
Die EU-Verordnungen MDR und IVDR stellen präzise Anforderungen an Importeure. Sie definieren auch, wer ein Importeur ist.
Nicht jede Einfuhr eines Produkts in die EU stellt einen Import dar. Und andererseits haben Firmen, die Medizinprodukte importieren, nicht nur die Anforderungen an Importeure zu erfüllen.
Sie finden weiter unten eine kostenlose Checkliste für Importeure zum schnellen Prüfen und Sicherstellen der Konformität.
1. Was ein Importeur ist
a) Definition des Begriffs „Importeur“
MDR und IVDR definieren einen Importeur wie folgt:
„jede in der Union niedergelassene natürliche oder juristische Person, die ein Produkt aus einem Drittland auf dem Unionsmarkt in Verkehr bringt;“
MDR Artikel 2 (33), IVDR Artikel 2
b) Definition des Begriffs „Inverkehrbringen“
„Inverkehrbringen“ heißt im Englischen „placing on the market“. Was darunter zu verstehen ist, erläutert die EU im sogenannten Blue Guide:
A product is placed on the market when it is made available for the first time on the Union market.
Blue Guide
Gemeint ist das Verfügbarmachen eines konkreten Produkts, das z. B. durch eine UDI-PI identifiziert werden kann, beispielweise ein EKG mit einer bestimmten Seriennummer. Gemeint ist nicht das Verfügbarmachen eines Produkttyps (wie das EKG „Cardiomaster NT“), der durch die UDI-DI identifiziert werden könnte.
Die Definition im Blue Guide stimmt mit der Definition aus MDR und IVDR gut überein:
„die erstmalige Bereitstellung eines Produkts, mit Ausnahme von Prüfprodukten, auf dem Unionsmarkt;“
MDR Artikel 2 (28), IVDR Artikel 2
Beachten Sie weitere wichtige Hinweise im Artikel zur Inverkehrbringung.
c) Abgrenzung zum Händler
Der Blue Guide stellt klar, dass nur Hersteller und Importeure Produkte in Verkehr bringen können.
The operation [Anm: gemeint ist das Inverkehrbringen] is reserved either for a manufacturer or an importer, i.e. the manufacturer and the importer are the only economic operators who place products on the market. When a manufacturer or an importer supplies a product to a distributor or an end-user for the first time, the operation is always labelled in legal terms as ‘placing on the market’. Any subsequent operation, for instance, from a distributor to distributor or from a distributor to an end-user is defined as making available.
Blue Guide
Ein Importeur kauft i. d. R. ein Medizinprodukt von einem Hersteller (außerhalb der EU) und verkauft es entweder an einen Händler oder einen Endabnehmer.
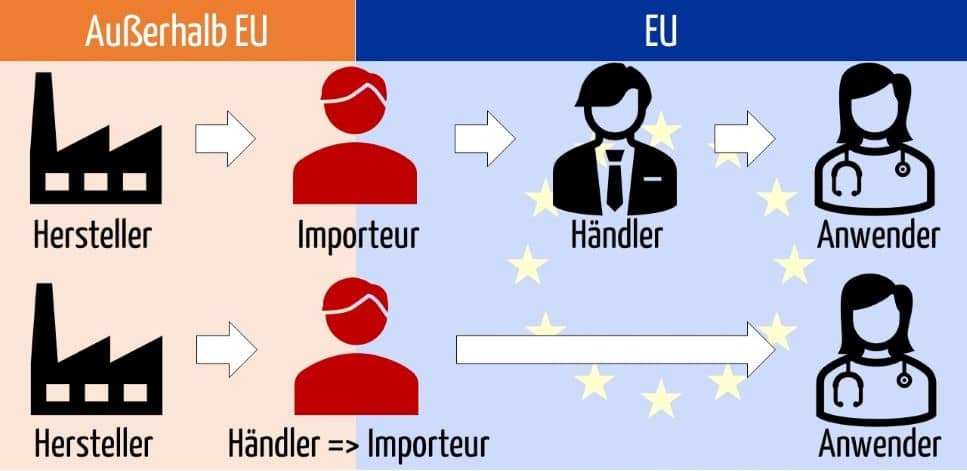
Die MDR unterscheidet zwischen Händlern und Importeuren. Dabei können Importeure als ein Sonderfall der Händler angesehen werden.
d) Abgrenzung zum Hersteller
Importeure, die Produkte verändern oder unter eigenem Namen in den Verkehr bringen, übernehmen Herstellerpflichten. Artikel 16 der MDR legt die Fälle fest, bei denen diese zusätzlichen Pflichten entstehen.
Der Artikel zu den Händlern nennt und erklärt die zusätzlichen Pflichten dieser Wirtschaftsakteure.
e) Wesentliche und nicht wesentliche Faktoren
Wesentlich ist, dass
- der Importeur seinen Sitz innerhalb der EU hat,
- eine Überlassung stattfindet (zwischen dem Importeur und einer weiteren natürlichen oder juristischen Person) und es ein Angebot oder eine Vereinbarung darüber gibt.
Nicht wesentlich ist hingegen, ob
- die Vereinbarung schriftlich oder mündlich erfolgt,
- die Überlassung entgeltlich oder unentgeltlich erfolgt,
- eine Übereignung, d. h. Übertragung von Eigentum stattfindet. Möglich sind auch die Übertragung des Besitzes oder sonstiger Rechte (ausgenommen Rechte des geistigen Eigentums), z. B. durch Verleihung.
f) Was nicht als Import zählt
Der Blue Guide nennt einige Beispiele, bei denen kein Import vorliegt:
- Ein Endverbraucher kauft ein Produkt außerhalb der EU und bringt es zum „Eigengebrauch“ in die EU.
- Ein Hersteller mit Sitz innerhalb der EU fertigt ein Produkt außerhalb der EU, aber unter dem Dach des eigenen QM-Systems, und bringt dieses danach in die EU.
- Ein Produkt wird zum Testen (Verifizieren, Validieren) in die EU gebracht.
- Ein Produkt wird zu einer Messe oder Ausstellung in die EU gebracht. Das setzt voraus, dass dieses Produkt entsprechend gekennzeichnet ist.
2. Was MDR und IVDR von Importeuren verlangen
a) Sich in der EUDAMED registrieren
Bevor Importeure Produkte importieren, müssen sie sich registrieren. Dies verlangen Artikel 30 und Artikel 31 sowie der Anhang VI der MDR. Die Anforderungen der IVDR sind identisch.
Die Eintragungen sind öffentlich sichtbar und müssen bei Bedarf aktualisiert und alle zwei Jahre bestätigt werden.
b) Sich auf der Produktkennzeichnung angeben (lassen)
Artikel 13 der MDR verlangt von den Importeuren, „auf dem Produkt oder auf seiner Verpackung oder auf einem dem Produkt beiliegenden Dokument ihren Namen, ihren eingetragenen Handelsnamen oder ihre eingetragene Handelsmarke, ihre eingetragene Niederlassung und die Anschrift anzugeben, unter der sie zu erreichen sind“.
c) Die Produkte prüfen
MDR und IVDR verpflichten die Importeure dazu, die Produkte zu prüfen:
- CE-Kennzeichnung ist vorhanden
- Konformitätserklärung ist ausgestellt
- Bevollmächtigter ist benannt
- Produkt ist MDR-/IVDR-konform gekennzeichnet
- Gebrauchsanweisung (so vorgeschrieben) liegt bei
- UDI ist für Produkt vergeben
Es gibt aber keine generelle Prüfpflicht. So muss der Importeur die Medizinprodukte nicht testen.
