank you for your fast reaction after the EU press release. We are already working on an own article about the new transitional periods.
To your question: As neither the EU Council nor the EU Parliament have made any changes on the amending Regulation proposed by the EU Commission on Oct. 14, 2021 this sentence will not change and it does not contradict what was discussed here. Thus for Produkts that will fall under class A which are not labeled as „sterile“ Manufacturers have to apply the IVDR from 26th of May next year. The same is true for products which are not yet and will not be conformity assessed under the IVD-Directive 98/79/EG before 26th of May 2022.
With best regards, Sebastian
Guten Tag Herr Grömminger,
In einem Beitrag hatten Sie folgendes geschrieben: „Das Produkt das von der längeren Übergangsfrist profitieren soll, muss zwei Kriterien erfüllen: Es muss noch vor dem 26. Mai 2022 CE-Konformitätserklärt sein und muss unter IVDR in Klasse B, C, D oder A-steril fallen. Je nach klasse gelten dann die unterschiedlichen Fristen“
Meine Fragen dazu lautet nun: Betreffen die verlängerten Übergangsfristen die „in Verkehr Bringung“ genauso wie die „Bereitstellung“? Können daher zukünftige Klasse B und C-Produkte, die bereits unter IVDD zugelassen sind (sonstige IVD bzw. Liste B, Anhang II) noch bis zum jeweiligen Ende der Übergangsfrist (z.B. 26. Mai 2026) unter IVDD-Konformität in Verkehr gebracht werden? Gilt das auch für neue Chargen des Produktes (z.B. neue Charge eines Immunoassays), die nach dem 26. Mai 2022 produziert wurden?
Vielen Dank und Beste Grüße
Sehr geehrte Frau Übner,
ja, mit der neuen Übergangsregelung ist nun auch explizit die Inverkehrbringung von Herstell-Chargen nach dem 26. Mai 2022 erlaubt.
In Ihrem Fall sollten Sie aber bereits über ein Zertifikat einer Benannten Stelle zumindest für das Produkt gemäß Anhang II Liste B unter der IVDD verfügen. dann gilt nur die Verlängerung der Übergangsfrist um 1 Jahr für die Inverkehrbringung: Statt 27. Mai 2024 nun bis 2025. Dazu muss Ihnen die benannte Stelle aber auch ein entsprechendes Zertifikat ausstellen. Wenden Sie sich daher an Ihre Benannte Stelle, um das konkrete Datum auszuhandeln.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Herr Dr. Grömminger,
bei uns stellt sich aktuell die Frage, ob Geräte und Reagenzien, die nach dem 26.05.2022 kein IVD mehr sind (da vom Hersteller nicht als IVD registriert), weiterhin in der Patientendiagnostik eingesetzt werden dürfen.
Sind hier besondere Regelungen z.B. interne Validierung etc. zu beachten?
Vielen Dank für Ihre kurze Rückmeldung.
Lieber Frau Seegerer,
ich kann Ihre Frage noch nicht vollständig überreißen. Ich versuche eine Interpretation:
Der Hersteller lieferte Ihnen bisher IVD-Produkte für das medizinische Labor. Er wird aber keine Neuzulassung (Konformitätsbewertung) unter der IVDR durchführen. Somit wird das Produkt bald keinen CE-IVD-Status mehr haben. Jetzt kommt es darauf an, ob das betreffende Produkt ein allgemeines Labor-Produkt ohne medizinische Zweckbestimmung ist. Dann wäre die Verwendung des nicht-IVD-Produktes nach Prüfung und Bewertung Ihrer labor-internen Validierugns-Dokumentation und einer Risikobewertung möglich. Wenn es sich jedoch um ein Produkt mit konkretem medizinischen Zweck gemäß IVD-Definition (z.B. Diagnose) handelt, sollten Sie sich nach einem alternativen CE-IVD-Produkt umsehen, sonst fallen Sie in die Hersteller-Verantwortlichkeit gemäß Artikel 16 der IVDR. Damit wäre auch Artikel 5 (5) für die Herstellung von In-House-Produkten für Sie relevant. Mehr Informationen dazu finden Sie hier: https://www.johner-institut.de/blog/regulatory-affairs/laboratory-developed-test-ldt/
Beachten Sie aber: Produkte, die in Betrieb genommen wurden, dürfen weiterhin verwendet werden. Bei Reagenzien, wird ihnen wahrscheinlich der Lagerbestand oder das Ablaufdatum bald zum limitierenden Faktor werden. Bei Geräten ist es jedoch ein wenig anders. Da hier die Lebensdauer des Produktes deutlich länger ist, können sie das in Betrieb genommene IVD-Gerät noch bis zum Ende der vom Hersteller angegebenen Lebensdauer verwenden, solange die Untersuchungsmethode noch dem Stand der Technik entspricht.
