
Die meisten Hersteller nutzen harmonisierte Normen, um die Konformität ihrer Produkte mit den grundlegenden Sicherheits- und Leistungsanforderungen nachzuweisen. Das gilt beispielsweise auch für Medizinproduktehersteller.
1. Normen und harmonisierte Normen
a) Definitionen und mehr
Die EU-Verordnung 1025/2012 definiert den Begriff harmonisierte Norm.
„eine europäische Norm, die auf der Grundlage eines Auftrags der Kommission zur Durchführung von Harmonisierungsrechtsvorschriften der Union angenommen wurde“
Normen sind Dokumente, die von nationalen oder internationalen Standardisierungskommissionen geschrieben werden, um den Stand der Technik zu dokumentieren. Sie beschreiben meist keine Best Practices oder gar den Stand der Forschung, sondern den Minimalkonsens, auf den sich das Normenkomitee einigen konnte.
Der Stand der Technik beschreibt, wie die sehr guten Unternehmen vorgehen. Hingegen beschreibt der Stand der Forschung, was theoretisch möglich ist.
Bestimmte Verschlüsselungsverfahren mit einer definierten Schlüssellänge entsprechen dem Stand der Technik. Sie stellen ein Niveau dar, das als ein professionell handelndes Unternehmen nicht unterschreiten sollte. Der Stand der Forschung kennt hingegen quantenphysikalische Verfahren zur Verschlüsselung und Entschlüsselung.
Lesen Sie hier mehr zur Unterscheidung von Stand der Technik und Stand der Forschung.

Die harmonisierten Normen hat die EU in ihren Amtsblättern veröffentlicht. Wenn Hersteller diese harmonisierten Normen einhalten, gehen z. B. Auditoren und Gutachter davon aus, dass die gesetzlichen Forderungen erfüllt sind, konkret: Die grundlegenden Anforderungen sind erfüllt.
b) Beweisführungskette
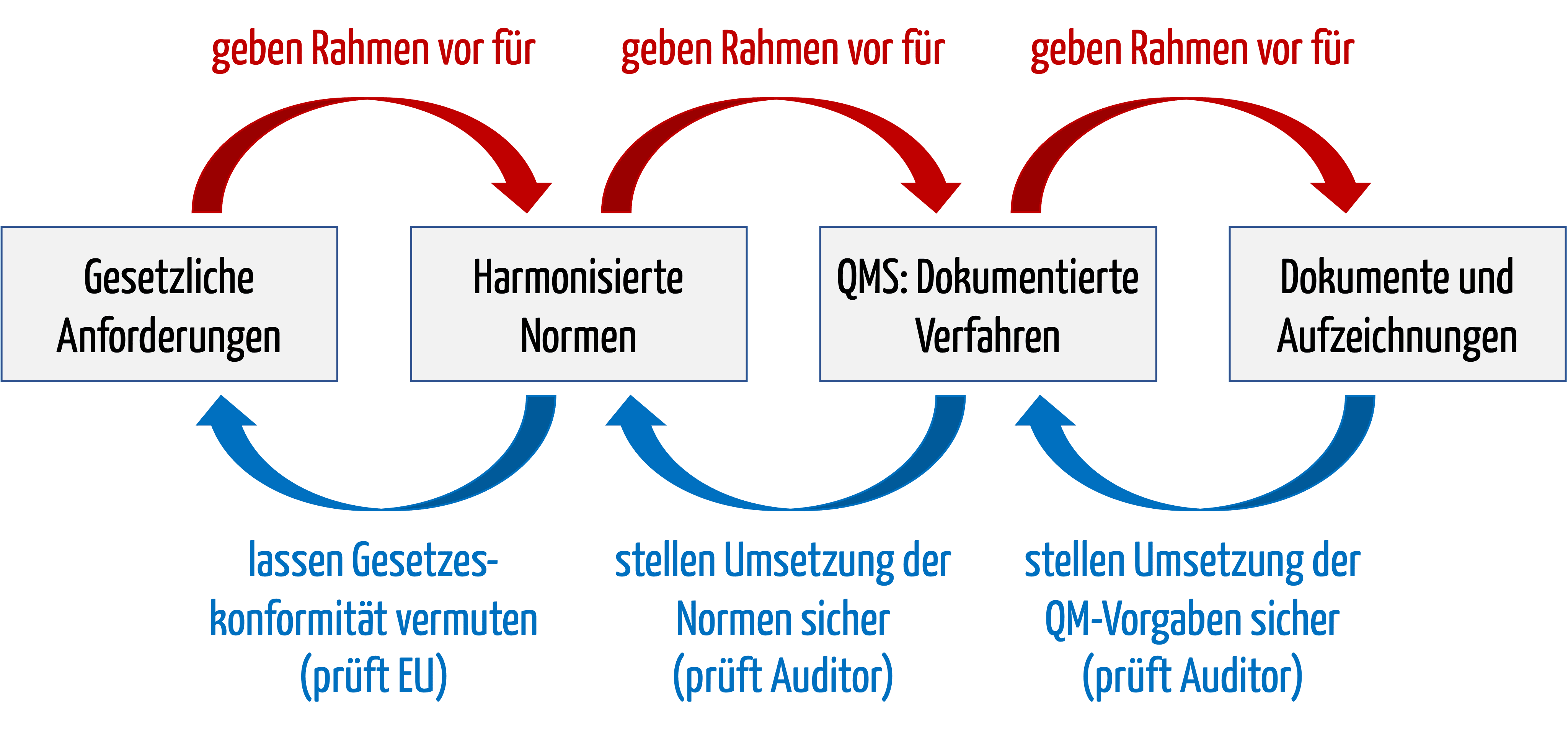
Diese Beweisführungskette lässt sich an einem Beispiel aufzeigen:
| Element der Beweisführungskette | Beispiel |
| Gesetzliche Forderungen | Grundlegende Anforderung der MDR in Anhang I an die Software-Lebenszyklusprozesse |
| Harmonisierte Norm | IEC 62304 |
| QMS und dokumentierte Verfahren | Verfahrensanweisung Software-Entwicklung bzw. der Software-Entwicklungsplan; beide verlangen u. a. ein Code-Review |
| Dokumente und Aufzeichnungen | Dokumentiertes Code-Review an Tag X, durchgeführt von Person Y mit dem Ergebnis Z |
Die Einhaltung der harmonisierten Normen ist nicht gesetzlich verpflichtend, weder in der EU noch bei der FDA. Allerdings werden sich Hersteller ohne die harmonisierten Normen schwer den Nachweis erbringen können, dass ihre Produkte dem Stand der Technik entsprechen und die gesetzlichen Anforderungen erfüllen. Das kann bei Audits und Gerichtsverfahren zum Problem werden.
c) Liste harmonisierter Normen für Medizinprodukte- und IVD-Hersteller
Zu der Liste der harmonisierten Normen zählen oder zählten im Kontext der Medizinprodukte:
- EN IEC 62304: Software-Lebenszyklus-Prozesse für Medizinprodukte
- EN IEC 62366-1: Anwendung der Gebrauchstauglichkeit auf Medizinprodukte