Umstritten ist derzeit, ob der Importeur eine 100-Prozent-Prüfung der oben genannten Aspekte durchführen muss. Die MDR nennt zwar im Gegensatz zu den Händlern keine Ausnahmeregelung (z. B. Stichprobenverfahren); man geht aber mit Bezugnahme auf den Blue Guide davon aus, dass eine 100-Prozent-Prüfung nicht gewollt war.
d) Zur Kommunikation zwischen Herstellern, Anwendern und Behörden beitragen
Die Importeure sind ein wesentlicher Knoten in der Kommunikation zwischen Herstellern, Anwendern und Behörden. Sie müssen dazu beitragen, dass diesen alle Informationen verfügbar sind, um die Sicherheit von Patienten zu gewährleisten bzw. bei Risiken schnell handeln zu können:
- Den Hersteller oder dessen Bevollmächtigen informieren, wenn er annimmt, dass das Produkt nicht die MDR erfüllt (Artikel 13(1) und Artikel 13(7))
- Bei schwerwiegender Gefahr die Behörden informieren (Artikel 13(2))
- Beschwerden von Kunden den Herstellern, dem Bevollmächtigte und den Händlern zur Verfügung stellen (Artikel 13(6) und Artikel 13(8))
e) Übergangsfristen
Die MDCG stellt in MDCG 2021-25 klar, dass die Wirtschaftsakteure ihren Pflichten nach MDR auch für die Legacy-Devices (Artikel 120, gültiges MDD-Zertifikat) nachkommen müssen. Die MDCG listet für jeden Wirtschaftsakteur die entsprechenden Artikel der MDR auf, die er beachten muss. Bei den Importeuren sind das die Anforderungen des Artikels 13:
- Artikel 13(2) letzter Abschnitt: Hier geht es um die Informationspflichten im Rahmen der Post-Market Surveillance.
- Artikel 13(4): Die Importeure prüfen, dass das Produkt in der EUDAMED registriert ist.
- Artikel 13(6): Die Importeure führen ein Register an Beschwerden.
- Artikel 13(7): Auch hier geht es um die Informationspflichten und das Mitwirken bei den Korrekturmaßnahmen.
- Artikel 13(8): Auch hier geht es um die Informationspflichten und das Mitwirken bei den Korrekturmaßnahmen.
- Artikel 13(10): Dieser Absatz betrifft die Kooperation der Hersteller mit den Behörden.
3. Was Importeure jetzt tun sollten
a) Checkliste nutzen
Importeure sollten die Checkliste für Importeure des Johner Instituts durchgehen. Damit gelingt es schnell und einfach, die Konformität mit den Anforderungen der MDR bzw. IVDR zu prüfen und mögliche Abweichungen zu beseitigen. Das verschafft Rechtssicherheit und vermeidet Sanktionen durch Behörden oder Gerichte.

Sie können die Checkliste hier herunterladen:
b) Sofort handeln
Für Importeure von Legacy-Produkten gibt es keine Übergangsfrist. Sie müssen seit dem 26.05.2021 die Anforderungen der MDR erfüllen.
c) Prüfen, ob weitere Anforderungen zu erfüllen sind
Wenn Importeure an den Produkten Änderungen vornehmen, dann müssen sie die Anforderungen des Artikels 16 befolgen. Das trifft beispielsweise zu, falls sie
- Produkte unter eigenem Namen in den Verkehr bringen,
- die Zweckbestimmung ändern oder
- die Gebrauchsanweisung übersetzen.
Unter diesen Umständen gelten auch für Importeure die UDI-Anforderungen, die die Leitlinie MDCG 2018-6 weiter ausführt.
4. Sonderfall Reimport von Medizinprodukten und IVD
a) Was Reimporte sind
Reimporte sind Importe von Waren, die zuvor exportiert wurden. Im Kontext dieses Artikels geht es um Medizinprodukte, die in die EU (re-)importiert werden, nachdem sie zuvor aus der EU exportiert wurden.
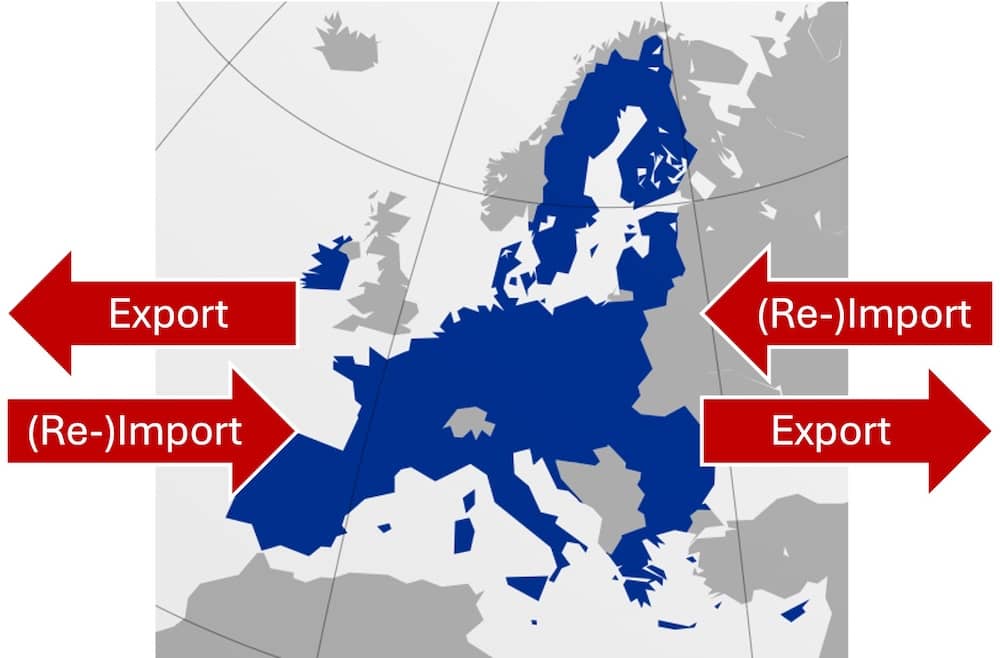
Reimporte sind bei Autos und Medikamenten üblich, weil Hersteller in verschiedenen Ländern unterschiedliche Preise erzielen können. Die Reimporteure möchten von diesen Preisdifferenzen profitieren.
b) Rechtliche Bewertung der Reimporte von Medizinprodukten/IVD
MDR und IVDR kennen das Konzept des Reimports nicht. Folglich unterscheiden sie nicht zwischen Importen und Reimporten von Medizinprodukten (bzw. IVD). Allerdings differenzieren sie zwischen dem erstmaligen Bereitstellen und dem nicht erstmaligem Bereitstellen. Deshalb sind zwei Fälle zu unterscheiden:
- Ein Medizinprodukt bzw. IVD wurden zuerst in der EU bereitgestellt, dann in ein Nicht-EU-Land exportiert und anschließend in die EU reimportiert. In diesem Fall unterliegt die importierende Person nicht den Importeurspflichten der MDR bzw. IVDR, da eine Inverkehrbringung in der EU bereits stattgefunden hat.
- Ein Medizinprodukt bzw. IVDR wurde direkt in ein Nicht-EU-Land exportiert und anschließend in die EU (re-)importiert und dort bereitgestellt, z. B. an einen Anwender verkauft. Damit liegt ein Import im Sinne der MDR bzw. IVDR vor. Der Importeur unterliegt den Importeurspflichten.