Mit herzlichen Grüßen,
Sebastian Grömminger
Guten Tag Herr Grömminger,
gemäß IVDR-Verordnung, müss den Barcode auf die Produkte (Klasse D) ab dem 26.06.2023 angebracht werden, ODER? Wenn ich jetzt ein Produkt (Klasse D) habe, welches noch eine gültige IVDD-Beschenigung bis zum Mai 2025 hat. ABER ich möchte es nur bis zu diesem Datum (Mai 2025) auf dem Mrkt behalten und dann nicht mehr nach IVDR. Die Frage jetzt, muss ich trotz ab dem 26.05.2023 den Barcode auf diese Produkte anbringen?
Vielen Dank im Voraus!
Mit freundlichen Grüße
Younis Skaik
Lieber Herr Skaik,
die Anbringung des UDI-Barcodes gilt nur für Produkte die unter der IVDR konformitätserklärt werden. Wenn das Produkt nie unter der IVDR konformitätsbewertet werden soll, ist diese Anforderung für Sie auch nicht anwendbar. Allerdings müssen Sie das Produkt als Legacy Device in der EUDAMED registrieren (Siehe MDGG 2019-5)
Beste Grüße,
Sebastian Grömminger
Lieber Herr Grömminger,
vielen Dank für die schnelle Rückmeldung! Ich wollte nur noch fragen, ob eine Deadline gibt, für die Anbringung des Barcodes von Legacy Devices (Klasse C oder D). Ich gehen davon aus, dass für Klasse C ist schon ab dem 26.05.2025?
Vielen Dank im Voraus!
Schöne Grüße
Younis Skaik
Lieber Herr Skaik,
Für Legacy Devices gilt diese Anforderung erst ab der Konformitätserklärung unter IVDR wenn das der Fall ist, müssen Sie für dein Klasse D-Produkt Ab 26. Mai 2023 und für ein Klasse C-Produkt ab 26. Mai 2025 die UDI-Codes anbringen Siehe Artikel 113 abs. 3 e) mit Bezug auf Art. 24 (4). Für das gemäß IVDD (98/79/EG) konformitätserklärte Produkt innerhalb der verlängerten Übergangsfristen können Sie freiwillig die UDI Codes anbringen.
Ich hoffe, ich konnte damit Ihre Frage klären.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Herr Prof. Johner,
im Pathologischen Labor kommen im Rahmen der Diagnostik regelmäßig Lichtmikroskope zum Einsatz. Richtet sich deren Einstufung in Risikoklassen grundsätzlich nach dem Untersuchungsziel mit dem höchsten Risiko für den Patienten? Im Falle einer Diagnostik eines ansteckenden Erregers, der eine lebensbedrohliche Krankheit mit hohem Verbreitungsrisiko verursacht, also pauschal in Klasse D, wenngleich die überwiegende Anzahl der mikroskopischen Beurteilungen in der Klasse C angesiedelt sind?
Ergänzend noch die Frage: Es ist nicht das Mikroskop, das die Diagnose stellt, sondern der Pathologe, also ein Mensch, der nicht IVD sein kann. Müsste nach dieser Überlegung nicht jedes Mikroskop in Klasse A einzustufen sein?