- EN ISO 14971: Anwendung des Risikomanagements auf Medizinprodukte
- EN IEC 60601-1: Programmierbare elektrische medizinische Systeme: Basissicherheit und wesentliche Leistungsmerkmale
- EN ISO 13485: Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke
- ISO 10993-Familie: Biokompatibilität und biologische Sicherheit für Medizinprodukte
Die vollständige Liste harmonisierter Normen findet sich auf der Webseite der EU.
Das Johner Institut unterstützt Hersteller bei der Überwachung von über 7.000 (harmonisierten) Normen und Gesetzen. Dafür stellt es Software bereit oder übernimmt den Überwachungsprozess.
2. Normen im Kontext von MDR und IVDR: Die Theorie
Die MDR kennt weiterhin das Konzept der harmonisierten Normen, die der „Beweisführung“ dienen dürfen. Sie schreibt in Artikel 8:
„Devices which are in conformity with the relevant harmonised standards, or the relevant parts of those standards, the references of which have been published in the Official Journal of the European Union, shall be presumed to be in conformity with the requirements of this Regulation covered by those standards or parts thereof.“
MDR Artikel 8
Neben den harmonisierten Normen gibt es auch Common Specifications. In Artikel 9 der MDR steht:
„where no harmonised standards exist or where relevant harmonised standards are not sufficient, or where there is a need to address public health concerns, the Commission […] may […] adopt common specifications (CS) in respect of the general safety and performance requirements set out in Annex I […]“
MDR Artikel 9
Das heißt, dass die Hersteller mehrere Beweisführungsinstrumente nutzen müssen.
Die Anforderungen der IVDR sind analog.
3. Normen im Kontext von MDR und IVDR: Die Praxis
a) Die Probleme mit den Normen
Inzwischen türmen sich so viele Probleme auf, dass sich die Frage nach deren künftiger Relevanz stellt. Beispiele für die Probleme sind:
Problem 1: Heterogene Qualität
Während zentrale Normen wie die zum Qualitätsmanagement (ISO 9001 und ISO 13485) solide sind, leidet die Qualität vieler Normen:
- Sie bilden nicht den Stand der Technik ab, sondern die Meinung der Autoren.
- Den Normen fehlt eine innere Konsistenz. Anforderungen erscheinen beliebig ausgewählt zu sein. Prozess- und Produktanforderungen werden nicht präzise unterschieden.
- Die Anforderungen sind so unscharf, dass ihre Überprüfung schwierig ist.
- Definitionen fehlen.
- Eine Abstimmung mit den Konzepten anderer Normen fehlt, was den Herstellern unnötige Aufwände aufbürdet.
Problem 2: Mangelnde Aktualität
Zur Qualität der Normen trägt auch deren Aktualität bei. Die IEC 62304 beispielsweise stammt in wesentlichen Teilen aus dem Jahr 2005. Erst zwei Jahre später wurde das erste iPhone veröffentlicht. Smartphones, Cloud-Computing, KI hatten damals nicht annähernd die Bedeutung wie heute. Der NSA-Skandal lag noch acht Jahre in der Zukunft. Soll solch eine Norm den Stand der Technik abbilden?