Aus Sicht von MDR und IVDR zählt ein Weiterverkauf eines Medizinprodukts, das zuerst in einem anderen EU-Mitgliedsstaat in den Verkehr gebracht wurde, in einen anderen EU-Mitgliedsstaat nicht als Reimport.
Beispiel: Ein deutscher Hersteller bringt sein Produkt in Frankreich in den Verkehr. Von dort wird es von einem Händler nach Deutschland verkauft, z. B. an ein deutsches Krankenhaus. Diesen Handel betrachten MDR und IVDR nicht als (Re-)Import.
c) Sicherheit beim Reimport von Medizinprodukten
Gelegentlich wird die Frage diskutiert, wie es um die Sicherheit von reimportierten Medizinprodukten bestellt sei. Es liegen keine verlässlichen Zahlen vor, die eine Aussage stützen, dass reimportierte Medizinprodukte unsicher seien.
Unabhängig davon, wie die Medizinprodukte in die EU gelangen: Alle unterliegen denselben gesetzlichen Anforderungen. Insbesondere die Medizinprodukte der Klassen IIa und höher (bzw. IVD Klasse B und höher) müssen ein Konformitätsbewertungsverfahren durchlaufen, an dem eine europäische Benannte Stelle beteiligt ist. Das schließt in der Regel eine Überprüfung des QM-Systems des Herstellers und der Technischen Dokumentation des Produkts ein.
5. Fazit
MDR und IVDR haben die Anforderungen an Importeure deutlich erhöht. Damit möchte die EU die Nachverfolgbarkeit der Produkte gewährleisten und die Gefahren für Patienten verringern, die von gefälschten und nicht-konformen Produkten ausgehen.
Für die Importeure bedeutet dies zusätzlichen Aufwand, insbesondere, um mit einem Qualitätssicherungssystem die gesetzeskonforme Prüfung der Produkte und die Kommunikation mit Behörden, Herstellern, Händlern und Dritten zu gewährleisten.
Die Checkliste des Johner Instituts hilft Ihnen, um schnell die eigene Konformität zu bestimmen, Abweichungen zu erkennen und diese zu beseitigen.
Änderungshistorie
- 2024-12-24: Kapitel 4 eingefügt
- 2021-10-25: Kapitel 2.e) mit den Übergangsfristen gemäß MDCG 2021-25 eingefügt
- 2023-07-10: Überarbeitung des Kapitel 1.e) Klarstellung zum Eigentumsübertrag als Voraussetzung



Sehr geehrte Damen und Herren,
folgender fikiver Fall:
Ein deutsches Unternehmen möchte Schnelltest aus China importieren.
Die Schnelltests zur Eigenanwendung haben das Konformitätsverfahren durchlaufen und besitzen ein CE Zeichen inkl. 4stelliger Nr der Benannten Stelle.
Können diese Schnelltests ohne Probleme und Auflagen importiert werden?
Oder gibt es an das deutsche Unternehmen Anforderungen die es als Importeur zu erfüllen gibt? Das Unternehmen hat zuvor keine IVD Produkte importiert.
Vielen Dank.
Die wesentlichen Anforderungen an die Importeure beschreibt dieser Artikel. Weitere Hinweise und Antworten finden Sie in meiner Antwort auf Ihre Frage unter dem Artikel zu den Medizinprodukten der Klasse 1.
Guten Tag Herr Johner
Eine Frage zu:
b) Sich auf der Produktkennzeichnung angeben (lassen): …oder einem dem Produkt beiliegenden Dokument ihren Namen….
Ist mit „beiliegendem Dokument“ auch ein Lieferschein gemeint, oder bezieht sich dieser Punkt auf mindestens die IFU?
Besten Dank für klärende Informationen
Jürgen Kaufmann
Guten Tag Herr Kaufmann,
das MCDCG 2021-12 sagt dazu: „‘Accompanying documentation‘ containing the importer’s details, may be separate from or
affixed to the individual device, as long as it accompanies the individual device throughout
the supply chain and reaches the end user…The importer may choose the appropriate accompanying
documentation, as long as it reaches the end user. Examples may include a sticker affixed to
the label or a leaflet. “
Beim Lieferschein ist die Frage, ob dieser wirklich den Endbenutzer erreicht und eine Verknüpfung mit dem jeweiligen Medizinprodukt gegeben ist.
Freundliche Grüße
Luca Salvatore
Guten Tag,
benötigen wir als Importeur von Klasse 1 Produkten wie Rollatoren ein QMS ?
Guten Tag,
Importeure von Medizinprodukten einschließlich Klasse I-Produkten benötigen im Regelfall kein QMS, ausgenommen die in Artikel 16 genannten Konstellationen (Importeur übernimmt Herstellerverantwortung, Importeur verpackt um oder übersetzt).
Freundliche Grüße
Luca Salvatore
Guten Tag Herr Johner,
wir sind Importeur und Händler von Produkten eines asiatischen Herstellers. Trotz umfangreicher Recherchen bleibt unklar, wann die Produkte als In Verkehr gebracht gelten.
Bereits nach erfolgreichem Durchlaufen der Zollabwicklung bzw. spätestens sobald die Produkte bei uns im Lager liegen. Oder sind die Produkte erst in dem Moment In Verkehr gebracht, wenn wir sie auf dem EU Markt (anderen Händlern oder Endkunden) zur Verfügung stellen, d.h. weiter verkaufen.
Wen betrachtet der Gesetzgeber in diesem Fall offiziell als Inverkehrbringer? Den Hersteller oder uns/den Importeur bzw. hängt dies vom vereinbarten Gefahrenübergang, also den vereinbarten Incoterms ab?
Die Frage stellt sich uns im Zusammenhang mit Legacy Devices, zumal dort das Datum der Inverkehrbringung relevant ist.
Herzlichen Dank für Ihre Antwort und freundliche Grüße
Andrea Kiecker
Guten Tag Frau Kiecker,
üblicherweise fällt der Zeitpunkt der Überlassung zum zollrechtlich freien Verkehr mit dem des Inverkehrbringens zusammen. Diese Erläuterung finden Sie im EU Blueguide (https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52016XC0726(02)&from=DE):
„Before they can reach the end-user in the EU, products coming from countries outside the EU will be presented to
customs under the release for free circulation procedure. The purpose of release for free circulation is to fulfil all import
formalities so that the goods can be made available on the EU market like any product made in the EU. Therefore when
products are presented to customs under the release for free circulation procedure, it can generally be considered that
the goods are being placed on the EU market and so they will need to be compliant with the applicable Union harmonisation legislation. However, it may also be the case that the release for free circulation and the placing on the market do
not take place at the same time. The placing on the market is the moment in which the product is supplied for
distribution, consumption or use for the purposes of compliance with Union harmonisation legislation. Placing on the
market can take place before the release for free circulation, for example, in the case of online sales by economic
operators located outside the EU, even if the physical check of the compliance of the products can take place at the
earliest when they arrive at the customs in the EU. Placing on the market can also take place after release for free
circulation. “
Laut Definition der MDR bringt immer der Importeur, im Falle von Herstellern in einem Drittland, das Produkt in der Union in den Verkehr.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
wenn es zu einem Importprodukt Ersatzteile gibt, müssen diese auch mit dem Importeur gekennzeichnet sein? Die Kennzeichnungsregeln der MDR gelten ja nicht für Ersatzteile- somit auch in diesem Fall?
Danke für Ihre Hilfe.
