
Mit dem UDI-System hat die EU eine Pflicht zur Identifikation und Registrierung von Medizinprodukten eingeführt, die weit über das unter der MDD noch Geforderte hinausgeht. Selbst für Standalone-Software fordert die Medical Device Regulation MDR eine UDI.
Lesen Sie hier, worauf Sie sich vorbereiten müssen.
1. Ziele des UDI-Systems
Die EU möchte Medizinprodukte schnell und einfach nachverfolgen können – vom Hersteller bis zum Anwender. Das erlaubt es, bei Zwischenfällen schnell zu reagieren, weil mithilfe des UDI-Systems
- Produkte einfacher zu identifizieren sind (bis hinunter auf das einzelne Gerät, den Batch oder eine Software-Version),
- die Standorte der Geräte nachvollziehbar und damit die Anwender schneller und spezifischer auffindbar sind,
- illegal vermarktete Medizinprodukte leichter identifiziert werden können.
2. Wie das UDI-System funktioniert
a) Organisation: Identifikationsnummer vergeben
Eine oder mehrere Organisationen sollen ein System betreiben, das UDIs vergibt und sicherstellt, dass diese einzigartig sind. Diese Organisationen werden Zuteilungsstellen genannt und müssen das über einen Zeitraum von mindestens 10 Jahren bleiben. Momentan sind folgende Organisationen benannte Zuteilungsstellen gemäß MDR Artikel 27 Absatz 2 / IVDR Artikel 24 Absatz 2:
- GS1
- Industry Business Communications Council (HIBCC)
- ICCBBA
- Informationsstelle für Arzneispezialitäten (IFA GmbH)
Oft wird der Begriff UDI Code verwendet. Diese Beschreibung ist aber unpräzise und lässt nicht unterscheiden, welcher Typ an UDI gemeint ist.
b) Hersteller: UDI an Produkten und Verpackungen anbringen
Bei den Zuteilungsstellen beziehen die Hersteller für jedes Medizinprodukt, aber auch für jede übergeordnete Verpackung (mit Ausnahme der Versandcontainer) eine eindeutige Identifikationsnummer. Einzig bei Sonderanfertigungen und Produkten zur klinischen Erprobung verzichtet die MDR auf die UDI für Medizinprodukte.
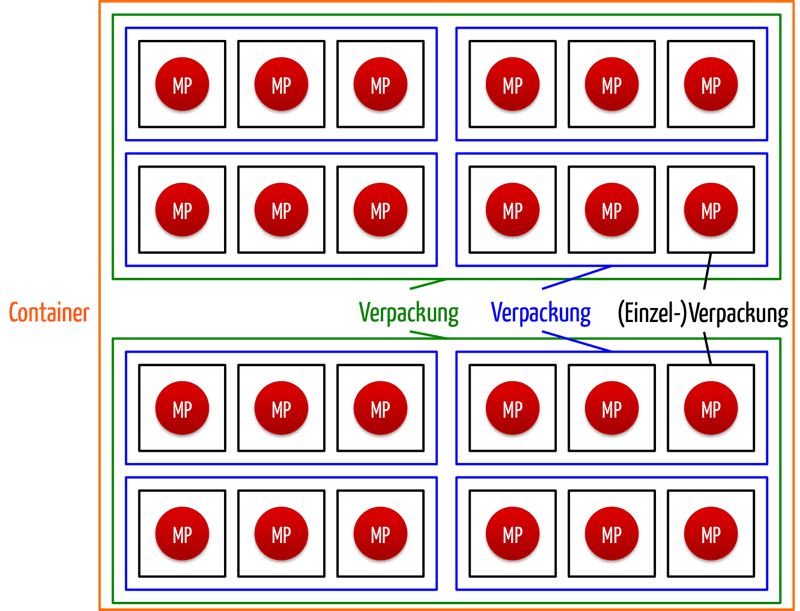
Die Hersteller müssen die Liste aller vergebenen UDIs für jedes Medizinprodukt als Teil der technischen Dokumentation pflegen und die sogenannte Basis-UDI-DI in der Konformitätserklärung angeben. Was die Basis-UDI-DI ist, erfahren Sie weiter unten.
c) Betreiber: UDIs der Medizinprodukte abspeichern
Die EU ermutigt die Mitgliedstaaten, auch die Betreiber dazu zu verpflichten, die UDIs der verwendeten bzw. gekauften Medizinprodukte zu speichern. Die MDR verpflichtet die sogenannten Gesundheitseinrichtungen, die abgegebenen oder bezogenen UDIs zu erfassen und zu speichern, sofern die bezeichneten Produkte zu den implantierbaren Produkten der Klasse III gehören. Diese Speicherung sollte vorzugsweise elektronisch geschehen (MDR, Artikel 27 (9)).
d) EU: UDI-Datenbank betreiben
Die EU soll eine Datenbank betreiben, in der alle UDIs für Medizinprodukte und IVD gespeichert werden. Sie legt fest, welche Attribute pro Medizinprodukt mindestens berücksichtigt werden müssen. Die Nutzung dieser Datenbank (zumindest der Zugriff) ist kostenfrei und öffentlich zugänglich.
Mehr zu dieser Datenbank, der sog. EUDAMED, finden Sie in unserem Fachartikel zur European Database on Medical Devices.
e) Hersteller: Informationen in UDI-Datenbank einpflegen und aktualisieren
Die Hersteller müssen die Basis-UDI-DI und die damit verbundenen Informationen wie UDI-DI, Produktversion, Einstufung (z. B. als steril oder zur Wiederverwendung geeignet) in der Datenbank abspeichern und aktuell halten.
3. Regulatorische Anforderungen
a) Überblick
Die EU formuliert die Anforderungen an das UDI-System in Artikel 27 (MDR), Artikel 24 (IVDR) sowie im Anhang VI (MDR und IVDR).
Der Anhang VI besteht aus mehreren Teilen:
- Teil A: Informationen, die bei der Registrierung des Produkts eingereicht werden müssen, u. a.
- Hersteller, Inverkehrbringer, Importeur
- Produkt (UDI, Typ, Haltbarkeitsdatum, Land der Inverkehrbringung, Klasse und „binäre“ Attribute wie „Single use“, „beinhaltet Gewebe“ oder „beinhaltet bestimmte Substanzen“)
- Teil B: Informationen, die in der UDI zu speichern sind. Diese sind teilweise deckungsgleich mit jenen aus Teil A.
- Teil C: Beschreibung des UDI-Systems, u. a.
- Definitionen
- Wann müssen welche UDIs wo aufgebracht werden? (Man unterscheidet z. B. UDI-DI und UDI-PI – dazu gleich mehr.)
- Menschen- und maschinenlesbare Identifikation
- Regeln für spezielle Produkttypen wie implantierbare Produkte, wiederverwendbare Produkte, Systeme, konfigurierbare Produkte, Software und im Fall der IVDR: Kits
b) UDI-DI und UDI-PI
Die MDR unterscheidet die beiden Unique Device Identifiers UDI-DI und UDI-PI:
- UDI-DI: Das ist die Device-Identifikation eines spezifischen Modells und dient in der UDI-Datenbank als Schlüssel. Beispielsweise haben alle Instanzen eines Defibrillators eines bestimmten Typs die gleiche UDI-DI.
- UDI-PI: Der Produktions-Identifier kennzeichnet jede einzelne Instanz eines Produkts bzw. eines Batches. Jeder Defibrillator hat somit eine eigene UDI-PI.
Lesen Sie weiter unten mehr zum Unterschied von Basis-UDI-DI und UDI-DI.
Die EU hat mehrere Dokumente veröffentlicht:
- 2013/172/EU: Empfehlung über einen gemeinsamen Rahmen für ein System einmaliger Produktkennung für Medizinprodukte in der Union
- UDI Helpdesk
- Durchführungsbeschluss (EU) 2019/939 zur Benennung der Zuteilungsstellen, die für den Betrieb eines Systems zur Zuteilung von eindeutigen Produktidentifikationen im Bereich der Medizinprodukte benannt sind
- Durchführungsbeschluss (EU) 2024/2120 zur Verlängerung der Benennung der Zuteilungsstellen, die für den Betrieb eines Systems zur Zuteilung von eindeutigen Produktidentifikationen (UDI) im Bereich der Medizinprodukte benannt wurden
c) Andere Rechtsbereiche
Die UDI für Medizinprodukte wird auch in anderen Ländern gefordert, beispielsweise von der FDA (s. u.). Das IMDRF beschreibt in einem Leitfaden ein System der einmaligen Produktkennung für Medizinprodukte.
4. UDI für Software
Für Standalone-Software gelten spezifische Regeln.
a) Vergabe einer neuen UDI-DI
Eine neue UDI-DI soll immer vergeben werden bei Änderungen an:
- „Performance“ und Effektivität
- Sicherheit
- Zweckbestimmung
- Interpretation von Daten
Als Beispiele nennt die MDR Änderungen an:
- Algorithmen
- Datenbankstrukturen
- Betriebssystem
- Architektur
- Benutzerschnittstelle
- Interoperabilität
b) Vergabe einer neuen UDI-PI
Hingegen wäre „nur“ eine neue UDI-PI notwendig bei kleinen Änderungen wie:
- Bugfixes
- Security Patches
- Benutzerschnittstelle (Wenn es nur um Usability, aber nicht um Verbesserung der Sicherheit geht.)
Man könnte als Hersteller sagen, dass alle Änderungen an der dritten Stelle der Versionsnummer zu einer neuen UDI-PI führen, alles andere zu einer neuen UDI-DI.
Eine Vergabe einer eindeutigen UDI-PI pro Installation wird nicht gefordert.
c) Labelling
Der Hersteller muss die UDI für Medizinprodukte darstellen:
- Wenn es physische Datenträger gibt (USB, DVD …), müssen diese sowohl menschen- als auch maschinenlesbare UDIs enthalten. Das Gleiche gilt für alle Umverpackungen.