Problem 3: Mangelnde Abdeckung der regulatorischen Anforderungen
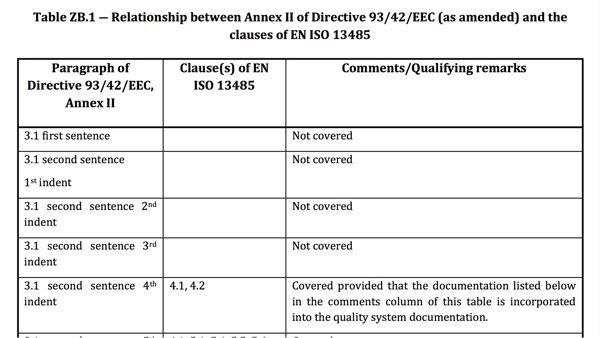
Selbst aktuelle harmonisierte Normen adressieren meist nicht alle Forderungen der EU-Verordnungen.
Die „Z-Anhänge“ verschaffen einen Überblick darüber, welche regulatorischen Anforderungen Hersteller bei Einhaltung der jeweiligen harmonisierten Norm als erfüllt vermuten lassen können und welche nicht.
Problem 4: Hohe Kosten
Normen kosten teilweise mehrere Hundert Euro. Das Johner Institut überwacht im Auftrag der Hersteller und Benannten Stellen über 7.000 regulatorische Dokumente. Ein nennenswerter Teil sind Normen. Auch wenn der typische Hersteller nur eine Teilmenge benötigt, ist die finanzielle Belastung nennenswert.
Die estnischen Standards (auch auf Englisch und mit harmonisierten Inhalten) kosten nur einen Bruchteil.
Problem 5: Zögerliche und unvollständige Harmonisierung
2017 trat die MDR in Kraft. Über sechs Jahre später ist nur ein Bruchteil der Normen harmonisiert, die unter der MDD und AIMD harmonisiert waren. Analog sieht es bei der IVDR aus.
b) Die Ursachen
Es gibt viele Ursachen, die zu diesen Problemen beitragen:
- Bürokratie
Manche Normengremien legen sich selbst mit Bürokratie still. Man ist mehr mit Regularien, internen Vorschriften, internationalen Abstimmungen und manchmal auch Streit beschäftigt als mit der eigentlichen Normenarbeit. - Mangelnde Ressourcen und Kompetenzen
Daher ist es nicht leicht, die Besten eines Fachs für die Normenarbeit zu begeistern. Dabei sind höchste Kompetenzen erforderlich: Domänenexpertise, wissenschaftliche Methodik sowie die Fähigkeit, abstrakte Modelle zu entwickeln und gleichzeitig die Praxisrelevanz und Umsetzbarkeit in der Praxis sicherzustellen. - Konkurrierende Präferenzen
Die EU-Kommission erweckt nicht den Eindruck, als würde sie den Normen noch die gleiche Bedeutung schenken wie in der Vergangenheit. Millionen gibt sie für HAS-Consultants aus, welche in meinen Augen das Prozedere eher verschleppen. Zudem hat sich die EU mit den Common Specifications eine Alternative zu den Normen geschaffen, die sie in den eigenen Händen hält.
c) Die Folgen für Hersteller und Benannte Stellen
Die Probleme mit den Normen führen dazu, dass sich Behörden und Benannte Stellen mal auf die Gesetze beziehen, mal auf die neusten Versionen der Normen, mal auf die harmonisierten Versionen der Normen, mal auf andere Best Practices und Leitlinien.
Für die Hersteller bedeutet dies:
- Rechtsunsicherheit in Audits und bei Zulassungen,
- mehr Aufwände für Nachbesserungen,
- damit verbunden höhere Kosten und somit
- eine längere Time to Market.
All dies geschieht in einem Wettbewerbsumfeld, das die Hersteller bis an oder über die Grenzen fordert.
d) Die Lösungsansätze
Um diese Probleme in Europa zu lösen, gibt es mehrere Ansätze. Wir sollten sie alle (!) verfolgen.
Zur ursprünglichen Idee zurückkehren
Sehr hilfreich wäre es, wieder voranzuschreiten und die Normen zu verbessern, zu aktualisieren, aufeinander abzustimmen und zu bezahlbaren Preisen anzubieten. Die Mitglieder der Normengremien arbeiten bereits unentgeltlich.
Die Wahrscheinlichkeit eines solchen Neustarts ist aber begrenzt, wenn der Wille fehlt und eine korrekt gelebte Bürokratie wichtiger erscheint als der Beitrag zum Wohl der Gesellschaft, z. B. der Patienten.
Dennoch oder gerade deshalb unsere Bitte: Engagieren Sie sich in den Normengremien!
Die Bedeutung von Normen für den Fortschritt erkennt und betont auch die EU.