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
die Anforderungen an die Angabe des Importeurs beziehen sich auf das Medizinprodukt (oder Zubehör) als Ganzes. Für Ersatzteile sehe ich nach MDR keine Verpflichtung der Angabe des Importeur, außer es handelt sich um „Zubehör“ im Sinne der MDR.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
vielen Dank für Ihre schnelle Antwort und Bestätigung meiner Annahme.
Viele Grüße
Christine Graß
Sehr geehrter Herr Salvatore,
die MDR verlangt nach Artikel 31(5), dass der Wirtschaftsakteur innerhalb bestimmter Fristen bestätigt, dass die Registrierungsdaten nach wie vor korrekt sind.
Meine Frage lautet: Gegenüber wem muss diese Bestätigung erfolgen und gibt es dafür einen formalen Ablauf?
Danke für Ihre Hilfe.
Mit freundlichen Grüßen
Reinhard Köhler
Sehr geehrte Frau Köhler,
die Bestätigung gemäß Artikel 31(5) soll über EUDAMED erfolgen. Zumindest ist dies so in der Funktionalen Spezifikation von EUDAMED angegeben. Ich vermute, dass man als Wirtschaftsakteur nach Ablauf des entsprechenden Zeitraums explizit zur Bestätigung aufgefordert wird vom System. Diesen Fall hatten wir allerdings leider bisher noch nicht. Ansonsten hilft vielleicht auch eine Nachfrage über den UDI/EUDAMED-Helpdesk.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
gibt es zu ihrer Antwort an Herrn Köhler vom 10.03.2023 neue Erkenntnisse? Wir hatten gestern eine Anfrage eines Herstellers, der diese Bestätigung abgeben wollte. Er hat sich in EUDAMED eingeloggt aber nirgends etwas gefunden, wo er seine Angaben bestätigen könnte. Lediglich Änderungen seiner Anzeige wären möglich.
Vielen Dank.
Freundliche Grüße
Martin W. Richter
Sehr geehrter Herr Richter,
ich habe mich eben nochmals mit einem ehemaligen Mitarbeiter des EUDAMED-Teams ausgetauscht. Nach seine Aussage ist aktuell keine Funktionalität dafür in EUDAMED vorgesehen. Somit müssten die Akteure dafür geeignete Verfahrensanweisungen und Aufzeichnungen implementieren, wodurch die Überprüfung sichergestellt wird (z.B. visuell in EUDAMED oder durch den Download der Daten aus EUDAMED im xml-Format und einem Vergleich mit den lokalen Daten). Ob eine entsprechende Funktionalität in EUDAMED zukünftig implementiert wird, wissen wir leider nicht.
Freundliche Grüße
Luca Salvatore
Vielen Dank für Ihre rasche Antwort.
Hallo liebes Johner Team,
müssen Importeure die Anforderung lt. MDR Artikel 13(3) bereits für Legacy Produkte einhalten?
Dh muss der Importeur bereits bei Legacy Produkten (MDD Konformität) Name und Anschrift auf dem Produkt/beiliegendem Dokument anführen?
Vielen Dank im Voraus für Ihre Rückmeldung.
Mit besten Grüßen
Sonja Holub
Hallo Frau Holub,
gemäß MDCG-Guidance (https://health.ec.europa.eu/system/files/2021-10/md_mdcg_2021_25_en_0.pdf) ist Artikel 13(3) nicht anwendbar für Legacy-Produkte.
Herzliche Grüße
Luca Salvatore
Guten Tag, bedeutet der Importeur (Beschrieb auf der Verpackung des Produktes) muss auch der zollrechtliche Importeur sein oder kann sich dieser auch vertreten lassen durch einen Distributor u.ä.? Ist der Importeur (Beschrieb auf der Verpackung) nur derjenige, welcher die Haftung übernimmt für alles oben genannte?
Besten Dank.
Sandra A.
Guten Tag,
der Importeur, der gemäß MDR angegeben werden muss, ist der Importeur im regulatorischen Sinne mit den Pflichten gemäß Artikel 13 der MDR.
Die MDR fordert nicht, dass dieser auch für die zollrechtlichen Angelegenheiten verantwortlich sein muss. Dies kann also eine andere Person übernehmen.
Freundliche Grüße
Luca Salvatore
Hallo,
wir möchten Schwangerschaftstest Streifen von einem chinesischen Hersteller importieren und direkt verkaufen. Ein gültiges Zertifikat vom TÜV Süd, CE0123, liegt vor. Ebenso eine gültige Konformitätserklärung vom Hersteller.
Wir möchten nun auf dem Produkt unsere Handelsmarke anbringen lassen, entweder direkt vom Hersteller oder durch Umpacken bei uns. Darf ich dann auf dem Produkt auch das Zeichen CE0123 anbringen oder nur noch CE ? Muß ich dann selbst auch eine Konformitätserklärung ausstellen ?
Bieten Sie Dienstleistungen zu diesem Prozes an ?
MfG
Hallo Herr Kern,
wenn der Originalhersteller noch als solcher auf dem Produkt angegeben wird und es eine Vereinbarung gibt zum Anbringen Ihrer Handelsmarke, dann bleibt der chinesische Hersteller auch Hersteller gemäß MDR. Demnach muss das entsprechende CE-Kennzeichen erhalten bleiben. Wenn der Originalhersteller allerdings nicht mehr erkenntlich sein soll, dann würden Sie die Herstellerrolle gemäß MDR übernehmen und müssten ein eigenständiges Konformitätsbewertungsverfahren durchlaufen gemäß MDR. In diesem Fall müssten Sie dann „Ihr“ CE-Kennzeichen anbringen nach erfolgreichem Abschluss.
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
das hieße dann konkret:
Der Hersteller hat ein CE Zertifikat einer benannten Stelle (z.B. CE0123), er bedruckt die Verpackung mit meiner Handelsmarke und dem CE0123 Zeichen.Ich muß jetzt nicht aktiv werden.
Wenn der Hersteller aber mir sein Originalprodukt mit seiner Handelsmarke und dem CE0123 Zeichen schickt und ich dieses Produkt umverpacke (ich stecke sein Originalprodukt in meine zusätzliche Umverpackung mit meinem Handelsnamen) – auf meiner Umverpackung steht dann die Adresse des Herstellers, des EU-REP und meine als Importeur. Ich selbst habe aber kein CE Zertifikat einer benannten Stelle. Darf ich dann trotzdem das CE Zeichen CE0123 auf meine Umverpackung aufdrucken oder nur das CE Zeichen ? Muß ich dann selbst ein Konformitätsbewertungsverfahren durchlaufen um das CE Zeichen anbringen zu dürfen ?
MfG
Kern
Hallo Herr Kern,
wenn Sie eigenmächtig die äußere Verpackung ändern, d.h. ohne Vereinbarung mit dem Hersteller bzw. unter dem QM-System des Herstellers, dann wäre Artikel 16 (2) (b) MDR anwendbar. In diesem Fall benötigen Sie gemäß Artikel 16 (3) und (4) MDR ein QM-System, welches von einer Benannten Stelle zertifiziert werden muss.
Freundliche Grüße
Luca Salvatore
Hallo Herr Johner,
Wir haben ein gültiges Zertifikat für unseren QMS nach 13485 , sind jedoch nicht zertifiziert als Hersteller.
Frage 1. ist es jedoch möglich unsere Adresse , Firmen Logo , Handelsmarke auf den Labels zusätzlich zu dem Original Hersteller auf Verpackung und Label auszudrucken?
kann man denn hier das Importeur Symbol lediglich benutzen
( dies würde schon in der Produktion bei dem Hersteller geschehen , also keine Umverpackung)
der Original Hersteller wäre weiterhin eindeutig erkennbar.
fallen dann die Herstellerpflichten auf uns ?