- Für die Anwender muss die UDI erkennbar sein, z. B. unter „About“ oder/und dem Splash-Screen. Eine Anzeige der maschinenlesbaren UDI auf dem Screen ist nicht gefordert.
- Bei Software ohne Benutzerschnittstelle müssen die Informationen über eine API abrufbar sein.
5. Basis-UDI-DI versus UDI-DI
a) Leitfäden
Die EU und ihre Medical Device Coordination Group (MDCG) haben mehrere Leitfäden publiziert, die Sie weiter unten unter „Aktuelles“ kommentiert finden.
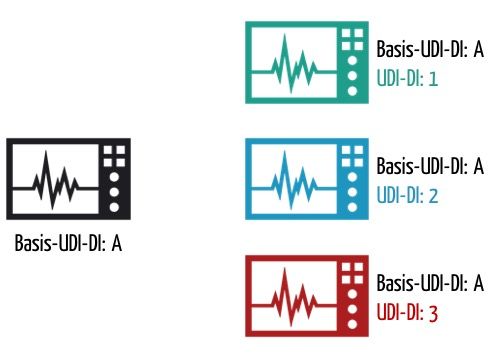
b) Was die Basis-UDI-DI ist
MDR und IVDR unterscheiden zwischen Basis-UDI-DI (Basic UDI-DI) und UDI-DI. Die Basis-UDI-DI soll eine Klammer für mehrere Varianten eines Medizinprodukts bilden. Beispiele für solche Varianten sind:
- Produkte mit unterschiedlichen Motorstärken
- Verschiedene Länderversionen (z. B. unterschiedliche Netzteile)
MDR und IVDR definieren die Basis-UDI-DI wie folgt:
Die Basis-UDI-DI ist die primäre Kennung eines Produktmodells. Es ist die Identifikation des Produkts, die auf Ebene der Gebrauchseinheit zugewiesen wird. Sie ist unabhängig von Verpackungseinheiten und gleichzeitig das wichtigste Ordnungsmerkmal für Datensätze in der UDI-Datenbank.
Die Basis-UDI-DI muss in den einschlägigen Bescheinigungen und EU-Konformitätserklärungen ausgewiesen werden.
Jede UDI-DI gehört zu genau einer Basis-UDI-DI.
c) Wo die Basis-UDI-DI verwendet wird
Sowohl MDR als auch IVDR verlangen, dass die Basis-UDI-DI in den folgenden Fällen angegeben wird:
- Auf der Konformitätserklärung gemäß Artikel 27 (6) (MDR) / Artikel 24 (6) (IVDR) und Anhang VI (MDR / IVDR). Allerdings muss die Konformitätserklärung auch die Produkte nennen, für die unter der gemeinsamen Basis-UDI-DI die Konformität erklärt wird.
- Bei der Eintragung der Produkte in die EUDAMED gemäß Artikel 29 bzw. Anhang VI der MDR / gemäß Artikel 26 der IVDR
- Im Kurzbericht über die Sicherheit und klinische Leistung gemäß Artikel 32 der MDR / Kurzbericht über Sicherheit und Leistung gemäß Artikel 29 der IVDR
- Auf den Freiverkaufszertifikaten gemäß Artikel 60 (MDR) / Artikel 55 (IVDR)
- In der technischen Dokumentation gemäß Anhang II (MDR/IVDR)
- In Vigilanz- und Post-Market Surveillance Reports, z. B. MIR und PSUR (MDCG 2021-19)
- Auf bestimmten EC-Zertifikaten, z. B. dem Zertifikat für die Bewertung der Technischen Dokumentation nach Anhang IX der MDR/IVDR (MDCG 2021-19)

Der schnellste und kostengünstigste Weg die UDI-Anforderungen konform umzusetzen!
Mit dem Auditgarant erhalten Sie ein Tool, um schnell UDI-Wissen aufzubauen und Dokumente mühelos zu erstellen. Unsere Vorlagen helfen dabei!
d) Welche Produkte unter einer Basis-UDI-DI zusammengefasst werden können
Übersicht
Gemäß der Leitlinie der UDI Working-Group enthalten die verschiedenen Basis-UDI-DIs/UDI-DIs die folgenden Datenelemente in der EUDAMED:
| Basic UDI-DI | UDI-DIs | UDI-DIs (container package DI) |
|
|
|
Tabelle 1: Die Pflichtfelder sind fett notiert; fett und kursiv sind die Felder, die verpflichtend sind, falls sie anwendbar sind. Unterstrichen sind die Datenelemente, bei denen Änderungen zu einer neuen Basis-UDI-DI/UDI-DI führen.
Details
Mehrere Varianten eines Medizinprodukts dürfen somit nur dann eine gemeinsame Basis-UDI-DI tragen, wenn die folgenden Felder übereinstimmen:
- Single Registration Number (SRN) des Herstellers
- Risikoklasse
- Implantierbar (j/n)
- Messfunktion (j/n)
- Wiederverwendbares chirurgisches Element (j/n)
- Aktives Medizinprodukt (j/n)
- Vorgesehen, um Arzneimittel zuzuführen oder zu entfernen (j/n)
- Medical Device Nomenclature Code (MDNC) (jetzt EMDN Code)
Im Falle eines IVD dürfen Hersteller nur dann eine gemeinsame Basis-UDI-DI tragen, wenn die folgenden Felder übereinstimmen:
- Wert der Basis-UDI-DI
- Single Registration Number (SRN) des Herstellers
- Risikoklasse
- Medical Device Nomenclature Code (MDNC) (jetzt EMDN Code)
Das heißt aber nicht, dass die Hersteller alle Produkte mit einer gemeinsamen Basis-UDI-DI versehen dürfen, deren o. g. Datenelemente gleich sind. Produkte dürfen laut MDCG 2018-1 v4 nur dann eine gemeinsame Basis-UDI-DI haben, wenn
- sie über die gleiche Zweckbestimmung verfügen,
- wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und
- wenn sich auch die Herstellung, also die Produktion im Wesentlichen nicht unterscheiden.
Nur wenn all diese Bedingungen erfüllt sind, erlaubt es die MDR, Produkte mit eigener UDI-DI zu einer Gruppe mit einer Basis-UDI-DI zusammenzufassen.
Weiterführende Informationen
Im MedTech Europe guidance for assigning Basic UDI-DI finden Sie auf Seite 5 ein detailliertes Flowchart. Dieses hilft Ihnen bei der Entscheidung, ob Sie eine neue Basis-UDI-DI vergeben müssen oder nicht. Beachten Sie auch die Erklärungen zu den jeweiligen Entscheidungen auf den Seiten 6 bis 8.
Interessanterweise müssen die Hersteller die „Medical Device Nomenclature“ auf Ebene der Produkte (UDI-DI) und nicht auf Ebene der Produktgruppe (Basis-UDI-DI) festlegen.
Diese Nomenklatur scheint granularer zu sein als die von der MDR eingeführte „Generic Device Group“ und die „Category of Devices“.
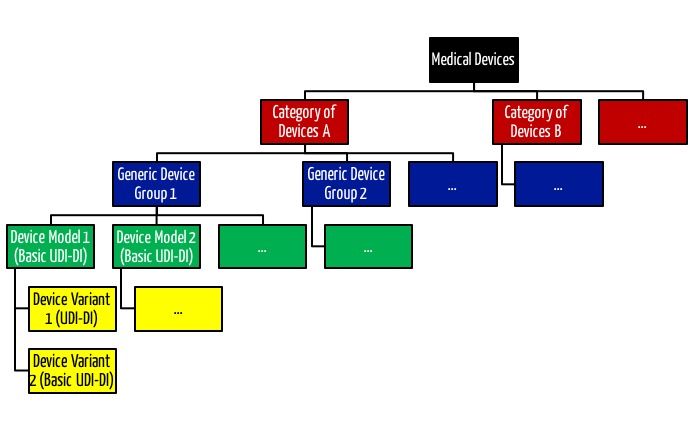
Lesen Sie hier mehr zum Thema Produktgruppen und Produktkategorien.
Sehen Sie sich dort auch das Video an, anhand dessen Sie diese Konzepte schnell verstehen werden.
6. Standards und Normen im Kontext der „UDI Codes“
Inzwischen gibt es noch eine Nomenklatur für Medizinprodukte, die für die EUDAMED genutzt werden soll. Zudem wurden einige Standards im breiteren Kontext der UDI veröffentlicht (kein Anspruch auf Vollständigkeit):
- ANSI/HIBC 2.4:2013 The Health Industry Supplier Labeling Standard: For Patient Safety & Unique Device Identification
- ISO/IEC 15415:2011-2-D Print quality standard (neue Version ist für 2024 geplant)
- ISO/IEC 15418:2016 Symbol data format semantics (GS1 application identifiers and ASC MH10 data identifiers and maintenance)
- ISO/IEC 15424:2008 Data Carrier Identifiers (including Symbology Identifiers) [IDs for distinguishing different barcode types]
- IISO/IEC 15434:2019 Syntax for high-capacity ADC media (format of data transferred from scanner to software, etc.)
- ISO/IEC 15459 Unique identifiers (note there are 6 parts to this standard)
- ISO/IEC 16022:2024 Data Matrix bar code symbology specification
- ISO 15223-1 Medical devices – Symbols to be used with medical device labels, labelling and information to be supplied – Part 1: General requirements (mehr Informationen im Fachbeitrag)
- GS1 DataMatrix-Leitfaden, den auch die FDA referenziert
- GS1 Leitfaden zur MDR und IVDR. Falls der Leitfaden dort nicht verfügbar ist, können Sie ihn hier herunterladen.