Effizienz auf Herstellerseite optimieren
Die Aufgaben der Hersteller bestehen aus mehr als „nur“ dem Nachweis der Konformität ihrer Produkte und Prozesse. Hier gibt es Optimierungsmöglichkeiten:
- Das Rad muss nicht jedes Mal neu erfunden werden: Bei Software lassen sich nicht nur Komponenten und Frameworks wiederverwenden, sondern auch ganze Backend-Services. Mehr darüber erfahren Sie in dieser Podcast-Episode.
- Viele Aufgaben lassen sich durch Automatisierung komplett vermeiden. Die Gesetzeskonformität lässt sich automatisiert nachweisen.
De-facto-Standards schaffen
Die Entscheidung über die Konformität von Produkten und Prozessen ist eine binäre: konform oder nicht konform. Sie ergibt sich aus vielen Mikro-Entscheidungen, etwa: Liegt die geforderte Liste an SOUPs vor? Ja oder nein?
Diese Entscheidungen kann man in Algorithmen abbilden – das tun wir. Wenn es eine Verständigung über diese Algorithmen gibt, ist ein de-facto-Standard geschaffen und damit Rechtssicherheit und schließlich die Basis für die Automatisierung.
4. Ist eine Zertifizierung notwendig?
Hersteller fragen sich, ob sie sich bzw. ihre Produkte nach einer Norm zertifizieren müssen. Die kurze Antwort lautet:
Wenn ein Konformitätsbewertungsverfahren (z. B. gemäß Anhang II der MDR bzw. Anhang IX der MDR) ein Qualitätsmanagementsystem verlangt, müssen Hersteller dieses QM-System von einer Benannten Stelle zertifizieren lassen. Im Erfolgsfall erhalten sie ein Zertifikat, das die Konformität mit dem entsprechenden Anhang (und meist auch der ISO 13485) bestätigt.
Bei der Überprüfung des QM-Systems prüfen die Benannten Stellen auch,
- ob die Hersteller konform mit den harmonisierten Normen wie der ISO 14971 oder IEC 62304 arbeiten bzw.
- ob die Vorgaben des QM-Systems ein Vorgehen verlangt, das mit diesen Normen konform ist.
Ein Zertifikat, das die Konformität mit den Anforderungen der ISO 14971, IEC 62304, IEC 62366-1 usw. bestätigt, ist weder verlangt noch üblich.
Zu den Ausnahmen gehören Normen wie die IEC 60601-1. Aber auch hier muss sich nicht der Hersteller zertifizieren lassen. Vielmehr ist es zielführend, ein Testlabor auszuwählen, das die Prüfungen konform mit dieser Norm bzw. Normenfamilie durchführt.
5. Welche Norm nutzen?
a) DIN oder EN oder ISO/IEC
Eine häufig gestellte Frage lautet: Welche Varianten der Normen soll ich heranziehen? Die DIN, die EN oder die ISO bzw. die IEC?
Anhand harmonisierter Normen können Hersteller den Nachweis führen, dass ihre Produkte oder Systeme den Anforderungen der europäischen Richtlinien bzw. Verordnungen genügen. Daher sind die europäischen Normen zu nutzen, also die EN-Normen. Die nationalen Normen, d. h. die Normen mit einem Präfix wie „DIN EN“, sind inhaltlich identisch und daher ebenfalls nutzbar.
Die internationalen Normen (ISO, IEC) verfügen im Gegensatz zu den „EU-Varianten“ nicht über die Z-Anhänge. Diese Z-Anhänge enthalten das „Mapping“ der regulatorischen Anforderungen (EU-Richtlinien, EU-Verordnungen) auf die normativen Anforderungen. Das heißt: Die Z-Anhänge beschreiben, welche regulatorischen Anforderungen vollständig, teilweise oder gar nicht durch die Norm abgedeckt sind.
Die Harmonisierung hat nichts mit der Nummerierung zu tun. Beispielsweise ist die DIN EN 14971 nicht die Norm zum Risikomanagement bei Medizinprodukten, sondern eine zu „Textilien – Maschenwaren – Bestimmung der Maschenzahl je Längeneinheit und Flächeneinheit“.
b) Welche Version der Norm?
Diskussion gibt es immer wieder zu den Übergangsfristen. Leider gibt es keine eindeutigen Regeln. Die folgenden Daumenregeln können helfen:
- Falls eine Norm harmonisiert ist, dann nutzen Sie diese Version.
- Wenn die Norm selbst eine Übergangsfrist nennt, dann beachten Sie diese.
- Andernfalls gehen Sie von einer Übergangsfrist von drei Jahren aus.
- Arbeiten Sie bei neuen Produkten mit den neuesten Versionen.
- Machen Sie bei Produkten, die sich bereits im Markt befinden, spätestens drei Jahre, nachdem eine neue Version veröffentlicht wurde, eine Gap-Analyse.
6. Was sind medizinische Normen?
Gelegentlich wird von „medizinischen Normen“ gesprochen. Doch es ist nicht definiert, welche Normen als „medizinische Normen“ zählen. Meist versteht man darunter:
- Normen im Kontext von Medizinprodukten, z. B. Normen in der Medizintechnik wie die IEC 60601-1 oder die ISO 7396-1 (Rohrleitungssysteme für Gase für pharmazeutische Anwendungen, für Medizinproduktgase …)
- Normen zu medizinischer / persönlicher Schutzausrüstung (PSA)
- Seltener: Normen im Kontext des Betriebs von Gesundheitseinrichtungen wie die DIN EN 15224
Daher sollte nicht von „medizinischen Normen“ gesprochen werden, sondern der jeweilige Kontext klargestellt werden.