Frage 2: bräuchten wir dann ein Zertifikat als Hersteller oder reicht es ein zertifiziertes QMS nach 13485 zuhaben.
Vielen Dank
Dora
Hallo Dora,
relevant ist in diesem Fall Artikel 16 (1) (a) MDR: „Bereitstellung eines Produkts auf dem Markt unter dem eigenen Namen, dem eigenen eingetragenen Handelsnamen oder der eigenen eingetragenen Handelsmarke, außer in den Fällen, in denen ein Händler oder Importeur eine Vereinbarung mit einem Hersteller schließt, wonach der Hersteller als solcher auf der Kennzeichnung angegeben wird und für die Einhaltung der nach dieser Verordnung für die Hersteller geltenden Anforderungen verantwortlich ist;“
Wenn Sie demnach mit dem Hersteller vereinbaren, dass dieser weiterhin auf dem Produkt als Hersteller gekennzeichnet ist und Sie zusätzlich eine Handelsmarke oder Logo angeben, dann würden die Herstellerpflichten nicht auf Sie fallen. Dafür benötigen Sie kein zertifiziertes QMS nach ISO 13485.
Die Informationen zum Importeur inkl. Symbol können Sie oder der Hersteller aufbringen. Diese muss allerdings nicht auf der Verpackung angebracht sein sondern z.B. auch auf einem beiligenden Dokument.
Herzliche Grüße
Luca Salvatore
Sehr geehrtes Johner-Team,
welche (Importeurs-) Kennzeichnung ist notwendig, wenn ein in der EU ansässiger Medizinproduktehersteller seine Produkte an verschiedene Händler und teilweise auch med. Endverbraucher vertreibt?
Muss bei jeder Lieferung an den entsprechenden Schweizer Händler auch der jeweilige Importeur aufgeführt/geändert werden, weil es keinen Alleinvertrieb in der Schweiz gibt?
Da die Angabe eines CH-Rep zwingend vorgegeben ist, muss überhaupt ein Importeur auf dem Produkt angegeben werden (mit der Annahme, dass verschiedene Händler und auch Endverbraucher das Produkt direkt beim EU Hersteller einkaufen)?
Danke im voraus.
Freundliche Grüße
Sehr geehrter Herr Wagner,
der entsprechende Importeur muss entweder auf dem Produkt, der Verpackung oder einem beiligenden Dokument angegeben werden. Zusätzlich muss der CH-Rep angegeben sein.
Detaillierte Informationen finden Sie in folgendem Merkblatt der Swissmedic: https://www.swissmedic.ch/dam/swissmedic/de/dokumente/medizinprodukte/mep_urr/mu600_00_016d_mb_pflichten_wirtschaftsakteure_ch.pdf.download.pdf/MU600_00_016d_MB_Pflichten_Wirtschaftsakteure_CH.pdf
Freundliche Grüße
Luca Salvatore
Sehr geehrtes Johner-Team
Vielen Dank für den sehr aufschlussreichen Artikel.
Wir möchten ein Medizinprodukt aus der Schweiz nach Europa exportieren. Nehmen wir an, das Produkt ist unter der MDR CE zertifiziert und ein EU-REP existiert. Um das Produkt in den Verkehr zu bringen braucht es einen Importeur.
Frage: Kann es theoretisch auch mehrere Importeure für ein und das selbe Medizinprodukt geben?
Freundliche Grüße
Stephan Gregorini
Guten Tag Herr Gregorini,
es ist möglich, dass mehrere Importeure das gleiche Medizinprodukt importieren. Jede Person, die ein Medizinprodukt von außerhalb der EU, innerhalb der EU in den Verkehr bringt, gilt als Importeur. Man wird also nicht explizit zum Importeur ernannt, sondern man ist es per Definition in der MDR, sobald man die genannte Tätigkeit ausführt.
Freundliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore,
vielen Dank für den aufschlussreichen Artikel und Ihre umfangreichen Antworten auf Nachfragen.
Nach meinem Verständnis ist es innerhalb eines multinationalen Hersteller-Konzerns möglich, eine einzige europäische Tochtergesellschaft als Importeur auszuwählen, welche dann (ohne selbst Eigentum an der Ware erworben zu haben), diese ohne Entgeltzahlung an andere EU Tochtergesellschaften weitergibt, welche die Ware dann in ihren lokalen Märkten an Verbraucher oder Distributoren veräußern. Es würde dann ausreichen, wenn die eine benannte EU Tochtergesellschaft die Anforderungen der MDR und IVDR für Importeure erfüllt. Nicht jede Landesgesellschaft müsste dann beispielsweise EUDAMED registriert sein. Ist dieses Verständnis des Importeur-Begriffs richtig?
Guten Tag Herr Stiller,
es ist möglich, einen zentralen Importeur auszuwählen, welcher für die Inverkehrbringung in die EU zuständig ist. Dies kann ohne Entgeltzahlung erfolgen. Wichtig ist aber, dass eine Vereinbarung besteht zumindest bezüglich „sonstiger (Verfügungs-)Rechte“.
Die weiteren Tochtergesellschaften würden in diesem Fall die Rolle des Händlers übernehmen. Für diese wäre in diesem Fall keine EUDAMED-Registrierung notwendig, allerdings wären die Händler-Pflichten nach Artikel 14 MDR zu beachten.
Herzliche Grüße
Luca Salvatore
Vielen Dank für Ihre klare und rasche Antwort. Eine kurze Nachfrage: ist es erforderlich, dass der Importeur die Ware zwischenzeitlich auch physisch in Besitz nimmt, oder wäre auch ein direkter Versand des nicht in der EU ansässigen Herstellers an die übrigen EU Tochtergesellschaften (die nicht formal Importeure sind) zulässig?
Eine physische Übergabe ist keine Voraussetzung. Allerdings muss der Importeur seinen Prüfpflichten gemäß Artikel 13 MDR nachkommen. Sicherlich kann dies delegiert bzw. ausgelagert werden. Die Verantwortung bleibt aber beim Importeur.
Guten Tag
Ich arbeite für eine schweizer Handelsfirma für Medizinprodukte in der Ophthalmolgie.
Wir haben einen Lieferanten – ebenfalls aus er Schweiz, bei dem wir Medizinprodukte in den Klassen 2a und 2b beziehen.
Wir verkaufen hauptsächlich in der Schweiz – aber auch in Österreich.
Wenn wir jetzt die Produkte des schweizer Herstellers in Österreich verkaufen möchten müssen wir ja auch Importeur für die Inverkehrbringung in die EU sein. Das sind wir aber nicht.
Der schweizer Hersteller hat eine Import-Firma für die Inverkehrbringung in die EU.
Meine Frage:
Können wir als schweizer Firma, in der Schweiz produzierte Produkte von der Importfirma des Herstellers beziehen, die Produkte somit in die Schweiz zurück holen und von hier aus in die EU verkaufen? Erlischt der Import der Importfirma bei der erneuten Einfuhr in die Schweiz?
Es wäre super, wenn Sie mir hier behilflich sein könnten.
Besten Dank und freundliche Grüsse
Guten Tag Herr Kreis,
Ihre Frage ist, basierend auf den gegebenen Informationen, nicht abschließend zu beantworten. Dazu wäre ein gründliche Analyse der konkreten Konstallation notwendig.