Lesen Sie hier mehr über die Standards UMDNS und GMDN.
Jede Zuteilungsstelle publiziert die Formate für die UDI (HRI und AIDC). Diese Spezifikationen können sie bei den Zuteilungsstellen kostenlos herunterladen. Bei der GS1 finden Sie hier eine Übersicht der Standards. Für Medizinproduktehersteller sind vor allem die Allgemeine GS1-Spezifikation (Stand 14. Mai 2022 in Version 22) sowie die GS1-GTIN-Vergaberegeln für das Gesundheitswesen (Stand 14. Mai 2022 in Version 8) relevant.
Die Zuteilungsstellen legen damit das Format des UDI Code fest.
7. Dokumente, die die UDI enthalten müssen
Die Anforderungen an die Verwendung der UDI sind über viele Artikel und Anhänge der MDR verteilt. Hier eine Übersicht:
| Englisch | Deutsch | Artikel, Verweis |
| Technical Documentation | Technische Dokumentation | Anhang II |
| Declaration of Conformity | Konformitätserklärung | Artikel 19, Anhang IV |
| Free Sales Certificate | Freiverkaufszertifikate | Artikel 60 |
| Summary of Safety and Clinical Performance (SSCP) | Kurzbericht über Sicherheit und klinische Leistung | Artikel 32 |
| Patient Information / Implant Card | Patienteninformation, Implantationsausweis | Artikel 18 |
| Periodic Safety Update Report (PSUR) | Regelmäßig aktualisierter Bericht über die Sicherheit | Artikel 86 |
| Reports regarding serious incident, Periodic Summary Report | Meldung von schwerwiegenden Vorkommnissen und Sicherheitskorrektur-Maßnahmen im Feld | Artikel 87 |
| Post-Market Clinical Follow-up (PMCF) Update Report | Bericht über die klinische Nachbeobachtung nach dem Inverkehrbringen | Anhang XIV Teil B |
| Field Safety Corrective Action (FSCA), Field Safety Corrective Notice | Sicherheitsrelevante korrektive Maßnahme im Feld, Sicherheitsinformationen | Artikel 87 |
| Repair, Refurbish, Maintenance Report | Reparatur-, Aufbereitungs- und Wartungsdokumente | optional |
| Logistic Documents (Delivery Note, Invoice, Production Order) | Logistikdokumente (Lieferscheine, Rechnungen, Fertigungsaufträge) | optional |
Danke an Martin Tettke von Berlin Cert für diese Übersicht.
8. Die UDI innerhalb Ihres Qualitätsmanagementsystems
Beachten Sie, dass Sie die Anforderungen der MDR/IVDR an die UDI in Ihr Qualitätsmanagementsystem (QMS) einbauen. Die Implementierung der UDI hat Auswirkungen auf verschiedene Teile Ihres QMS, beispielsweise auf die Produktion (Anbringung und Verifizierung der UDI-Träger), den Einkaufsprozess oder Ihr System für die Rückverfolgbarkeit.
Die Medical Device Coordination Group (MDCG) hat ein Guidance-Dokument publiziert, das auf die Integration der UDI in das Qualitätsmanagementsystem einer Organisation eingeht (MDCG 2021-19). Nutzen Sie diese Guidance als Checkliste bzw. Anleitung für die (korrekte) Integration aller UDI-Anforderungen in Ihr Qualitätsmanagementsystem.
9. Aktuelles (in zeitlich absteigender Reihenfolge)
Oktober 2024: Master UDI
Wie oben beschrieben, unterscheiden die MDR und IVDR die UDI-DI, die Basis-UDI und die UDI-PI. Die EU plant jetzt zusätzlich eine „Master UDI-DI“. Sie möchte damit für wenige Produktgruppen wie Kontaktlinsen die Aufwände für das Verwalten der UDIs verringern.
Sie können demnächst Feedback zu dieser Initiative geben und ggf. Produktgruppen vorschlagen, bei denen Sie ebenfalls Nutzen und Aufwand in einem ungünstigen Verhältnis sehen.
April 2022
In unserem Micro-Consulting häufen sich die Nachfragen zur sogenannten „Master UDI-DI“. Nach unseren letzten Informationen entwirft die EU einen Rechtsakt, der eine solche Master UDI-DI möglich machen soll. Mit dieser Master UDI-DI sollen stark individualisierte Produkte mit eindeutigen klinischen Ähnlichkeiten zusammengefasst werden können. Das Problem bei diesen stark individualisierten Produkten (z. B. Brillengläser) ist nämlich, dass der hohe Individualisierungsgrad zu sehr vielen UDI-DI Einträgen in der EUDAMED führt. Diese unverhältnismäßig große Zahl von Einträgen bringt weder einen regulatorischen noch einen sicherheitstechnischen Nutzen; deshalb hat die EU diese Initiative ins Leben gerufen.
Die MDCG hat bereits zwei Leitlinien zu dem Thema veröffentlicht:
- MDCG 2021-09: MDCG Position Paper on the Implementation of UDI requirements for contact lenses, spectacle frames, spectacle lenses & ready readers
- MDCG 2020-18: MDCG Position Paper on UDI assignment for Spectacle lenses & Ready readers
Februar 2020
Der Beitrag zu den Übergangsfristen bei der MDR geht auch auf die Übergangsfristen für die UDI und die UDI-Träger ein.
April 2019: Weitere Veröffentlichung der MDCG
Die MDCG schlägt vor, dass die EUDAMED so geändert wird, dass die Hersteller „legacy products“ auch ohne Basis-UDI-DI und UDI-DI in der EUDAMED registrieren können:
[The] MDCG considers it appropriate to adapt the Eudamed design to allow the registration of legacy devices in Eudamed in the absence of a (Basic) UDI-DI.
Quelle: MDCG 2019-05
Eine weitere Veröffentlichung der MDCG (MDCG 2019-04) betrifft den Zeitpunkt, zu dem die EUDAMED verpflichtend ist. Diese Klarstellung ist notwendig, weil der Artikel 123 missverständlich ist. Weiteres finden Sie im Artikel zur EUDAMED.
Oktober 2018: Veröffentlichungen der MDCG
Die Medical Device Coordination Group MDCG hat im Oktober gleich fünf Dokumente publiziert, die wir Ihnen hier kurz vorstellen wollen:
MDCG 2018-3 „Guidance on UDI for systems and procedure packs“
Das erste der neuen Dokumente adressiert die Systeme und Behandlungseinheiten. Es wiederholt die Definitionen der Begriffe und nennt eine Ausnahme: Stellt eine natürliche oder legale Person mehrere Produkte (z. B. Medizinprodukte) speziell für einen bestimmten Kunden „zusammen“ zur Verfügung, würde man dies nicht als Inverkehrbringung bezeichnen.
Weitere Klarstellungen sind:
- Eine natürliche oder legale Person, die Systeme oder Behandlungseinheiten in den Verkehr bringt, muss sich registrieren lassen.
- Sie muss für dieses System bzw. für diese Behandlungseinheit eine Basis-UDI-DI vergeben und die zugehörigen Attribute in der EUDAMED hinterlegen.
- Die Systeme und Behandlungseinheiten benötigen eine eigene UDI-DI und UDI-PI.
Das Dokument MDCG 2018-4 geht genauer auf die eben genannten Attribute ein.
MDCG 2018-5 „UDI Assignment to Medical Device Software“
Was der Sinn dieser Publikation ist, erschließt sich auch nach mehrfachem Lesen nicht. Das Dokument wirkt, als ließen die Autoren den Leser an ihrem eigenen Erkenntnisprozess teilhaben. Die Ergebnisse sind banal. Die Anforderungen bezüglich der UDI für Standalone-Software formuliert die MDR (und IVDR) bereits ausreichend klar:
- Das Konzept der Basis-UDI-DI gilt auch für Software. Die Basis-UDI-DI gruppiert Software mit sehr ähnlicher Zweckbestimmung.
- Eine Änderung der Basis-UDI-DI führt auch zu einer neuen UDI-DI.
- Eine Änderung des Software-Namens führt ebenfalls zu einer neuen UDI-DI.
- Das Gleiche gilt für nennenswerte Änderungen an der Software wie Zweckbestimmung, Kontraindikationen, neue Sprachen (Anwender-, nicht Programmiersprachen).
- Nur bei kleinen Patches darf die UDI-DI unverändert bleiben, und nur die UDI-PI ist zu ändern.
MDCG 2018-6 „Clarifications of UDI related responsibilities in relation to Article 16 of the MDR and IVDR“
Artikel 16 der MDR bzw. IVDR trägt den Titel „Fälle, in denen die Pflichten des Herstellers auch für Importeure, Händler oder andere Personen gelten“. In diesen Fällen sind die Wirtschaftsakteure (z. B. Händler, Importeure) verpflichtet, die Anforderungen der MDR zu erfüllen, die sich sonst an die Hersteller wenden. Dies schließt die Anforderungen an die UDI und die Registrierung dieser Wirtschaftsakteure ein.
Die MDCG stellt fest, dass die Wirtschaftsakteure von dieser Pflicht befreit sind, wenn es eine entsprechende Vereinbarung mit dem Hersteller gibt und dieser als solcher auf der Kennzeichnung angegeben ist.
Weshalb es dieser „Clarification“ bedarf, ist nicht ganz klar. Im Artikel 16(1)a) ist alles gesagt.
MDCG 2018-7 „Provisional considerations regarding language issues associated with the UDI database“
Die MDCG stellt fest, dass im Wesentlichen nur drei (der öffentlichen) Datenelemente in der EUDAMED einen Freitext erlauben. Da die EUDAMED u. a. das Ziel verfolgt, den europäischen Bürgern Transparenz zu verschaffen, stellt sich die Frage, in welcher Sprache diese Freitexte zu hinterlegen sind.