7. Fazit und Zusammenfassung
Harmonisierte Normen sollten Herstellern und Benannten Stellen helfen, ein gemeinsames Verständnis zu entwickeln, wie die Anforderungen der MDR und IVDR erfüllt werden sollen.
Leider stockt die Harmonisierung der Normen, sodass dieses Ziel nur teilweise erfüllt wird. Dennoch sollten Hersteller einschlägige Normen nutzen. Diese beschreiben den Stand der Technik und helfen nachzuweisen, dass regulatorische Anforderungen erfüllt sind.
Änderungshistorie
- 2025-04-16: In Kapitel 1.c) Normen sowie Link auf Webseite der EU eingefügt
- 2024-12-19: Kapitel 6 „Was sind medizinische Normen?“ eingefügt
- 2024-05-08: Einleitung und Kapitel 1.a) neu geschrieben. Tabelle in Kapitel 1.b) eingefügt. Redaktionelle Änderungen, um die Bereiche besser zu trennen, welche alle Hersteller in Europa betreffen bzw. welche für Medizinprodukte spezifisch sind.
- 2023-09-06: Kapitel 2 zerlegt. Kapitel 3 neu eingefügt.
- 2023-04-03: Beitrag komplett überarbeitet und aktualisiert. Die Abschnitte „Aktuelles“ und „Probleme“ entfernt und, wo notwendig, in den Text integriert.
- 2022-06-09: In der Sektion „Aktuelles“ den Abschnitt „Juni 2022“ eingefügt.
- 2022-03-16: In der Sektion „Aktuelles“ den Abschnitt „Januar 2022“ eingefügt.
- 2021-07-27: In der Sektion „Aktuelles“ den Abschnitt „Juli 2021“ eingefügt.
- 2021-04-26: In der Sektion „Aktuelles“ den Abschnitt „April 2021“ eingefügt.
- 2020-11-08: Neuen Entwurf für einen Standardization Request ergänzt. Text diesbezüglich angepasst.



Sehr geehrter Herr Professor Dr. Johner,
Ihre Anregungen zu Übergangsfristen für den Fall „Was mache ich, wenn es keine (aktuelle) harmonisierte Norm gibt?“ finde ich sehr gewagt.
1. Es gibt meines Wissens sehr wenige Normen, die selbst eine Übergangsfrist definieren. Im Normalfall ersetzt eine neue Version der Norm die alte Norm. Übergangsfristen sind regulatorische Themen, die dann von z.B. der EU, FDA; Health Canada über die Instrumente der Harmonisierung, Recognition etc. abgebildet werden
2. Ein neues Produkt auf den Markt zu bringen, wenn eine neue Norm (=Stand der Technik) schon publiziert ist und diese nicht zu berücksichtigen (ggf. als Gap-Analyse wie beschrieben) halte ich für grob fahrlässig
und 3. für Produkte die bereits im Markt sind, spielen neue Normen erstmal gar keine Rolle. Für Produkte die unverändert In-Verkehr gebracht werden (ich nehme an, dass sie diese meinen) ist eine Übergangsfrist von 5 Jahren wiederum auch sehr lang. Für technische Normen sind drei Jahre Übergangsfrist, das was in den meisten Fällen auch bei der Harmonisierung angegeben wurde.
Grundsätzlich kann die Empfehlung in Hinblick auf den Umgang mit neuen Normen und deren Übergangsfristen nur lauten: Besprechen Sie das mit ihrer benannten Stelle. Nur ihre benannte Stelle kann diese Frage beantworten.
Sehr geehrter Herr Suchi,
danke für Ihre wertvollen Rückmeldungen! Zu Ihren Punkten:
Ihrer Empfehlung, die anzuwendenden Normen mit der benannten Stelle abzusprechen, stimme ich ausdrücklich zu.
Herzlichen Dank für Ihre Rückmeldung und die Möglichkeit, dadurch Dinge noch klarer darzustellen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ich bezog mich auf die „Anregungen“ bzw. „Daumenregeln“ im genannten Abschnitt:
Eine weitere Diskussion betrifft die Übergangsfristen. Leider gibt es keine ganz klaren Regeln. Aber die folgenden Anregungen können vielleicht helfen:
– Wenn die Norm selbst eine Übergangsfrist nennt, dann nehmen Sie die.
– Andernfalls nehmen Sie für neue Produkte drei Jahre.
– Und für Produkte, die bereits im Markt sind, hält man fünf Jahre für akzeptabel.
Mit freundlichen Grüßen,
Andreas Suchi
Sehr geehrter Herr Prof. Johner
Was macht denn die FDA mit ihren Recognized Consensus Standards anders – oder sogar besser…?