Herzlichen Dank für Ihre Antwort und beste Grüße nach Konstanz,
Johannes Hädrich (Freiburg)
Lieber Herr Hädrich,
vielen Dank für Ihre spannende Frage. Bei der Mikroskopie ist es das Zusammenspiel von Probenpräparation, Färbemethode, Mikroskopie und ggf. Software-Auswertung und der Pathologe oder die Pathologin, der/die das Ergebnis im Patientenkontext interpretiert. Jedes Produkt, das dabei eingesetzt wird muss entsprechend der Zweckbestimmung und des mit dem Produkt verbundenen Risiko klassifiziert werden. Es kann durchaus sein, dass das Mikroskop, wenn es ein allgemeines Instrument für die Diagnostik ist und „nur“ die Technologie zur mikroskopischen Bildvergrößerung ermöglicht in Klasse A fällt über Regel 5. Wenn die Färbemethode je nach Analyt, der gefärbt wird mit einer lebensbedrohlichen Erkrankung assoziiert wird, die ggf. sogar ein hohes Verbreitungsrisiko hat, ergeben sich daraus Risiken bei falschen Ergebnissen. Diese Färbemethoden fallen daher je nach Zweckbestimmung in höhere Klassen. Alternativ oder ergänzend kann bzw. sollte eine Auswerte-Software, die eigenständig Ergebnisse interpretiert auch in eine höhere Klasse fallen, entsprechend ihrer Zweckbestimmung.
Ich hoffe, ich konnte Ihre Frage damit beantworten.
Herzliche Grüße,
Sebastian Grömminger
Lieber Herr Grömminger,
vielen Dank für Ihre informative Antwort die noch einmal aufzeigt, dass das Einstufen in Risikoklassen nicht immer so straight-forward zu sein scheint, wie wir Anwender dies vielleicht hoffen mögen.
Im Rahmen einer Tumordiagnostik (dass ein Tumor vorliegt, weiß ich vorab allerdings nicht immer, je nach klinischer Verdachtsdiagnose !) im pathologischen Labor käme dem reinen Färbeprozess gewiss ein Risiko der Klasse C zu. Die abschließende mikroskopische Beurteilung des Befundes wird ausschließlich durch den Pathologen vorgenommen. Nun könnte das Mikroskop vielleicht nicht fokussiert oder ohne Beleuchtung oder bei unzureichender Vergrößerung verwendet werden (alles ziemlich unwahrscheinlich). Wenn wir allerdings von einem z.B. im Sinne der DIN EN ISO/IEC 17020 funktionsbereiten Gerät (Mikroskop) ausgehen, was ich für selbstverständlich halte, dann kann nur dem Pathologen selbst ein Fehler bei der Diagnose unterlaufen. Das Risiko eines fehlerhaften Befundes wäre bei Vorliegen eines Tumors, der nicht erkannt wird, weil es dem Pathologen z.B. an Erfahrung mangelte, oder er den Tumor falsch einordnet, entsprechend erhöht. Nur ist eben der Pathologe kein IVD.
Ich käme hier zu dem Schluss, dass Produkte, die im Rahmen dieser Tumordiagnostik verwendet werden, der Klasse C zuzuordnen sind, nicht aber das letztlich zur Diagnose verwendete Mikroskop. Würden Sie mir zustimmen?
Mit besten Grüßen
Johannes Hädrich
Lieber Herr Hädrich,
das ist absolut korrekt, der Pathologe ist der kritische Faktor. Produkte mit einem besonders hohen klinischen nutzen können Risiken, die durch menschliche Fehler entstehen deutlich reduzieren, wenn sie denn nach dem Stand der Technik entwickelt und hergestellt werden. Ob die Färbelösung dann zwingend Klasse C sein muss, hängt von Ihren Eigenschaften und der Zweckbestimmung ab. Es ist ein riesengroßer Unterschied, ob der Pathologe ein DAPI-stain oder eine HER2-FISH macht und für welchen Zweck, bei welcher Indikation und aus welchem Probenmaterial das erfolgt. Das Mikroskop bleibt in Klasse A, wenn es rein der Bilderfassung dient. Wenn aber eine Software (Zubehör) zum Mikroskop der Interpretation der Ergebnisse dient, geht das meist nicht mehr.