Generell ist die Frage, ob die „Importfirma“ diese Produkte überhaupt in der EU in den Verkehr gebracht hat, wenn diese für den Export in die Schweiz gedacht sind. Somit hätte vermutlich in Ihrem Fall keine Inverkehrbringung stattgefunden. Wenn Sie direkt an EU-Endbenutzer verkaufen, dann handelt es sich um Fernabsatz. In diesem Fall kann es sein, dass es keinen Importeur in der EU gibt. Falls Sie an einen Händler in der EU verkaufen, würde dieser die Rolle des Importeurs einnehmen. Schauen Sie sich dazu das Beispiel 3 im MDCG 2021-27 an (https://health.ec.europa.eu/system/files/2021-12/mdcg_2021-27_en.pdf). Beachten Sie auch die Verordnung EU 2019/1020 und den zugehörigen Leitfaden: https://ec.europa.eu/docsroom/documents/44908/attachments/2/translations/en/renditions/native. Dort finden Sie auf Seite 7 einen Entscheidungsbaum für verschiedene Konstellationen.
Freundliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore,
wir sind Importeur von Medizinprodukten. Unsere importierten Artikel sind mit der Adresse der Herstellers und unserer Firma als Importeur gekennzeichnet. Nun besteht die Forderung, dass zwingend der EC REP mit in die Kennzeichnung aufgenommen wird. Verlangt die MDR wirklich , dass alle drei auf der Markierung erscheinen müssen?
Mit freundlichen Grüßen
S. Wolthusen
Guten Tag Frau Wolthusen,
die MDR fordert in der Tat in Anhang I 23.2 d) die Angabe des EC REP in der Kennzeichnung. Für die Angabe des Importeurs gibt es verschiedene Optionen (siehe Artikel 13 (3) MDR).
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
ich benötige eine Abklärung zum Thema Parallelimport von Medizinprodukten Klasse 2a in der MDR und hoffe Sie können mir behilflich sein.
Beispiel 1:
Originalprodukte des Herstellers mit Ursprung in der EU und bereits durch den Hersteller direkt in den Verkehr gebracht, werden aus einem Drittland bezogen, nach Deutschland re-importiert und innerhalb der EU weiterverkauft.
Beispiel 2:
Originalprodukte des Herstellers mit Ursprung in CH und bereits durch den Hersteller via EC Rep in DE direkt in den Verkehr gebracht, werden aus einem Drittland bezogen, nach Deutschland re-importiert und innerhalb der EU weiterverkauft.
In beiden Beispielen werden keine Änderungen am Produkt oder der Verpackung vorgenommen.
Welcher Wirtschaftsakteur bin ich in dieser Konstellation?
Händler, da Hersteller Ware bereits in Verkehr gebracht hat und keinerlei Änderung an Label getätigt werden oder Importeur, da Ware zwar EU-Ursprung ist aber wieder in die EU re-importiert wurde?
Besten Dank
Guten Tag,
in beiden Fällen müsste man sich die Konstellation im Detail anschauen, um eine korrekte Aussage treffen zu können (z.B. ob die Produkte wirklich in der EU bereits in Verkehr gebracht wurden oder nur für den Export bestimmt waren).
Im Falle, dass die Produkte tatsächlich in der EU bereits in den Verkehr gebracht wurden und nun re-importiert werden, sehe ich sie in beiden Fällen in der Rolle des Händlers. Denn laut Definition bezeichnet einen Importeur „jede in der Union niedergelassene natürliche oder juristische Person, die ein Produkt aus einem Drittland auf dem Unionsmarkt in Verkehr bringt;“ Und diese Inverkehrbringung ist bereits durch den Hersteller (im ersten Fall) bzw. EC-Rep (in der Rolle des Importeurs im 2. Fall) selbst erfolgt. Das MDCG 2021-27 sagt z.B. aus: „. It is not possible however, to have multiple importers of the same individual device.“ Und im EU-Blue Guide steht: „Nach den Harmonisierungsrechtsvorschriften der Union kann jedes einzelne Produkt nur einmal auf dem Unionsmarkt in Verkehr gebracht werden“
Herzliche Grüße
Luca Salvatore
Besten Dank Herr Salvatore.
Gibt es hierzu definierte Guidelines oder „Rechtssprechungen“ auf die man sich beziehen kann?
Ich wäre Ihnen dankbar wenn wir uns hierzu im Detail austauschen könnten und bitte um Kontaktaufnahme via Email sodass ich Sie kontaktieren kann.
Vielen Dank.
Hallo liebes Johner-Team,
durch Strukturänderungen im Unternehmen (Schweizer Medizinproduktehersteller) soll unsere Firma (Tochterfirma mit Sitz in Deutschland) jetzt Importeur gemäß MDR werden und ein Mitarbeiter von uns wird Bevollmächtigter gem. MDR. Unser Unternehmen ist 13485 zertifiziert. Brauchen wir ein MDR (oder Anhangszertifikat für Produkte) oder ist die Zertifizierung ISO 13485 ausreichend?
Eine zweite Frage: betrifft die Kennzeichnung. Die Produkte müssen neu mit der Adressen unserer Firma (als Importeur) gekennzeichnet werden. Da ein Mitarbeiter PRRC werden soll, muss seine Privatadresse auch angegeben werden?
Liebe Frau Zechel,
als Importeur benötigen Sie kein MDR-Zertifikat. Auch ein ISO 13485-Zertifikat ist nicht zwingend notwendig, aber sicherlich hilfreich. Die Angabe des PRRC auf der Kennzeichnung ist nicht notwendig. Allerdings muss dieser in EUDAMED bei der Hersteller- bzw. EC-Rep Registrierung angegeben werden.
Freundliche Grüße
Luca Salvatore
Hallo liebes Johner-Team,
mit großem Interesse habe ich die Informationen zum Importeur gelesen. Aufgrund einer speziellen Anfrage sind wir uns leider etwas unsicher bzgl. der Kennzeichnung für den folgenden Fall: Wir (Sitz in DE) sollen für ein drittes Unternehmen mit Sitz in der EU Medizinprodukte (Klasse I) importieren. Das Layout gibt im groben der Auftraggeber vor. Muss unser Unternehmen als Importeur auf den Verpackungen stehen (wovon wir ausgehen) und zusätzlich auch der Auftraggeber auf der Verpackung genannt werden? Da wir nicht der Hersteller im Sinne der MDR sind, ist der Hersteller sowie dessen Bevollmächtigter ebenfalls auf den Verpackungen gekennzeichnet.
Über eine Antwort freue ich mich sehr und bedanke mich bereits im Voraus.
Lieber Herr Mühlhausen,
zunächst muss geklärt werden, ob Sie als Importeur gemäß MDR gelten, d.h. für die Inverkehrbringung in der EU verantwortlich sind. Logistik- und Transportdienstleister (3PLs) sind beispielsweise im Regelfall keine Importeure im Sinne der MDR (siehe auch https://health.ec.europa.eu/system/files/2023-12/mdcg_2021-27_en.pdf, Frage 9 und 10).
Falls Sie Importeur gemäß MDR sind, müssen Ihre Kontaktdaten und Anschrift „auf dem Produkt oder auf seiner Verpackung oder auf einem dem Produkt beiliegenden Dokument…“ angegeben werden. Ihr Auftraggeber wird vermutlich die Rolle des Händlers einnehmen. Dieser muss nicht zwingend auf der Kennzeichnung angegeben werden.