Beste Grüsse,
Markus Angst
Die FDA weißt den Consensus Standards eine noch geringere Rolle zu. Die Ohnmacht gegenüber der Behörde, die das Grundprinzip der Trennung von Legislative, Exekutive und Judikative verletzt, ist auch ein gewaltiger Kritikpunkt. Das eine macht das andere nicht besser.
Dass die FDA bessere Experten zur Verfügung hat, ist vielleicht Spekulation. Sie beteiligt sich aber aktiv an der Erstellung von Normen. Das empfinde ich als konstruktiver.
Sehr geehrter Herr Johner,
ich finde es toll, dass Sie Ihre Beiträge so fleißig updaten. Könnten Sie uns noch die Quelle angeben für die Liste der EU für normen die Sie plant zu harmonisieren? Leider finde ich dazu nichts im Netz. Das wäre super toll!
Danke vorab.
Mit freundlichen Grüßen Jennifer Szymkiewicz
Sie finden das auf den Seiten der EU: https://ec.europa.eu/docsroom/documents/36104?locale=en
Ich werde den Link ergänzen, danke für den Hinweis!
Guten Tag,
leider funktioniert der Link nicht mehr und ich lande auf einer Seite 404.
Ggf. handelt es sich nur um ein temporäres Problem … oder kennen sie einen neueren Link?
Sehr geehrter Herr Kreuzer,
vielen Dank für den Hinweis! Ich vermute, dass es sich um ein temporäres Problem gehandelt hat. Ich habe den Link geprüft und er müsste eigentlich funktionieren. Könnten Sie es erneut versuchen und mir Rückmeldung geben?
Liebe Grüße
Tea Bodrusic
Guten Tag,
inzwischen funktioniert der Link wieder.
War also falscher Alarm…
Vielen Dank für die zügige Antwort!
Johannes Kreuzer
Es scheint, als würde die Liste der Normen, welche zur Harmonisierung im Rahmen der MDR / IVDR vorgsehen ist, länger werden.
Im Netz (https://polit-x.de/documents/3170588/europa/deutsch/europaische-kommission/ec-allgemein/mitspracherecht-komitologieakte-2020-03-12-entwurf-eines-durchfuhrungsbeschlusses-der-kommission-uber-einen-normungsauftrag-an-das-europaische-komitee-fur-normung-und-das-europaische-komitee-fur-elektrotechnische-normung-in-bezug-auf-medizinprodukte-zur-unterstutzung-der-verordnung-eu-2017745-des-europaischen-parlaments-und-des-rates-und-auf-in-vitro-diagnostika-zur-unterstutzung-der-verordnung-eu-2017746-des-europaischen-parlaments-und-des-rates) findet sich ein Entwurf
„Europäische Kommission COM-AC_DR(2020)D066402-01
Entwurf eines Durchführungsbeschlusses der Kommission über einen Normungsauftrag an das Europäische Komitee für Normung und das Europäische Komitee für elektrotechnische Normung in Bezug auf Medizinprodukte zur Unterstützung der Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates und auf In-vitro-Diagnostika zur Unterstützung der Verordnung (EU) 2017/746 des Europäischen Parlaments und des Rates“
Die Liste ist von ca. 30 auf ca. 40 Seiten angewachsen. U.a. finden sich auf der Liste zur Anpassung / Fortschreibung jetzt die meisten Normen der Reihe 80/60601-2-x, der Reihe 80001-x, 82304-1, etc.
Auf einer Liste neu zu erstellender Normen finden sich zahlreiche ISO und IEC-Normen für die noch kein EN-Äquivalent existiert, z.B. ISO 20417, IEC 80001-5, aber auch IEC TR 60601-4-5, …
Als Datum für die Annahme ist weitgehend Ende Mai 2024 angegeben.
Großartig, danke, lieber Herr Hagen!
Ich werde das gleich im Instituts-Journal mit aufnehmen und am Wochenende auch den Artikel aktualisieren. Danke, dass Sie helfen, diesen auf dem neusten Stand zu halten!
Herzliche Grüße, Christian Johner
Bitte COMMISSION IMPLEMENTING DECISION (EU) 2022/757 of 11 May 2022
https://eur-lex.europa.eu/eli/dec_impl/2022/757/oj mit aufnehmen.
Wird häufig übersehen und ist gerade bzgl. EN ISO 14971:2019 wichtig!
Vielen Dank für die Info, lieber Herr Jurkiewicz! Den Hinweis nehmen wir gleich mit auf.
Hallo,
es wird empfohlen die europäischen Normen, also die EN Normen zu nutzen. Meines Wissen kann ich diese Normen aber gar nicht kaufen, sondern nur die nationalen Normen. Darf ich denn in meiner Standards-List auf eine Norm verweisen (bspw. EN ISO 13485) die in meinen Unternehmen gar nicht vorliegt, da ich eigentlich mit der DIN EN ISO 13485 oder der BS EN ISO 13485 gearbeitet habe (und deren Besitz ich auch vorweisen kann)?