Gerne können wir uns Ihren Fall genauer anschauen und bei der Klassifizierung unterstützen. Schreiben Sie uns gerne an.
Mit besten Grüßen,
Sebastian Grömminger
Sehr geehrter Herr Prof. Johner,
Albrecht Stenzinger schreibt in seinem sehr interessanten Beitrag „Einfluss der neuen In-vitro-Diagnostik-Regulation (IVDR) der Europäischen Union auf die Pathologie. Was ist wichtig?“ https://doi.org/10.1007/s00292-020-00867-9 folgendes:
„Interessant für die Pathologie ist in diesem Kontext, dass alle Tests, die in ihrem „intended purpose“ (beabsichtigten
Zweck) das Wort „Krebs“ beinhalten, automatisch in die Risikoklasse C (oder höher) einsortiert werden. Zudem fallen
nun auch generell Reagenzien der histologischen Verarbeitung, zum Beispiel Pufferlösungen, histologische Färbungen
und sogar Formalin, formal in den Regulationsbereich der IVDR und werden hier dann der Klasse A zugeordnet.“
Wenngleich mir Regel 5 bekannt ist, erscheint mir dies dennoch widersprüchlich. Wenn eine Färbelösung, z.B. Hämatoxilin-Eosin, im Rahmen einer Tumordiagnostik verwendet wird, müsste sie nicht in Klasse C eingestuft werden?
Beste Grüße
Johannes Hädrich
Lieber Herr Hädrich,
Wenn ein Hersteller ein Produkt für die Krebsdiagnostik vorsieht und diesen „Claim“ setzen möchte, ja dann fällt das Produkt in Klasse C. Es ist aber eine strategische Entscheidung des Herstellers das zu tun. Eine Pufferlösung, die alleine dafür dient, Probenmaterial auf die nachfolgende Untersuchung vorzubereiten, auch wenn diese Untersuchung am Ende der Krebsdiagnose dient, muss das kein Klasse-C-Produkt sein, sondern fällt meist Klasse A über Regel 5 – sofern die Zweckbestimmung entsprechend formuliert ist. Andersherum könnte man ja auch behaupten, jeder DNA-Extraktions-Kit dient der genetischen Untersuchung und wäre damit auch Klasse C. Auch das ist unnötig und kann über Regel 5 in Klasse A gehalten werden. Das sieht auch die MDCG so (https://ec.europa.eu/health/system/files/2022-01/md_mdcg_2020_guidance_classification_ivd-md_en.pdf). Ich gehe sogar einen Schritt weiter und sage, bestimmte Puffer sind gar kein IVD. Sie fallen nicht unter die Definition eines IVD und damit auch nicht unter die IVDR und das kann auch für Hämatoxilin-Eosin gelten. Lesen Sie gerne dazu diesen Artikel: https://www.johner-institut.de/blog/regulatory-affairs/for-research-use-only-ruo/ In Kapitel 4 geht es um den allgemeinen Laborbedarf.
Wir unterstützen Hersteller und medizinische Labore bei diesem Vorgehen gerne mit der IVD-Zulassungsstrategie oder mit strategischen Workshops.
Kommen Sie gerne auf uns zu.
Herzliche Grüße,
Sebastian Grömminger
Lieber Herr Grömminger,
herzlichen Dank für Ihre kompetenten, nachvollziehbaren und vollständigen Antworten auf meine Fragen, die mir den Grundgedanken der Klassifizierungen nun viel besser vermitteln und in der Sache tatsächlich sehr gut weitergeholfen haben!
Mit besten Grüßen
Johannes Hädrich
.
Sehr geehrter Herr Prof. Johner,
ich habe 2 Fragen im Zusammenhang mit der IVDR:
1. Inwiefern muss bei Verwendung von Pipettierrobotern die IVDR beachtet werden? Wir benutzen eine an sich IVDR konforme Methode, die bisher manuell pipettiert wurde und möchten das Ganze nun gerne automatisieren. Muss dann der Gesamtprozess als in house Verfahren validiert werden und wenn ja, in welchem Umfang?