Die MDCG schlägt vor, diese Daten sowohl in Englisch als auch in den Sprachen der Länder vorzusehen, in denen das Produkt verkauft wird. Das Gleiche gilt auch für die Warnungen. Die MDCG erwägt, dass auch allgemein verständliche Symbole in Betracht gezogen werden könnten.
Fazit
Wie sehr diese Dokumente wirklich „Guidance“ geben, lässt sich hinterfragen. Die Dokumente erläutern nicht, welche Unklarheiten es tatsächlich gibt, die es aufzulösen gilt. Auch die Auswahl der Themen können Außenstehende nicht nachvollziehen.
Sind das wirklich die größten Probleme, die wir in der Auslegung von MDR und IVDR haben? Wäre es nicht dringender, die API und das Format für die „Bulk-Uploads“ festzulegen oder zumindest Empfehlungen dafür zu geben? Wie sollen die Hersteller sonst rechtzeitig die IT-Systeme auf die regulatorischen Anforderungen bezüglich EUDAMED und UDI umstellen?
März 2018: Veröffentlichungen der MDCG
Die MDCG hat sich zur Basis-UDI-DI Gedanken gemacht. Die entsprechenden Dokumente sind oben vorgestellt.
- MDCG 2018-1: Leitfaden zur Basic UDI-DI und UDI-DI
- MDCG 2018-2: Anforderungen an eine künftige Nomenklatur (à la GMDN oder UMDNS): Dass diese Nomenklatur kostenfrei sein soll, ist sicher sinnvoll. Wenn die EU aber nicht weiter ist, als es diese Veröffentlichung vermuten lässt, dürfte es noch dauern.
- UDIWG-2: Beschreibung der Basic-UDI-DI und UDI-DI Daten: Hier hat die MDCG zwei Folien der UDI Working Group in schlechter Qualität kopiert. Das Dokument wird als offiziell betrachtet. Wir haben Ihnen den Inhalt lesbar und verständlich aufbereitet.
10. UDI aus Sicht der FDA
Die FDA verlangt eine eindeutige Nummer für Medizinprodukte, die Unique Device Identification (UDI). Bei physischen Produkten mag das ja gut funktionieren. Aufkleber auf der Verpackung bzw. am Gerät sind kein Problem. Doch bei Software? Wo will man auf einem virtuellen Produkt eine UDI anbringen? Man könnte die DVDs eindeutig labeln, damit wären aber alle Downloads verboten.

Zum Glück hat die FDA die UDI requirements dahingehend konkretisiert, dass „unlike most devices, software will only have to exhibit a means of displaying the UDI […] it can also be within the software itself“.
Es genügt also, die UDI innerhalb der Software anzuzeigen. Wenn es aber darum geht, diese Nummer mit einem Kunden in Verbindung zu bringen (um ihn z. B. bei einem Produktrückruf kontaktieren zu können), ist das allein nicht ausreichend. Ein Download ohne Authentifizierung bzw. Registrierung ist deshalb nicht sinnvoll.
Mögliche Lösung: Sie können bei der Registrierung eine eindeutige UDI vergeben und in der Software eintragen, um sie beispielsweise unter „Help > About“ anzuzeigen.
11. FAQ zur UDI
11.1 Allgemeines
- F: Gelten die UDI-Übergangsfristen der MDR auch für Vergabe von UDIs?
A: Nein, die UDI-Übergangsfristen gemäß Artikel 123 MDR gelten explizit für die Anbringung der UDIs auf dem Produkt und den Verpackungsebenen. - F: Sind die UDIs auch für das Konformitätsbewertungsverfahren nach MDD erforderlich?
A: Nein, die MDD fordert keine UDIs.
11.2 Basis-UDI-DI
- F: Wo überall muss der Hersteller die Basis UDI-DI angeben?
A: Registrierung in EUDAMED, Konformitätserklärung, Kurzbericht zur Sicherheit und klinischen Leistung, technische Dokumentation, Freiverkaufszertifikate. Die Basis UDI-DI wird weder auf dem Produkt noch auf den Verpackungsebenen angebracht. Somit wird für die Basis UDI-DI kein maschinenlesbares Format benötigt. - F: Wir vertreiben Medizinprodukte der Klasse IIb als nicht steril bzw. steril verpackt. Außer der Sterilisation sind alle Spezifikationen und Prozessschritte der Fertigung identisch. Kann ich für diese Produkte nur eine BASIC UDI-DI vergeben oder werden zwei gefordert – jeweils für nicht steril bzw. steril ausgelieferte Implantate?
A: In diesem Fall würde es Sinn ergeben, eine gemeinsame Basis UDI-DI zu vergeben für die beiden Varianten steril/nicht steril. Die Informationen zu „labeled as sterile“ geben Sie auf Ebene der Produkt-UDI-DIs in EUDAMED an und nicht auf Ebene der Basis-UDI-DI. - F: Müssen alle Produkte, welche zu einer Basis-UDI-DI zusammengefasst werden, in einer gemeinsamen technischen Dokumentation beschrieben werden?
A: Eine Vorgabe, dass zu einer Basis-UDI-DI nur genau eine technische Dokumentation existieren darf, können wir aus der MDR nicht herauslesen. Somit sollte es möglich sein, mehrere technische Dokumentationen zu führen, welche auf dieselbe Basis-UDI-DI verweisen.
11.3 Vergabe
- F: Wenn ich die UDI-DI mittels GS1 vergebe, kann ich mit der ersten Zahl die Verpackungseinheit definieren (z. B. 0 für ein Stück, 2 für zehn Stück …). Muss ich das machen oder kann ich auch neue UDI-DIs vergeben, da ich auch eine andere REF-Nummer habe?
A: Das ist optional. Sie können auch komplett neue UDI-DIs vergeben. - F: Wie ist mit dem Zubehör eines Medizinprodukts zu verfahren?
A: Zubehör im Sinne der MDR benötigt eine eigene UDI. - F: Unser Medizinprodukt enthält Verschleißteile, die regelmäßig getauscht werden. Wie werden die UDIs für solche Ersatzteile vergeben?
A: Verschleiß- bzw. Ersatzteile benötigen keine eigene UDI, solange es sich nicht um Zubehör im Sinne der MDR handelt.
11.4 UDI-Anbringung und UDI-Träger
- F: Ist es ausreichend, die Datamatrix auf die Produkte aufzubringen, oder ist es verpflichtend, zudem die Zahlencodes zur direkten Lesbarkeit aufzubringen? Falls ja, wo finde ich diese Vorgabe als Referenz in der MDR?
A: Die MDR fordert die Angabe sowohl der maschinenlesbaren als auch der menschenlesbaren Form in Anhang VI 4.1: „Der UDI-Träger (AIDC- und HRI-Darstellung der UDI) wird auf der Kennzeichnung oder auf dem Produkt selbst sowie auf allen höheren Ebenen der Produktverpackung angebracht.“ Versandcontainer gelten nicht als höhere Verpackungsebene.
Bei erheblichem Platzmangel ist nur die maschinenlesbare Form (z. B. DataMatrix) anzugeben: „4.7. Gibt es erhebliche Probleme, beide Formate — AIDC und HRI — auf der Kennzeichnung unterzubringen, so ist nur das AIDC-Format auf der Kennzeichnung zu verwenden.“ - F: Können sowohl lineare Barcodes als auch 2D-Barcodes verwendet werden? Oder muss man sich für eine Art entscheiden und diese dann konsequent durchziehen?
A: Es können für unterschiedliche Produkte auch unterschiedliche UDI-Träger und -Zuteilungsstellen verwendet werden.
11.5 Sonderfall Standalone-Software
- F: An welcher Stelle muss die UDI bei Standalone-Software erscheinen und in welcher Form?
A: Gemäß MDR Anhang VI, 6.5.4 kann die UDI innerhalb der Software angegeben werden, z. B. „in einem für den Nutzer leicht zugänglichen Fenster in einem leicht lesbaren reinen Textformat angezeigt, wie z. B. im Infofenster mit Systeminformationen oder im Startfenster.“ Darunter fallen z. B. ein Hilfe-Menü, eine About-Box oder ein Splash-Screen. Die UDI muss dabei nur im menschenlesbaren Format angegeben werden. - F: Was muss die UDI-PI bei Software enthalten?
A: Gemäß Anhang VI 6.5.1 der MDR wird die Software-Identifikation als Herstellungskontrollmechanismus betrachtet und in der UDI-PI angegeben. Somit enthält die UDI-PI typischerweise mindestens die SW-Versionsnummer.
11.6 Sonderfall Implantate
Frage: Unterliegen nicht steril in Verkehr gebrachte Implantate den Anforderungen der UDI-Direktmarkierung?
Die Anforderung zur direkten Kennzeichnung des Produkts mit dem UDI-Träger besteht nach meinem Verständnis nur für wiederverwendbare Produkte, bei denen zwischen den Anwendungen am Patienten eine Reinigung, Desinfektion, Sterilisation oder Aufbereitung erforderlich ist. Wie sind in diesem Zusammenhang nicht steril in Verkehr gebrachte Implantate (Platten, Schrauben, Nägel etc.) zu betrachten?
Diese Implantate sind nicht zur Wiederverwendung vorgesehen, allerdings ist vor der ersten Anwendung natürlich eine Aufbereitung erforderlich. Weiterhin werden diese Implantate üblicherweise in Lagerungssystemen, z. T. zusammen mit dem notwendigen Instrumentarium, sterilisiert und zur Operation bereitgestellt. Auf diesen Systemen befinden sich meist eine Vielzahl an Varianten der Implantate, von denen nur ein Teil genutzt wird. Der Rest wird dann der erneuten Aufbereitung zugeführt, um bei der nächsten Operation wieder zur Verfügung zu stehen, genauso wie die Instrumente, bei denen die Pflicht zur Direktmarkierung zweifelsfrei besteht.