Liebe Frau Tschirner,
Das stimmt, Sie können immer nur die IEC Version als englische Version direkt kaufen. Die EN Version können Sie bei den EU Ländern kaufen. Viele Länder bieten die Norm zweisprachig an. Die länderspezifischen EN Normen (BS, DIN, SNV etc.) dürfen sich textlich nicht unterscheiden. Daher ist es auch akzeptabel, dass Sie in Ihrer Normentabelle die EN Version nennen und dann die DIN oder BS Version im Haus haben.
Liebe Grüsse, Mario Klessascheck
Harmonisierte Norm ist MDR sprechende Norm?
Wenn nicht harmonisierte Norm die Testung immer als stand anfordet, ohne unwichtiges wording von MDR zu haben. Macht alles ueberhaupt Sinn?
Das heistst doch dass das nicht EU land alles falsch mit nicht harmonisierter Norm macht? 🙂
Liebe Frau Seitz,
Ich bin nicht sicher, ob ich den Kontext Ihrer Frage richtig verstanden habe. Sie fragen, ob für ein Produkt Konformität zur MDR erklärt werden kann, wenn der Hersteller ausserhalb der EU mit einer nicht Norm geprüft hat, die nicht harmonisiert ist. Stimmt das? Wenn Sie ein Produkt in der EU in Verkehr bringen möchten, und eine Norm für den Nachweis mit den Anforderungen der MDR verwenden möchten (Die Verwendung einer Norm ist freiwillig.) sollten Sie auch die europäische Version der Norm als die EN Version verwenden. Nicht alle EN Normen sind harmonisiert, aber dennoch anwendbar. Erstellen Sie für Ihr Dossier eine Liste mit den angewendeten Normen für das Produkt und zeigen Sie welche Anforderungen Sie damit erfüllen.
Konnte ich Ihre Frage beantworten?
Liebe Grüsse, Mario Klessascheck
Sehr geehrtes Johner Team,
wir führen aktuell eine leidige Diskussion mit unserem NB was die Referenzierung von angewendeten Normen betrifft. Oftmals sind diese ja nicht immer zu 100% anwendbar. Muss bei der Angabe, welche Normen zur Anwendung kommen, immer mit angegeben werden, ob diese ganz oder nur in Teilen angewendet werden? Und für den Fall, dass Teile der Normen nicht angewendet werden, sind diese Teile explizit anzuführen bzw. ist dann jeweils zu begründen, wieso dieser Teil nicht angewendet wurde bzw. nicht relevant ist oder gibt es hier einen pragmatischeren Ansatz? Gibt es eine Vorgabe wie zwischen voll und teilweise unterschieden werden kann? Herzlichen Dank für Ihre wertvolle Unterstützung!
Beste Grüße
Stefan Scheider
Lieber Herr Schneider,
mit der Frage sind Sie nicht allein. Wenn Sie über eine Norm Konformität erklären wollen, müssen Sie angeben, welche Teile für die Konformitätsvermutung relevant sind. Bei harmonisierten Normen finden Sie die Angaben in den Z-Anhänge. Der pragmatische Weg wäre, dass Sie in der GSPR-Checkliste den Status „vollständig“ oder „nicht vollständig“ angeben und im Prüfbericht zu einer Norm (insbesondere bei Teilen, die Sie selbst prüfen) die nicht anwendbaren Abschnitte mit „NA“ kennzeichnen. So macht es zum Beispiel auch die IEC 60601-1, was weitläufig akzeptiert wird. Wenn Sie eine neuere oder ältere Norm als eine harmonisierte Norm verwenden, begründen Sie das am besten in der Liste der angewendeten Normen oder Sie erstellen eine Gap-Analyse.
Liebe Grüsse, Mario Klessascheck
Sehr geehrtes Johner Team,
mittlerweile sind mehrere Normen mit der MDR harmonisiert, wie z.B. EN 14971. Wie können wir die Konformität mit MDR harmonisierten Normen für Legacy Devices, die noch unter MDD geführt werden, nachweisen? In diesem Fall ist es schwierig zu sagen, dass die Norm vollständig angewendet wird. Ein weiteres Beispiel ist die EN 15223, die Symbole für UDI definiert, Legacy Devices sind noch nicht verpflichtet, UDI zu verwenden.
Sehr geehrte Frau Haßelfeldt,
auch für Legacy Devices müssen Sie sicherstellen, dass diese dem Stand der Technik entsprechen. Das bedeutet, dass Sie die neuen Versionen verfolgen und über eine Gap-Analyse sicherstellen sollten, dass sie keine wesentlichen Anforderungen übersehen. Sie sollten somit prüfen, welche Anforderungen Sie nicht erfüllen, und abschätzen, ob dadurch keine inakzeptablen Risiken entstehen.
Im Fall der ISO 14971 sind das v.a. die Anforderungen an die Produktions- und Post-Produktionsphase. Insbesondere letztere ist durch die MDR auch für Legacy Produkte gefordert und zu beachten. Für die Post-Market Surveillance gelten die MDR-Anforderungen.
Für die Legacy Produkte gelten die UDI Pflichten nicht. Lesen Sie gerne mehr zu den Übergangsfristen hier.
Beste Grüße, Christian Johner
Vielen Dank für Ihre Antwort.