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
unser Lieferant aus den Vereinigten Staaten hat seinen EU-Repräsentanten auch zum Importeur ernannt. Diese Firma soll komplett die Pflichten des Importeurs gemäß der MDR übernehmen, aber nicht die Medizinprodukte physisch importieren (wir werden sie direkt in den USA beim Hersteller bestellen und ausgeliefert bekommen). Nun ergeben sich folgenden Fragen:
1. Ist dieses Konstrukt zwischen dem Hersteller und seinem EC-Repräsentanten / Importeur überhaupt rechtlich gültig?
2. Werden wir dadurch von unseren Pflichten als tatsächlicher Importeur befreit und nur die Pflichten eines Händlers erfüllen müssen?
Vielen Dank und beste Grüße
Guten Tag Herr Cecek,
generell wird ein Importeur im Sinne der MDR nicht ernannt. Abhängig ob eine (natürliche/juristische) Person ein Medizinprodukt aus einem Drittland in der EU in den Verkehr bringt, übernimmt diese Person per Gesetz die Rolle des Importeurs. Es gibt allerdings Anbieter, welche die „regulatorische“ Importeursrolle für Händler in der EU übernehmen, ähnlich in Ihrem Fall. Dabei muss aber sichergestellt werden, dass in Ihrem Fall der EU-Bevollmächtigte dennoch die Produkte erstmalig bereitstellt. D.h. es muss eine Übertragung des Eigentums, Besitzes oder sonstiger Rechte zwischen dem Hersteller und dem Bevollmächtigten stattfinden und nicht zwischen Ihnen und dem Hersteller. Ansonsten würden Sie u.U. die Rolle des Importeurs einnehmen. Ich empfehle, diese Konstellation vertraglich abzusichern und im Idealfall einen Fachanwalt einzuschalten, um rechtlich auf der sicheren Seite zu sein.
Im Falle, dass der Bevollmächtigte tatsächliche die Rolle des Importeurs einnimmt und Ihnen dieser die Produkte bereitstellt, würden Sie die Rolle eines Händlers einnehmen. Somit müssten Sie die Pflichten der Händler beachten (Artikel 14 MDR) und nicht die des Importeurs (Artikel 13 MDR).
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
ich habe eine etwas ungewöhnliche Frage. Wir sind ein Importeur von Klasse I Medizinprodukten. Für den hypothetischen Fall, dass ein in DE ansässiges Unternehmen mit 13485 Zertifizierung auf uns zu käme mit der Bitte, in seinem Namen Produkte zu importieren, müsste unser Unternehmen dann als Importeur auftreten? Ich verstehe die MDR dahingehend, denn nach Artikel 13 Absatz 3 unser Unternehmen würde die Waren ja erstmals im Geltungsbereich der MDR veräußern (an den Hersteller = Auftraggeber) und somit in Verkehr bringen. Verstehen Sie das auch so oder habe ich einen Denkfehler? Über eine Antwort freue ich mich sehr.
Vielen Dank im Voraus.
Guten Tag Herr Mühlhausen,
die entscheidende Frage wäre, ob in dieser Konstellation eine Inverkehrbringung durch Sie stattfindet und ob die Produkte aus einem Drittstaat eingeführt werden. Voraussetzung ist u.a., dass eine Übertragung von „Eigentum, Besitz oder sonstiger Rechte“ hinsichtlich des Produkts auf Sie stattfindet. Falls Sie in diesem Fall in Verkehr bringen, würden Sie die Rolle des Importeurs einnehmen. Unklar aus Ihrer Frage ist allerdings, warum Ihr Auftraggeber (das in DE ansässige Unternehmen?) Hersteller ist, wenn doch das Produkt aus einem Drittstaat importiert wird.
Freundliche Grüße
Luca Salvatore
Sehr geehrtes Johner team,
wie sehen die Pflichten bei SaMD Herstellern außerhalb der EU (z.B Schweiz) aus, bei denen die Software direkt über einen Appstore in der EU in Verkehr gebracht werden. Einen EU-Rep braucht es natürlich. Braucht es aber im Falle dieses Direktvertriebes über den Appstore auch einen Importeur?
Viele Grüße,
S. Letter
Guten Tag Herr Letter,
das ist eine sehr spannende Frage, die viel diskutiert wird. Bisher gibt es meines Wissens leider noch keine offizielle Stellungnahme der EU-Kommission trotz vieler Anfragen genau zu diesem Thema.
Generell wird ein Importeur nicht benannt, wie beispielsweise ein EU-Rep. Die Rolle ergibt sich vielmehr aus der Konstellation der Lieferkette. Die Kommission stellt in Verordnung EU 2019/1020 zur Marktüberwachung (nicht Medizinprodukte-spezifisch) klar, dass es Konstellationen geben kann, bei denen Produkte außerhalb der EU direkt an Endverbraucher abgegeben werden (Fernabsatz), ohne dass ein Importeur in der EU vorhanden ist. Es gibt allerdings auch die Ansicht, dass die App Stores die Rolle der Importeure bzw. Händler einnehmen (siehe z.B. COCIR Impact Paper on Software: https://fhi.nl/app/uploads/sites/70/2020/06/2017-05-12-COCIR-EU-RA-FG-COCIR-MDR-Impact-Papers-SOFTWARE-31P-kopie.pdf oder https://www.nature.com/articles/s41746-023-00754-6). Diese müssten ihren Sitz dann allerdings in der EU haben. Wiederrum gibt es weitere Meinungen, welche die Endbenutzer als Importeure ansehen. Diese Ansicht teilen wir nicht, da Endbenutzer wie z.B. Gesundheitseinrichtungen im Regelfall die Produkte in Betrieb nehmen und nicht in den Verkehr bringen gemäß MDR-Definition.
Inzwischen gibt es auch unabhängige Anbieter, welche die Rolle des EU-Importeurs unter der MDR/IVDR formell übernehmen.
Unabhängig davon muss jeder Einzelfall separat betrachtet werden. Für mehr Rechtssicherheit empfehlen wir, die jeweilige Konstellation immer von einem Fachanwalt prüfen zu lassen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Johner
Für ein Schulprojekt stellt sich uns folgende Frage worauf wir bis jetzt keine schlaue Antwort erhalten haben:
Darf jeder, jedes Produkt importieren, oder haben gewisse Firmen (Importeure / Speditionen) das alleinige Recht darauf, ein Artikel zu importieren? EU oder Schweiz? Wenn ja wie funktioniert das? Oder wie heissen solche Gesetze/Patente?
Besten Dank!
Freundliche Grüsse
Sehr geehrter Marty,
da hätte möglicherweise eine KI Ihnen die Antwort zaubern können :).
Es gilt zwei Dinge zu unterscheiden:
D.h. der Schutz vor unerwünschten Importeuren schaffen eher EU-Gesetze als Patente.
Beste Grüße, Christian Johner
Hallo Herr Salvatore,
wir sind ein Medizinprodukteunternehmen und in der EUDAMED als Hersteller und Importeur registriert. Als Importeur beziehen wir Medizinprodukte aus UK.
Nach Artikel 13 (4) MDR muss der Importeur prüfen, dass das Produkt registriert ist. Wir müssen diese Registrierung mit unseren Daten ergänzen. Erfolgt in EUDAMED das Linking eines non-EU Herstellers zum Importeur durch den non-EU-Hersteller oder durch den Importeur
Hallo Frau Hauptmann,
das Linking zum non-EU-Hersteller erfolgt durch den Importeur. Dazu benötigen ein Linker-Profil in EUDAMED. Eine Anleitung finden Sie hier: https://health.ec.europa.eu/system/files/2022-09/md_user_guide_actor_module_en.pdf (Seite 29).