Die unsterilen Implantate sind also nicht wiederzuverwendende Produkte, die aber immer wieder aufbereitet werden. Ich würde nun sagen: Man müsste sie nicht mit UDI direkt markieren, da das Hauptkriterium die Wiederverwendung ist. Dies steht aber vermutlich im Gegensatz zu der Intention, ein System zum Tracking der Aufbereitungszyklen bereitzustellen?
Nun zur konkreten Frage: Unterliegen nicht steril in Verkehr gebrachte Implantate, wie oben beschrieben, den Anforderungen der UDI-Direktmarkierung?
Antwort
Die direkte Kennzeichnung ist in der Tat nur bei wiederverwendbaren Produkten vorgeschrieben, bei denen zwischen den Anwendungen eine Aufbereitung notwendig ist. Bei Implantaten muss die UDI nur auf der direkten Verpackung angegeben werden. Allerdings fordert die MDR, dass die UDI von implantierbaren Produkten vor der Implantation identifizierbar ist (Anhang VI, 6.1.3 MDR). Dies wäre in diesem Fall nicht mehr gegeben, sodass in dieser Konstellation vermutlich nur eine Direktkennzeichnung sinnvoll erscheint.
11.7 Sonderfall Systeme und Behandlungseinheiten
- F: Wenn ich eine Produktkombination im Sortiment habe, welches unter MDR ein System ist, so muss ich dieses System mit einer UDI kennzeichnen. Welche UDI-PI (von welchem Produkt) muss dann in der UDI auf der Verpackung des Systems stehen? Die von dem Produkt mit dem kürzesten Ablaufdatum?
A: Die UDI des Systems ist eine eigene. Somit wird eine eigenständige UDI-PI vergeben. Diese könnte z. B. das „Herstellungsdatum“ des Systems oder eine Chargennummer enthalten. - F: Was ist als „Convenience Kit“ im Sinne von UDI zu verstehen (US-FDA)?
A: Hierzu gibt es eine ausführliche Beschreibung mit mehreren Beispielen in folgendem Guidance-Dokument der FDA: https://www.fda.gov/media/95120/download
11.8 USA
- F: In der EUDAMED werden keine UDI-PI eingegeben, was ist mit GUDID?
A: In der GUDID werden ebenfalls keine UDI-PIs eingetragen. - F: Ist die „amerikanische UDI“ (GTIN) auch für den Europäischen Wirtschaftsraum anwendbar nach Inkrafttreten der MDR?
A: Es gibt in dem Sinne keine amerikanische UDI. GS1 mit GTIN ist auch in der EU unter der MDR als UDI-Zuteilungsstelle akkreditiert. Somit ist eine GTIN zur UDI-DI-Umsetzung auch innerhalb der EU möglich.
11.9 Ihre Frage und die Antwort darauf fehlen?
Die EU hat einen Helpdesk eingerichtet.
Das Johner Institut hilft Herstellern dabei, die regulatorischen Anforderungen an die UDI schnell und sicher zu erfüllen. Insbesondere kurze Fragen beantworten wir im Rahmen unseres Micro-Consultings regelmäßig und sogar kostenfrei.
Nehmen Sie einfach Kontakt mit uns auf!
Änderungshistorie
- 2025-02-11: Kapitel 3.b) um weiterführende Informationen ergänzt, Kapitel 3.c) hinzugefügt
- 2024-10-24: Im Abschnitt 6. die Updates der Normen ISO/IEC 16022:2024 und ISO/IEC 15415 aktualisiert
- 2024-10-16: In Kapitel 9 Hinweise zur Master-UDI ergänzt. Zwischenüberschriften in Kapitel 5.d) ergänzt
- 2022-05-20: Komplett überarbeitet
- 2021-07-05: Link zu EU Helpdesk ergänzt



Das scheint weitgehend mit dem US/FDA Konzept konform zu gehen.
Wird es zwei Datenbanken geben?
Definitiv ja.
Gibt es bereits eine Organisation die eine UDI Datenbank in der EU führt, an der man sich anmelden kann?
Sehr geehrter Herr Funke, die Antwort ist derzeit leider ebenso kurz wie klar: Nein. Ich vermute, dass die EUDAMED, an der man seit Jahrzehnten arbeitet, eher ein Projekt vom Typ „Hauptstadtflughafen“ wird.
Hallo Herr Johner,
das hört sich ja dann eher langwierig an. Wie setzt man dann die Anforderung des novellierten Gesetzes um? Baut man sich hilfsweise einen eigenen Code? Oder meldet man sich bei der FDA an und verwendet den? Verwendet man folgenden Dienst oder ist das ein Fake? http://www.egibcc.com
Gruss A. Funke
Sorry http://www.ehibcc.com
Es gibt derzeit keine Möglichkeit und keine Pflicht, sich anzumelden. Die Daten pflegen ausschließlich die nationalen Behörden, im Falle Deutschlands das http://www.dimdi.de.
Die Datenbanken der FDA und EU haben nichts miteinander zu tun. Leider.
Ok. Verstanden, aber wie komme ich an den uniquen Herstellercode. Kann ich da einen wählen? Wohl kaum, wer vergibt diesen?
Denn Code vergeben die noch nicht existierenden Organisationen. Daher können derzeit die UDI-Anforderungen nicht erfüllt werden. Das könnte die MDR in Teilen oder Gänze verzögern.
Guten Tag Herr Prof. Johner,
wenn ich das lese frage ich mich, welche Nummern, Kürzel etc. denn aktuell in Deutschland verwendet werden um ein Medizinprodukt eindeutig zu kennzeichnen? Und gelten diese Nummern dann nur für Medizinprodukte die hier Produziert werden oder auch für alle MP die eine Zulassung für Deutschland, bzw. die EU haben möchten?
Die Product-Identification wird künftig über die Eudamed vergeben. Soweit ist man aber noch nicht. Die Product Identification vergeben Sie als Hersteller. Das sind „means a series of numeric or alphanumeric characters“.
„Eine Vergabe einer eindeutigen UDI pro Installation wird also nicht gefordert.“ – dies ist zu dem System, was viele bereits mit eindeutigen Seriennummern führen, ein gewaltiger Rückschritt mit Zusatzaufwand. Ich frage mich, wieso man sich nicht an solchen Beispielen orientiert hat und die UDI nach nem gewissen Code, den jeder sich selbst aufbauen kann, aufbaut. Z.B. wie eine EAN, nur „fortschrittlicher“
Wer darf die UDI-Kennzeichnung neu aufbringen wenn diese nicht mehr lesbar ist oder wenn zur Reparatur die UDI entfernt werden muss?
Bei der Reparatur von chirurgischen Instrumenten wird immer die komplette Oberfläche aufgebarbeitet, dabei geht die UDI verloren.
Wenn man vorher die Informationen ausgelesen hat, kann man dann auch als nicht Hersteller die UDI Kennzeichnung wieder aufbringen?
Vielen Dank
Da gibt es einige Missverständnisse. Die issuing agencies (FDA) bzw. Zuteilungsstellen/ issuing entities in der MDR sind zwar nicht alle definiert. Die Übergangsbestimmungen der MDR Art. 120 (12) sagen aber: „Bis die Kommission gemäß Artikel 27 Absatz 2 die Zuteilungsstellen benannt hat, gelten GS1, HIBCC und ICCBBA als benannte Zuteilungsstellen“. Also sicher nicht eigene Codes definieren, das wäre sinnlos.
Da die FDA auch GS1 und HIBCC gibt es da kaum Probleme. Anders bei der zentralen Datenbank (FDA: GUDID). Hier ist in Europa eine separate Datenbank im Aufbau (Eudamed 3).
Quintessenz: wer sich für USA gut aufgestellt hat, muss sich für Europa in erster Näherung keine Sorgen machen.
Sehr geehrter Herr Riedwyl, danke für Ihren Hinweis.
Worin liegen die kritisierten Missverständnisse? Ich war mir nicht bewusst, geschrieben zu haben, dass die Hersteller Codes selbst vergeben.
Ihren wichtigen Hinweis auf den Artikel 120(12) habe ich gerne ergänzt. Das ist sehr hilfreich. Vielen Dank!
Lieber Herr Johner, nicht Sie, sondern ein Fragesteller hat vermutet, dass man eigene Codes „erfinden“ muss (mindestens habe ich das so verstanden). Ein weiteres Missverständnis: Die UDIs werden nicht von der EUDAMED3 vergeben, sondern die Informationen zu einer spezifischen UDI werden vom Hersteller in der EUDAMED gepflegt. Der Hersteller ist für die Verwaltung seines „Codeblockes“, den er vorgängig bei GS1 oder HIBC bekommt, selbst zuständig. Wird gleich laufen wie mit der GUDID der FDA. Freundliche Grüsse Hansjörg Riedwyl, ISS
Ist mein Verständnis richtig, dass jedes Medizingerät (z.B. Laser) einen UDi haben muss. So wie eine Reisepass den Inhaber eindeutig identifiziert?
So ist es, das versuchte ich zu artikulieren.
Richtig, allerdings sind gemäss der Final Rule der FDA gewisse Ausnahmen möglich (z.B. bei sehr kleinen Devices, oder bei Multipacks, die mehrere Devices enthalten und das Multipack immer beibehalten wird (z.B. „Spender“ von Spritzennadeln, jede Nadel ist ein MD , aber nur die Umverpackung muss zwingend den UDI enthalten). Achtung: Aussage trifft auf USA zu, die Regelungen (insbesondere was Ausnahmen betrifft) für die EU sind noch nicht abschliessend bekannt.
Guten Morgen,
wie verhällt sich das nun mit Zubehör?
Braucht jedes Zubehör einen eigenen UDI?