2. Wenn vom Hersteller eines Kits nicht ausdrücklich auf eine bestimmte Methode zur Nukleinsäure-Extraktion verwiesen wird, kann dann einfach frei ein Extraktionsverfahren gewählt werden (natürlich ebenfalls IVDR-konform) oder muss ich die Kombination dennoch validieren?
Vielen Dank im Voraus und viele Grüße,
Anika Penzel
Liebe Frau Penzel,
Zu Ihrer 1. Frage:
Sofern der Assay, den Sie im Labor benutzen, ein CE-markiertes IVD ist und Sie nicht von der Zweckbestimmung und der Gebrauchsanweisung abweichen, stellen Sie kein In-house IVD her und fallen selbst somit nicht unter die IVDR. Sollte der Hersteller des CE-IVD jedoch die Nutzung von Pipettierrobotern in der Gebrauchsanweisung ausschließen, würden Sie bei Benutzung eines Roboters unter die IVDR fallen und hätten ein In-house IVD. Nichts desto trotz müssen Sie natürlich auch bei Verwendung nach Herstellervorgaben über eine Verifizierung des Gesamtprozesses die sichere Verwendung des Roboters bestätigen, wie es auch ein QM-System nach ISO 15189 bzw. Rili-BÄK einfordert.
Zu Frage Nummer 2:
In diesem Fall können Sie frei wählen. Sie müssen natürlich sicherstellen, dass die DNA die geforderte Qualität aufweist. Und auch hier müssen Sie den Gesamtprozess zumindest verifizieren.
Lesen Sie zu In-house IVD auch gerne unseren entsprechenden Artikel.
Liebe Grüße,
Ulrich Hafen
Sehr geehrter Hr. Johner,
Könnten Sie uns bitte kurz mitteilen, woraus sie in der IVDR abgeleitet haben, dass eine Zertifizierung des QM-Systems durch eine Benannte Stelle für (unsterile) Klasse A Produkte nicht erforderlich ist (s. o.) „Alle Hersteller benötigen ein QM-System, u. a. für die Entwicklung, Produktion und Überwachung der Produkte im Markt. Mit Ausnahme von Produkten der Klasse A ist eine Zertifizierung notwendig“.
Wir konnten bezüglich der Bewertung des QM-Systems in der IVDR keine Ausnahme finden.
Vielen Dank im Voraus für Ihre Bemühungen.
Grüße
Hartmut Simon
Sehr geehrter Herr Simon,
wahrscheinlich sind Sie auf einen Fehler gestoßen. Ich wollte diese Aussage nicht getroffen haben. Können Sie mir helfen, diese zu finden?
Im Beitrag finde ich diese Aussage:
Mit Ihrer Hilfe bekommen wir das sicher schnell gelöst.
Besten Dank und herzliche Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielleicht kann ich helfen. Die betreffende Aussage findet sich hier (ganz oben auf dieser Seite, unter 3.):
3. Überblick über die Anforderungen der IVDR
Nachdem der Aufbau und die Struktur der IVDR nun bekannt sind, gehen wir im Folgenden auf die Anforderungen der Hersteller (3 a)) und auf die Anforderungen der anderen Wirtschaftsakteure wie Händler, Importeure und Labore (3 b)) ein.
a) Anforderungen an die Hersteller selbst und deren IVD
Tabelle 3 zeigt die produktübergreifenden Anforderungen an die Hersteller von IVD. Strebt der Hersteller für eines seiner IVD eine Konformität nach IVDR an, muss er ein Qualitätsmanagementsystem etablieren und über den gesamten Lebenszyklus der Produkte aufrechterhalten. Hier gilt die ISO 13485 als harmonisierte Norm zu beachten.
Produktübergreifende Anforderungen
Anforderung der IVDR
(Mit Links zu Fachartikeln) Erläuterung
Qualitätsmanagementsystem Alle Hersteller benötigen ein QM-System, u. a. für die Entwicklung, Produktion und Überwachung der Produkte im Markt. Mit Ausnahme von Produkten der Klasse A ist eine Zertifizierung notwendig.