Herzliche Grüße
Luca Salvatore
Guten Tag,
vielen Dank für diesen Artikel.
Wir haben folgende Situation: Unser Unternehmen in Deutschland ist Legal Manufacturer eines IVD-Produktes.
Die Produktion wird nun an eine Schwesterfirma in Japan ausgelagert, als outgesourcter Prozess, contract manufacturing, so dass das Unternehmen mit Sitz in Deutschland Legal Manufacturer bleibt. Ich gehe davon aus, dass der physische manufacturer auch das CE Zeichen in Japan anbringen wird.
Was ist dann beim Importieren zu beachten?
Wird dann der Legal Manufacturer gleichzeitig zum Importeur?
Freundliche Grüße
Stephan Schäfer
Guten Tag Herr Schäfer,
da Ihr Unternehmen in Deutschland Hersteller gemäß MDR bleibt, gibt es in dieser Konstellation keinen Importeur im Sinne der MDR. Im EU-Blueguide 2022 ist ein ähnliches Szenario geschildert (4.2.2.4):
„Befindet sich der Hersteller innerhalb der EU (handelt es sich also um ein in der EU ansässiges Unternehmen, das sich durch die Angabe seines Namens und seiner Anschrift auf dem Produkt selbst zum Hersteller erklärt), obwohl die Produkte außerhalb der EU hergestellt werden, gilt dieses Unternehmen als Hersteller, der das Produkt auf dem Unionsmarkt in Verkehr bringt, selbst wenn die eigentliche Einfuhr durch ein anderes Unternehmen erfolgt. In diesem Fall gibt es keinen Einführer im Sinne der entsprechenden Definition; es reicht also aus, lediglich die Anschrift des Herstellers anzugeben.“
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
vielen Dank für den aufschlussreichen Artikel!
Allerdings ist mir die Funktionalität des Importer Actors in EUDAMED noch nicht ganz klar.
Sehe ich es richtig, dass eine Verlinkung mit einem Fremdhersteller in EUDAMED in dem Fall erfolgen soll, wenn man ein Produkt dieses Herstellers in Verkehr bringt?
Und gibt es eine Funktionalität in EUDAMED um der Anforderung in MDR Artikel 13(4) nachzukommen, oder erfolgt das bisher formlos?
Besten Dank!
Freundliche Grüße
Jan-Luca Dreves
Hallo Herr Dreves,
ganz genau. Sie verlinken in EUDAMED den non-EU Hersteller, wenn Sie dessen Produkte in der EU in den Verkehr bringen und somit die Importeur-Rolle einnehmen. Das Überprüfen, ob die Produkte, welche Sie importieren, registriert sind (gemäß Artikel 13(4)), erfolgt formlos bzw. gemäß Ihrer Vorgaben.
Freundliche Grüße
Luca Salvatore
Guten Tag Liebes Johner-Team,
wir verkaufen neben unseren Markenprodukten auch Private-Label- Produkte an deutsche Handelsunternehmen.
Wir treten dabei als Händler auf; die Hersteller produzieren meist in Asien.
Wenn auf einem Eigenmarken- Medizinprodukt nur steht: „hergestellt für“ (und es folgt der Name des Handelsunternehmens) + der EU REP + der eigentliche Hersteller in Asien, erübrigt sich dann die Nennung des Importeurs (das wären praktisch wir) oder muss dieser zusätzlich angegeben werden?
Wir als Markenhersteller wollen ungern auf der Verpackung genannt werden.
Vielen Dank für Ihre Einschätzung.
Freundliche Grüße
Jürgen Sitter
Guten Tag Herr Sitter,
Artikel 13(3) MDR fordert die Angabe des Importeurs. Allerdings überlässt die MDR dem Importeur, wo diese Angabe gemacht wird. Möglich wäre:
– Auf dem Produkt selbst
– Auf der Verpackung
– Auf einem Begleitdokument
Weitere Details finden Sie im MDCG Guidance-Dokument 2021-27 (https://health.ec.europa.eu/system/files/2023-12/mdcg_2021-27_en.pdf), insbesondere Fragen 6-8.
Freundliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore,
bitte gestatten Sie mir eine Rückfrage.
Wenn auf der Private Label-Verpackung steht „hergestellt für: xy GmbH“ und dieses Handelsunternehmen hat seinen Sitz in Deutschland, wird es dann automatisch zum legalen Hersteller? So hat es ein Consultant interpretiert. Dementsprechend müsste man keinen Importeur angeben, da das in Deutschland sitzende Handelsunternehmen, unter dessen Marke der Artikel auch vertrieben wird, ausreichen würde.
Vielen Dank für Ihre Rückmeldung hierzu.
Freundliche Grüße
Jürgen Sitter
Guten Tag Herr Sitter,
das Unternehmen müsste explizit als Hersteller auf dem Label erkenntlich sein (schwarzes Fabriksymbol) und das produzierende Unternehmen dürfte nicht mehr als Hersteller genannt sein. Das Handelsunternehmen müsste in diesem Falle auch sämtliche Herstellerpflichten erfüllen gemäß MDR, welche u.a. ein QM-System verlangt sowie eine Technische Dokumentation zum Produkt. Bei Produkten höher als Klasse I wäre eine Benannte Stelle einzuschalten.
Freundliche Grüße
Luca Salvatore
Guten Tag,
in der MDCG 2021-27 Rev.1 gibt es drei Beispiele für Hersteller/Importeur konstellationen, die versuchen zu erklären wann ein IMporteur notwendig ist.
Dabei stellte sich für mich die Frage: Wenn der Hersteller A (nach IVDR) in einem Drittland beheimatet ist und eine Produktionsseite bei einem anderen Unternehmen B innerhalb der EU hat, wird dann ein Importeur benötigt? Das Produkt wird ja nicht impoprtiert, es ist schon innerhalb der EU hergestellt.
Besten Dank
Guten Tag Herr Breß,
ob das Produkt in der EU produziert wird oder nicht, spielt zunächst eine untergeordnete Rolle. Relevant ist der Sitz des Herstellers gemäß IVDR und wie die Produkte in den Verkehr gebracht werden. Falls der Hersteller im Direktvertrieb direkt an die Anwender verkauft, dann wird es vermutlich keinen Importeur geben. Falls aber über einen lokalen Händler in den Verkehr gebracht wird, würde dieser die Rolle des Importeurs einnehmen.
Freundliche Grüße
Luca Salvatore
Buongiorno, stimmen Sie zu, dass MDR Artikel 13 (3) Kennzeichnung nicht für Legacy Devices gilt. MDCG 2021-215 Rev1 beschreibt „In addition, the obligations of economic operators set out in the following provisions should also apply to economic operators with respect to ‘legacy devices“…… ➢ for importers: Article 13(2), 2nd subparagraph, (4), (6)-(8), (10); ?
Bis bald
Francesco
Lieber Herr Fantoni,
ich stimme Ihnen vollständig zu. Die anwendbaren MDR-Pflichten der Importeure beschränken sich bei den Legacy Devices auf die von Ihnen aufgeführten Absätze im Artikel 13. Das hängt damit zusammen, dass auch Hersteller von Legacy Devices bereits die Anforderungen der MDR an das Post-Market Surveillance, die Registrierung von Wirtschaftsakteuren und Produkten sowie die Vigilanz erfüllen müssen. Demzufolge werden auch die anderen beteiligten Wirtschaftsakteure verpflichtet, die Anforderungen diesbezüglich bereits für Legacy Devices umzusetzen.
Herzliche Grüße,
Manuela Reinhold