Es gibt ja verschiedene sicherheitsrelevante Verbrauchsmittel bei denen es durchaus Sinn ergibt, allerdings ein Wasserschlauch der zum Befüllen eines Gerätes mitgeliefert wird da wäre es durchaus übertrieben.
Die MDR schreibt hierzu in Anhang VI Teil C Absatz 3.6 „Jede Komponente die als Produkt gilt und und für sich genommen kommerziell verfügbar ist wird eine UDI zugewiesen.“ Daraus schließe ich, wenn ich den Schlauch als Ersatzteil anbiete, braucht er eine UDI?
Ich freue mich über eine fundierte Antwort.
Sehr geehrter Herr Winstel,
das ist eine ausgezeichnete Frage. Leider ist die Übersetzung nicht ganz gelungen. Im englischen Text heißt es „Each component that is considered to be a device…“.
Der Schlauch ist weder ein Medizinprodukt noch ein Zubehör. Daher ist er kein Device. Ersatzteile sollten auch nicht mit Komponenten gleichgesetzt werden.
Der Schlauch braucht also keine UDI, es sei denn Sie bieten ihn als Zubehör an.
Viele Grüße, Christian Johner
Sehr geehrter Prof. Dr. Johner,
werden die UDI Vorgaben für alle Medizinprodukte einheitlich sein, somit auch für Klasse 1 Produkte?
Im Voraus vielen Dank für Ihre Antwort.
MfG
Die MDR unterscheidet bei den Anforderungen nicht nach der Klassifizierung. Wie die zeitliche Abfolge der Einführung der EUDAMED sein wird, ist derzeit noch offen.
Sehr geehrter Prof. Dr. Johner,
wie ist es bei Medizinprodukten (Scheren, Nadelhalter) die im Klinischen Bereich bereits im Bestand sind?
Müssen diese oder können diese noch mit der UDI gekennzeichnet werden, wenn ja wer könnte dies tun?
Vielen Dank
Mit freundlichen Grüßen
S.Fuchs
Produkte, die bereits in den Verkehr gebracht werden, müssen nicht mit einer UDI gekennzeichnet werden.
Produkte, die unverändert weiterverkauft werden, müssen ab 2020 bzw. nach Ablauf der Übergangsfrist neu „zugelassen“ werden und damit die Anforderungen der MDR einschließlich UDI erfüllen.
Sehr geehrter Herr Prof. Dr. Johner,
vielen Dank für Ihre Antwort, können denn Instrumente ohne UDI weiter am Patienten benutzt werden?
Mit freundlichen Grüßen
S. Fuchs
Sehr geehrte Frau Fuchs,
bereits in Verkehr gebrachte Produkte dürfen Sie weiter nutzen auch ohne UDI. Falls das nicht Ihr Punkt ist, dann schreiben Sie mir einfach. E-Mail ist oben auf Webseite zu finden.
Beste Grüße, Christian Johner
Sehr geehrter Prof. Johner,
meine Frage bezieht sich auf das Thema der UDI für medizinische Verbrauchsmaterialien der Klassen I und IIa. Ist die Vorgabe so zu interpetieren, dass z. Bsp. für sterile OP-Handschuhe die UDI sowohl auf dem Produkt als auch auf der Sterilbarriere und sämtlichen weiteren Umverpackungen ausgewiesen werden soll?
Auch hier würde sich aus meiner Sicht eine Verfehlung des eigentlichen Zieles der UDI erweisen.
Die MDA schreibt im Anhang VI, Teil C:
Die Gebrauchseinheit-DI ist wie folgt definiert:
D.h. Sie müssen nicht auf Ebene des OP-Handschuhs eine UDI aufbringen.
Vielen Dank für die gute Übersicht. Eine Frage zur Basis-UDI-DI: Ich habe verstanden, dass nur eine gemeinsame Basis-UDI-Di vergeben werden darf, wenn u.a. die Risikoklasse als auch die Subkategorie zur Klassifizierung (implantierbar, aktiv, etc.) identisch sind. Wie verhält sich dies bei IVDs?
Es ist genau wie Sie sagen. Die Attribute, die Sie nennen, müssen übereinstimmen. Das ist eine notwendige aber nicht hinreichende Voraussetzung für eine gemeinsame UDI. Die Anforderungen aus der IVDR an die UDI sind größtenteils identisch mit denen der MDR.
Guten Tag Herr Dr. Johner,
ich komme durch die vielen theoretischen Erklärungen gerade etwas ins Straucheln.
Wir wenden bereits UDI an und als Beispiel so eine Art Klarschrift
+EBRE DI 0/$$7 LOT Prüfziffer
DI ist bei uns die REF
PI ist die LOT
Ich verstehe noch nicht so ganz wie ich dafür nun eine Basis-UDI-DI generieren soll bzw. bei einer Änderung (z.B. Zweckbestimmung) die DI ändern soll.
Ich hoffe, ich konnte mein Problem verständlich wiedergeben.
Beste Grüße S. Bochtler
Sehr geehrte Frau Bochtler,
ich bin nicht ganz sicher, ob ich die Frage schon verstanden habe. Dennoch wage ich eine Antwort:
Zum einen gibt es keine Pflicht, eine Basis UDI DI zu bestimmen. Falls Sie das doch machen, muss diese nicht auf dem Gerät stehen. Die Basis-UDI-DI wird von einer noch zu implementierenden Stelle vergeben, die bestimmen sie nicht selbst.
Bei einer Änderung der Zweckbestimmung gibt es eine neue UDI-DI. Es wird nicht die alte geändert.
Vielleicht konnte ich etwas helfen.
Beste Grüße, Christian Johner
Guten Tag Herr Rohner
In Kapitel 5. sollte es wohl GMDN statt GMDS heissen.
Freundliche Grüsse
Peter Egli
Sie haben natürlich Recht, lieber Herr Egli! Der Fehler ist behoben.
Danke für Ihren Hinweis! Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
ersetzt der UDI-Träger, in welcher Form auch immer er aufgedruckt ist, einen bereits vorhandenen Barcode?
Vielen Dank für Ihre Antwort!
Freundliche Grüße,
Tobias Riesinger
Sehr geehrter Herr Riesinger,
es gab bisher keine Pflicht für einen Barcode. Aber Sie können einen Barcode für das UDI-Label nutzen.
Beste Grüße,
Christian Johner
Hallo Herr Prof. Dr. Johner,
vielen dank für ihre Antwort! Ich habe mich aber leider missverständlich ausgedrückt.
Ich wollte wissen, ob ein UDI-Träger im AIDC Format einen bereits vorhandenen AIDC-Träger ersetzt.
Ich habe nochmal nachgelesen und laut MDR muss der UDI-Träger deutlich erkennbar sein. Er ersetzt den anderen Träger nicht.
Ich hätte noch Fragen zur Gebrauchseinheit-DI. Diese muss ja nur dann verwendet werden wenn mehrere Produkte des selben Modells zusammen verpackt sind. Wo wird diese DI aufgedruckt? Anstelle der UDI-DI auf der Verpackung der Produkte? Oder muss sie zusätzlich aufgedruckt werden?
Vielen Dank für Ihre Antwort!
Beste Grüße,
Tobias Riesinger
Hallo Herr Prof. Johner,
es ist Annex VI zur EU MDR den Sie meinen.
MfG
Elke Calamia
Sie haben absolut Recht, liebe Frau Calamia!
Ich hatte die erste Version des Beitrags geschrieben, als die MDR im Entwurf vorlag und die UDI-/EUDAMED-Anforderungen noch im Anhang V beschrieben waren. Damals fanden sich die PMS-Anforderungen in Anhang IIa.
Dank Ihrer Hilfe konnte ich die Nummerierung auf den aktuellen Stand bringen. Danke dafür!
Viele Grüße, Christian Johner
Sehr geehrter Herr Dr. Johner,
Ab wann muss die UDI eingeführt werden? Erst wenn man den Umstieg von der MDD auf die MDR macht? Oder bereits 2020, unabhängig davon, ob man das MDR Zertifikat hat?
Mit freundlichen Grüßen
Mario Isler
Lieber Herr Isler,
die UDI ist eine Forderung der MDR, nicht der MDD. Sie hat aber nichts mit dem Zertifikat zu tun. Auch ohne Zertifikat (bei Klasse I Produkten) gibt es die Anforderung.
Allerdings ist noch nicht klar, ob die Behörden die Nummern rechtzeitig vergeben können. D.h. Sie müssen die Anforderungen erst dann erfüllen.
Viele Grüße, Christian Johner
Hallo Herr Prof. Johner,
wie Sie schreiben, ist die UDI eine Forderung der MDR und nicht der MDD. Regulär kann ich ein Medizinprodukt noch bis 06 .Mai. 2024 nach der MDD in Verkehr bringen. Danach verlieren die Zertifikate der Benannten Stellen nach MDD Ihre Gültigkeitkeit. Was bedeutet dies aber für die UDI? Ist ein Medizinproduktehersteller, der seine Produkte bis 06.05.2024 nach der MDD in Verkehr bringt, eine UDI für ein Klasse III – Produkt zum 26.05.2020 schon in der Eudamed-Datenbank zu hinterlegen.
Mit freundlichem Gruß
Johannes Beuth
Sehr geehrter Herr Beuth,
danke für diese anspruchsvolle Frage. Die MDR schreibt:
Damit greifen auch die Pflichten u.a. für die Periodic Safety Update Reports. Diese sind aber in der EUDAMED zu hinterlegen gemäß Artikel 86. Denn dort heißt es:
Da die UDI der Schlüssel in der Datenbank ist, wüsste ich keinen Weg, wie man das Produkt ohne eine UDI weiter in den Verkehr bringen kann.
Viele Grüße, Christian Johner.