Beste Grüße,
Karen Haßelfeldt
Lieber Herr Hasselfeldt,
Wenn eine neue Norm veröffentlicht wird, sollten Sie prüfen, inwieweit Ihre altrechtlichen Produkte davon betroffen sind. Bei diesen Produkten würden Sie die relevanten Abschnitte der Norm in die Checkliste der grundlegenden Anforderungen zuordnen. Diese Checkliste muss jedoch nur aktualisiert werden, wenn eine Änderung in der Norm auch eine Anpassung am Gerät oder in der Gebrauchsanleitung erforderlich macht. Falls beispielsweise für ein bestimmtes Gerät noch keine UDI-Pflicht besteht, wäre die entsprechende Norm nicht anwendbar. Diese Information können Sie ebenfalls in der Liste der angewendeten Normen dokumentieren. Es ist auch nicht zwingend erforderlich, dass eine Norm in ihrer Gesamtheit angewendet wird. In diesem Fall, würden Sie das ebenfalls in der MDR-Checkliste oder MDD-Checkliste vermerken.
Liebe Grüsse, Mario Klessascheck
Vielen Dank für Ihre Antwort.
Beste Grüße,
Karen Haßelfeldt
Liebes Johner-Team,
wir sind auf der Suche nach Standards, die in Australien für die Konformitätsbewertung von Medizinprodukten gelten, auf die „Therapeutic Goods (Conformity Assessment Standard for Quality Management Systems) Order 2019“ (https://www.legislation.gov.au/F2019L00426/latest/versions) gestoßen. Dort wird aber bisher nur die ISO 13485 für QMS bei Medizinprodukten gelistet und noch einige Standards für steril ausgelieferte Medizinprodukte. Im dazugehörigen ´replacement explanatory statement´ schreibt die TGA: „[…] also recommended that the former Order be replaced with a more comprehensive CASO that references the EU’s harmonised standards (for the purposes of affording a presumption of conformity with the EU’s requirements) and the recognised consensus standards published by the United States’ Food and Drug Administration.“ Bisher ist eine solche Veröffentlichung/Liste mit weiteren anerkannten Standards von der TGA jedoch nicht erschienen, oder wissen Sie hierzu evtl. mehr?
Vielen herzlichen Dank für Ihre immer hilfreiche Unterstützung!
Beatrice Dachsel
Liebe Frau Dachsel,
Meines Wissens gibt es noch keine relevanten Standard Orders. Sie können daher die international anerkannten Normen weiterhin verwenden. Wenn Sie zu einer Norm oder zu einem Essential Principle eine spezifische Frage haben, verbinde ich Sie gern mit meiner Kollegin, die in Neuseeland unsere Tochtergesellschaft führt.
Liebe Grüsse, Mario Klessascheck
Guten Tag
Unter 1 c) ist eine Liste von (möglichen) harmonisierten Normen aufgeführt. Meines Wissens sind davon nur EN ISO 13485 und EN ISO 14971 mit der MDR harmonisiert. Die EN IEC Normen gibt es so (noch) nicht, unter der MDD waren die Normen als EN 62304 / 62366-1 / 60601-1 harmonsiert (keine 1:1 Übernahme der IEC, darum nur als EN).
Herzlichen Dank, Herr Egli!
Sie haben völlig Recht! Die Normen sind erst für die Harmonisierung vorgesehen. Das hängt z.T. in den Normengremien, weil man dort die „Z-Anhänge“ erstellen müsste. Diese Arbeit ist etwas ermüdend und manchmal auch etwas politisch.
Das meines Erachtens noch größere Problem ist, dass die Normen z.T. völlig veraltet sind. Hier stehen sich die Normengremien teilweise selbst im Weg.
Das traurige Ergebnis ist, dass die Hersteller unter einer noch größeren Rechtsunsicherheit leben müssen. Dieses Mal kann man nicht alles der EU in die Schuhe schieben…
Nochmals vielen Dank!
Beste Grüße, Christian Johner
Guten Tag,
wir führen in unserer List of Applied Standards aktuell die harmonisierten Normen sowie die aktuelleren Versionen davon. Wir führen auch im Fall einer Normenaktualisierung eine GAP-Analyse zur harmonisierten Version durch. Nun stellt sich immer wieder die Frage, ob diese GAP-Analyse ausreichend ist, oder ob wir mit jeder neuen Normenversion die Konformitätsbewertung neu durchführen müssten. Wie sehen Sie das?
Liebe Frau Esslinger,
Die Gap-Analyse ist ausreichend. Kleiner Tipp. Bewerten Sie jede Änderung in der Norm unter in die Aspekte: a) textliche Klarstellung, b) Änderung der Kennzeichnung mit Einfluss auf das Produkt, c) Änderung einer Spezifikation mit Einfluss auf Produkt und bestehende Prüfergebnisse und d) Neue Anforderung mit Einfluss auf Produkt. In den Fällen b) bis d) können Deltaprüfungen notwendig sein. Wenn keine der Änderungen auf Ihr Produkt zutrifft, dann reicht die Gap-Analyse als Protokoll aus.
Hilft Ihnen das weiter?
Liebe Grüsse, Mario Klessascheck