Mit besten Grüßen, Johannes Hädrich
Sehr geehrter Hr. Prof. Johner,
wie schon von Hr. Hädrich beschrieben, findet sich diese Aussage weiter oben auf dieser Webseite unter dem Punkt „produktübergreifende Anforderungen“. Uns interessiert insbesondere, ob für unsterile Klasse A Produkte auf die Zertifizierung des QMS verzichtet werden kann. Nach unserer Interpretation der IVDR ergibt sich dieses aus Artikel 48 (10) in Verbindung mit Artikel 17. Für sterile Klasse A Produkte träfe dieses allerdings nicht zu.
Grüße
H. Simon
Lieber Herr Simon, lieber Herr Hädrich,
vielen Dank für Ihre Kommentare und Fragen bezüglich unserer Überarbeitung des Fachartikels zur IVDR.
Wir haben uns die Textpassage zu den Anforderungen an das QM-System von Herstellern mit Produkten der Risikoklasse A noch einmal im Detail angesehen und haben den Abschnitt, wie folgt beschrieben, präzisiert.
Grundsätzlich stellt die IVDR nach Artikel 10 Absatz 8 an alle Hersteller die Anforderung ein Qualitätsmanagementsystem zu etablieren und danach zu arbeiten. Wie Herr Simon bereits richtig geschildert hat, führt Artikel 48 Absatz 10 weiter aus, dass die Konformität von Klasse A Produkten selbst durch den jeweiligen Hersteller durch eine EU-Konformitätserklärung nach Artikel 17 erklärt wird. Dabei ist keine Benannte Stelle involviert.
Aber Achtung, für sterile Produkte der Klasse A muss der Hersteller, die in Anhang IX oder Anhang XI genannten Verfahren anwenden und eine Benannte Stelle für das Konformitätsbewertungsverfahren hinzuziehen. Deren Involvierung ist jedoch begrenzt auf die Aspekte, die mit der Herstellung, der Sicherung und der Aufrechterhaltung der sterilen Bedingungen des Produkts zusammenhängen (siehe auch Anhang XII Kapitel I, Nr. 7).
Aus diesen Anforderungen folgt, dass Hersteller von Klasse A IVD ein QM-System etablieren und leben müssen, die Konformität ihrer Produkte aber selbst erklären. Eine Konformitätsbewertung des QM-Systems durch eine Benannte Stelle erfolgt nicht. Eine Behörde kann beispielweise im Rahmen der Marktüberwachung das QM-System eines Klasse-A-Herstellers prüfen.
Unabhängig von einer Konformitätsbewertung durch eine Benannte Stelle, kann ein Hersteller sein QM-System durch einen Dienstleister gemäß ISO 13485 zertifizieren lassen. Diese ISO 13485-Zertifizierung dient manchen Herstellern als Qualitätsmerkmal. Im alltäglichen Sprachgebrauch kommt es manchmal zur unpräzisen Begriffsverwendung – so ist auch uns das passiert.
Wir haben die Textpassage nun angepasst und mit folgendem Wortlaut präzisiert: „Mit Ausnahme von unsterilen Produkten der Klasse A wird das QM-System von den Benannten Stellen im Rahmen des Konformitätsbewerbungsverfahren geprüft. Bei sterilen Produkten der Klasse A beschränkt sich die Involvierung einer Benannten Stelle auf „die Aspekte der Herstellung, die mit der Erreichung und dem Erhalt des sterilen Zustands zusammenhängen“ (s. IVDR, Art. 48 (10) und Anhang XII, Kapitel I, Nr. 7.)“
Mit Einführung der IVDR wird ein deutlich größerer Anteil der QM-Systeme von Herstellern während des Konformitätsbewertungsverfahren durch Benannte Stellen geprüft, daher kann die Zahl der (zusätzlichen) Zertifizierungen des QM-Systems gemäß ISO 13485 abnehmen.
Ich hoffe ich konnte Ihre Frage bezüglich des QM-Systems von Klasse A IVDs beantworten. Sollten Sie weitere Fragen zu Ihrer unternehmensspezifischen Umsetzung haben, nutzen Sie gern unser Kontaktformular für eine passgenaue Unterstützung.
Herzliche Grüße,
Kai Eder