Hallo Herr Dr. Johner,
auf der DIMDI Webseite habe ich gelesen:
„Wo und wie kann ich einen Unique Device Identifier (UDI) beantragen?“
„Den UDI vergeben nicht die Behörden und auch nicht die Eudamed Datenbank, sondern dieser wird von den sog. Zuteilungsstellen vergeben, mit denen Sie bereits zum jetzigen Zeitpunkt Kontakt aufnehmen können.
Diese Zuteilungsstellen sind in Artikel 120(12) MDR aufgelistet: GS1, HIBCC und ICCBBA. Die Verordnung finden Sie auf den Webseiten der Europäischen Union.
– GS1 (Global Standards One)
– HIBCC® – Health Industry Business Communications Council
– ICCBBA – abgeleitet von International Council for Commonality in Blood Banking Automation
– MDR – Europäische Verordnung für Medizinprodukte (PDF, 1,6 MB)“
Gibt es Erfahrungen mit den 3 Stellen, die UDI vergeben?
GS1 z.B. schreibt auf ihrer Webseite, ab Ende Mai 2019 mit der Vergabe der UDI zu starten. Ist es empfehlenswert, dem in naher Zukunft nachzukommen?
MfG
N. Geerkens
Lieber Herr Geerkens,
ich kenne die Stellen nicht aus eigener Erfahrung. Die GS1 ist ein großer Player.
Die Beantragung der UDIs wird meines Erachtens schnell gehen. Wichtiger ist wahrscheinlich, die anderen UDI-Anforderungen zu erfüllen. Das kann sehr aufwändig sein, wenn z.B ERP-Systeme anzupassen sind.
Sobald ich neuere Informationen habe, werde ich diese wieder bekanntgeben.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
wie verhält sich die UDI bei konfigurierbaren Produkten? In dem Anhang der MDR heißt es:
6.4.1. Die UDI wird dem konfigurierbaren Produkt in seiner Gesamtheit zugeteilt und als UDI des konfigurierbaren Produkts bezeichnet.
6.4.3. Eine UDI-PI des konfigurierbaren Produkts wird jedem einzelnen konfigurierbaren Produkt zugeteilt.
6.4.5. Jede Komponente, die als Produkt gilt und allein kommerziell verfügbar ist, bekommt eine gesonderte UDI zugewiesen.
Bedeutet dies, dass einige Produkte mit zwei verschiedenen UDIs markiert werden müssten? Eine eigene UDI für das eigentliche Medizinprodukt (was z.B. auch einzeln verkauft wird) und eine für die Konfiguration in der das Medizinprodukt beteiligt ist?
Mit freundlichen Grüßen
Timo Janssen
Hallo Herr Johner,
Wie ist es mit Software in einem Medizinprodukt (keine Stand alone Software). Gilt für die ebenfalls die Passage über Software? Muss hier jede interne Sofware Version (mehrere pro Gerät) im UDI-PI koodiert sein? Muss hier bei jedem größeren Sw Update die UDI-DI geändert werden? ZB muss hier dann nachträglich im Feld jemand die Geräteaufkleber tauschen?
Danke für die vielen praktischen Tipps
Mit freundlichen Grüße
Angelika K.
Sehr geehrte Frau K.,
eine interne Software benötigt ebenso wie andere Komponenten keine eigene UDI. Insofern muss bei einem Update auch nichts geändert werden insbesondere keine Geräteaufkleber.
Allerdings müssen Sie unabhängig von der UDI Thematik immer nachvollziehen können, in welchem Produkt welche Version der Software verbaut ist.
Beste Grüße, Christian Johner
Hallo Herr Johner,
wie interpretieren Sie das Requirement Annex VI Part C 3.5 zusammen mit den spezifischen Regeln für die Software-Produkte? Beispiel: der Hersteller entschließt sich aus welchen Gründen auch immer eine Seriennummer auf das Label zu schreiben. Muss dann 3.5 zwingend angewendet werden und die UDI-PI die Seriennummer enthalten, oder überschreibt das Requirement 6.5. als spezifischeres Requirement die 3.5, und die UDI-PI muss nur die neue minor software revision berücksichtigen?
VIelen Dank & freundliche Grüße
Olaf Kessel-Deynet
Sehr geehrter Herr Kessel-Deynet,
danke für Ihre Frage!
Ich bin nicht ganz sicher, ob ich diese vollumfänglich verstehe. Sie sagen, dass der Hersteller eine Seriennummer auf die standalone Software schreibt?
Falls diese SN für alle Produkte gleich wäre, entspräche sie der UDI-PI. Falls er für jede Software-Installation eine eigene SN hat, ist die Software nicht binäridentisch. Damit wäre es sogar eine andere UDI-DI. Das wäre aber ein Missbrauch des Gedankens.
Die SW-spezifischen Teile des Annex VI überschreiben wie Sie sagen die vorderen Teile bzw. deuten die „Seriennummer“ bei Software um.
Viele Grüße, Christian Johner.
Hallo Herr Johner,
ist die Anbringung eines UDI Codes am Medizinprodukt zwingend oder reicht ein HRI, für den europäischen Raum und aber auch für die USA.
Und unterscheiden sich die Übergangsfristen für die Anbringung eines Codes zwische EU (MDR) und USA(FDA) in Abhängigkeiten Risikoklassen ?
Freue mich über eine kurze Rückantwort.
Schönen Gruß
Mathias Flügel
Sehr geehrter Herr Flügel,
danke für Ihre Frage! Ich vermute Sie beziehen sich mit dem „UDI Code“ auf den AIDC. Dieser ist anzugeben, falls möglich auch der HRI. D.h. der HRI reicht nicht:
Die Antwort zu den Unterschieden der Übergangsfristen abhängig von den Risikoklassen wäre nicht ganz kurz. Weil ich gerade im Urlaub bin, schaffe ich es daher nicht. Nutzen Sie gerne unser Micro-Consulting.
Beste Grüße, Christian Johner
Hallo Herr Johner,
wie verhält es sich bei Medizinprodukten (z.b. rotierende Instrumente) die aufgrund ihrer geringe größe und Maße (1,6mm Durchmesser, ca. 19mm Länge) kaum Platz für eine lesbare UDI bieten? Wenn ein solches Produkt in Verpackungseinheiten zu á 6 Stück verpackt wird, gilt dann die Gebrauchseinheit-UDI-DI? Unabhängig vom wahnsinnigen Aufwand, wäre es kaum lesbar… Welche Möglichkeit bietet sich dann regulatorisch?
Vielen Dank!
LG Tobias Bauer
Sehr geehrter Herr Bauer,
die MDR schreibt in Anhang I Abschnitt 23.1 b)
Ist das nicht genau das, nach was Sie fragen?
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für Ihre ausführlichen Erklärungen.
Ich habe verstanden, dass die Basis UDI-DI von den benannten Stellen GS1, HIBCC und ICCBBA bezogen werden soll um diese dann in die EUDAMED einzutragen.
Meine Frage:
Muss ich als Medizinprodukte Herstelller die UDI-DI und UDI-PI ebenfalls von den benannten Stellen beziehen oder kann ich diese als Hersteller selbst generieren?
Bietet es mir Vorteile diese (neben der Basis-UDI) von den benannten Stellen zu beziehen?
Außerdem sind mir noch folgende Punkte unklar:
Bei einem Medizinprodukt mit mehreren für die Funktion notwendige Komponenten (Bsp. Schlauchsystem oder ähnliches) mit eigenen CE-Kennzeichen, wie werden die einzelnen Komponenten bzw. das gesamte System gekennzeichnet?
Gibt es eine Basis-UDI für das Gesamte System:
Besipiel: Beatmungsgerät (insgesamt)
Komponenten: Schlauch, Beatmungseinheit, Monitor etc.
Benötige ich für die Komponenten ebenfalls eine Basis UDI?
Oder besitze ich eine Basis-UDI für das System Beatmungsgerät und der Schlauch, sowie die Beatmungseinheit und der Monitor werden jeweils mit DI und PI differenziert?
Ich hoffe ich konnte meine Frage vertsändlich ausdrücken.
Beste Grüße
Sehr geehrte Frau D.,
die Basis-UDI-DI bzw. die UDI-DI vergeben die Zuteilungsstellen. Die UDI-PI vergeben Sie als Hersteller.
Die UDI bekommt das Produkt, nicht Komponenten des Produkts. Beispielsweise bekäme das Netzteil eines Beatmungsgeräts keine eigene UDI-DI.
Wenn Sie Schläuche und Beatmungsgeräte als einzelne Medizinprodukte verkaufen, dann bekommen diese jeweils eigene UDI-DIs. Falls sie Schläuche und Beatmungsgeräte zusätzlich als System/Behandlungseinheit vertreiben, bekommt diese/s System/Behandlungseinheit ebenfalls eine UDI-DI.
Konnte ich helfen? Falls nicht, haken Sie gerne nach.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für Ihre sehr hilfreichen Beiträge.
Was mir weiterhin Kopfzerbrechen bereitet, sind offene Punkte bzgl. der Eintragung der UDI in EUDAMED.
1. Wann muss diese für Produkte mit gültigen MDD-Zertifikaten (Ablaufdatum < 26.05.2020) erfolgen? In den Übergangsbestimmungen (Artikel 120, (3)) ist nur von einer Registrierung von Produkten und Wirtschaftsakteuren zum Geltungsbeginn der MDR die Rede. Schaut man sich den Artikel 29 (Registrierung von Produkten) an, dann heißt es dort, man muss die UDI in die Datenbank eintragen, bevor das Produkt in Verkehr gebracht wird, also ab dem 26.05.2020. Die VO widerspricht doch
sich selbst!
2. Zusätzlich steht in Artikel 123, dass eine 18 monatige Übergangsfrist zur Übertragung der Daten in EUDAMED besteht, deren Beginn wiederum um 6 Monate versetzt werden könnte falls EUDAMED noch nicht soweit ist zum Geltungsbeginn.
Die Ausarbeitungen der zuständigen NAKI UG sind auf dem Stand von Februar 2018, was wohl kaum den aktuellen Stand der Dinge abbildet
Vlt. können Sie da etwas Licht ins dunkle bringen.
Mit freundlichen Grüßen
Konstantin Frank
Sehr geehrter Herr Frank,
die Antwort auf die Verfügbarkeit der EUDAMED weiß ich auch nicht, weil derzeit noch nicht ganz klar ist, wann die Datenbank in welcher Funktion bereit steht. Die Tatsache, dass es bereits ein Testsystem gibt, wenn gleich auch ein sehr eingeschränkt arbeitendes, lässt aber hoffen, dass dies zum Mai 2020 der Fall ist. Damit müssten die Daten Ende November in der EUDAMED erfasst sind.
Beste Grüße, Christian Johner
Korrektur: habe mich verschrieben, es soll heißen: Ablaufdatum > 26.05.2020
Hallo Herr Dr. Johner,
nachdem wir unsere Funktion als PLM in Vorbereitung auf die MDR nun aufgeben, fungieren wir ab Mai 2020 nur noch als Händler bzw. Importeur und liefern Medizinprodukte direkt an private Verbraucher. In dieser Funktion spielt die Rückverfolgbarkeit anhand der UDI die wesentliche Rolle.
Es ist klar, dass wir in unserem Verantwortungsbereich der Lieferkette die Rückverfolgbarkeit sicherstellen müssen.
Unklar für mich ist jedoch, wie das eine Apotheke macht, wenn es einen Rückruf von Verbandmitteln, Inkontinenzprodukten oder ähnlichen OTC-Medizinprodukten gibt? Und wie gewährleisten das die Drogeriemärkte und der LEH (REWE etc.)?
Daneben ist mir auch noch nicht klar, wie genau die verschiedenen Informationen der UDI-PI codiert werden. In einem Dokument des IMDRF (IMDRF/UDI WG/N7Final:2013: Ch. 8) habe ich gefunden, dass bei GS1 nach der (01) die GTIN kommt, nach der (17) das expiration date und nach (10) die LOT.
Ist dieses Prinzip allgemein gültig? Oder müssen wir jeden unserer ca. 200 Zulieferer (Hersteller) fragen, wie er gedenkt, seine UDI-PI zu codieren?
Unique regards – A. Hürster
Sehr geehrter Herr Hürster,
danke für Ihre wichtige Frage! Hierzu ein paar Gedanken, die vielleicht in Summe Ihre Frage beantworten:
Wenn die Antwort nicht verständlich oder ausreichend ist, haken Sie gerne nach.
Beste Grüße, Christian Johner.
Sehr geehrter Herr Johner,
eine Verständnisfrage zur UDI-DI/UDI-PI:
Muss die UDI-DI auf jede Verpackungsebene aufgedruckt werden.
Unsere Produkte sind jeweils eins ein einer Packung und diese in Blistern (Aussen- und Innenblistern) verpackt? Würde in dem Fall für uns bedeuten:
Auf die Aussenpackung, den Aussenblister und den Innenblister?
Vielen Dank
Peter Postert
Sehr geehrter Herr Postert,
hier hängt es davon ab, ob das Außen- und Innenblister als eine Verpackung definiert wird. Dann wäre es nur auf der äußersten aufzubringen.
Ich weiß, dass meine Antwort etwas kurz ist, aber ohne besseres Verständnis der Verpackung und deren Zweck fällt es mir schwer, spezifischer zu antworten.
Viele Grüße, Christian Johner
Hallo Herr Johner
Unsere Produkte sind Konfigurierbar, in dem unterschiedliches Zubehör verwendet werden kann, wenn ich es aus der MDR richtig interpretiere bekommt jedes Zubehör eine UDI-DI die dann der Basis-UDI-DI zugeordnet. Diese Zubehör ist jedoch an unterschiedlichen Geräten (mit unterschiedlichen Zweckbestimmungen) verwendbar.
Daher meine Frage: Kann eine UDI-DI unterschiedlichen Basis-UDI-DI’s zugeordnet werden ?
Sehr geehrter Herr Gabele,
danke für Ihre Frage! Um diese zu beantworten, müssten wir den Begriff der Zuordnung in diesem Kontext einheitlich verstehen. Ein Zubehör mit seiner UDI-DI kann natürlich verschiedenen Produkten mit verschiedenen Basis-UDIs zugeordnet werden. Aber das Zubehör hätte nicht die gleiche Basis-UDI-DI. Den die gleiche Basis-UDI-DI dürfen nur Produkte haben, „to connect devices with same intended purpose, risk class and essential design and manufacturing characteristics“. Das ist bei Zubehör und dem „verbundenen“ Produkt nicht der Fall.
Viele Grüße, Christian Johner
Ich habe einen Anhaltspunkt darauf gefunden, warum EUDAMED immer eine Basis-UDI und eine Varianten UDI verlangt und es nicht möglich ist, nur die UDI zu verwenden.
Wenn die Basis-UDI als Identifier in EUDAMED fungieren soll, dann muss dies funktionieren, egal ob ein Unternehmen Varianten für ein Produkt hat oder nicht.
Die GS1 Germany Organisation hat ein Whitepaper zur GTIN-Vergabe für konfigurierbare Produkte veröffentlicht. Darin ist die zentrale Forderung formuliert, dass ein konfigurierbares Produkt selbst niemals gleichzeitig eine Konfigurationsinstanz sein darf!
Da die Basis-UDI in EUDAMED als zentrale Indentifikationsnummer diese „konfigurierbaren Produkte“ repräsentieren soll, muss sie dass immer tun und sicherstellen, dass dahinter keine Konkreten Modelle stecken.
Andernfalls müsste EUDAMED unterschiedliche Datenmodelle für Registrierungen mit und ohne Varianten bereitstellen, was sicherlich eine unverhältnismäßig komplexe Systemarchitektur hervorbringen würde.
Unterm Strich wird es also eine Frage der EUDAMED-Systemarchitektur und damit der Identifizierbarkeit/Rückverfolgbarkeit sein, warum immer Basis-UDI und UDI gebraucht werden.
Das Whitepaper GS1 Standards für Variantenreieche Artikel (CSA), kann man auf der Seite der gs1-germany erwerben. Ich kann es sehr empfehlen, wenn man in die Welt der konfigurierbaren Produkte einsteigen möchte.
Hallo an alle,
gibt es mittlerweile jemanden mit Erfahrungen zu GS1 und HIBC? Mir erschließt sich nicht warum GS1 sich gerade in den USA so durchgesetzt hat (zumindest ist das mein Eindruck)
Welchen Vorteil bietet GS1 gegenüber HIBC überhaupt?
Sehr geehrter Herr Schweizer,
danke für die ausgezeichnete Frage! Ich gestehe, dass ich noch nicht ausreichend viele Rückmeldungen bekommen habe, um Ihnen fundiert antworten zu können. Das tut mir leid. Hoffentlich ändert sich das bald — auch wenn die Verschiebung der EUDAMED etwas den Druck aus dem System genommen und damit die Beschäftigung mit genau solchen Fragestellungen verzögert hat.
Sobald ich mehr weiß, werde ich es hier berichten.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich habe mich in den letzten Tagen mit den Begriffen Produktkategorie, generische Produktgruppe und dem CND Code beschäftigt. Wie Sie bereits erwähnt haben, müssen die Hersteller die „Medical Device Nomenclature“ auf Ebene der Produkte (UDI-DI) festlegen. Aus dem MDCG 2019-13 Dokument geht hervor, dass eine generische Produktgruppe als die 4te Ebene des EMDN (bzw. CND) Codes definiert wird. In Ihrem Beitrag „Generische Produktgruppe versus Produktkategorie“ stellen Sie, wie ich finde, sehr schön und plausibel die Hierarchie aus Produktkategorie, generischer Produktgruppe, Basis-UDI-DI und UDI-DI dar. Demnach würde ich die 5te Ebene des CND Codes der Basis-UDI-DI zuordnen. Ein 6tes Level, das dann der UDI-DI zugeordnet werden könnte, wie es in der UDI Datenbank gefordert wird, ist längst nicht immer vorhanden, so wie ich das verstehe (bitte korrigieren Sie mich, wenn ich falsch liege). Wie soll der Hersteller mit dieser Situation umgehen? Oder verstehe ich alles völlig falsch und es gibt eine ganz simple Lösung?
Sehr geehrter Herr Krämer,
danke für Ihre Feedback!
Ihrer Schlussfolgerung würde ich nicht in jedem Fall folgen, weil die Basis-UDI-Di nur Produkte umfassen darf mit „essential design and manufacturing characteristics“. Diese sind aber keine Klassifizierungsmerkmale bei der EMDN, weder auf der 5. noch auf der 6. Ebene.
Ihre Überlegungen halte ich für sehr wichtig, weshalb ich plane, den Beitrag um diesen Aspekt zu ergänzen.
Besten Dank auch dafür!
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich habe eine kleine Frage zu Abschnitt 7 (Dokumente, die die UDI enthalten müssen). Welche UDI ist hier genau gemeint, ich gehe davon aus, dass hier nur von der Basic-UDI-DI gesprochen wird. Interpretiere ich das richtig?
Viele Grüße
Patricia Kirchner
Sehr geehrte Frau Kirchner,
das hängt vom Dokument ab. Wenn es eine technische Dokumentation ist, reicht die Basis-UDI-DI. Wenn es ein Service-Bericht ist, brauchen wir sogar die UDI-PI, weil wir das spezifische Produkt identifizieren wollen. In den meisten Fällen ist es aber, wie Sie sagen, die Basis-UDI-DI. Ich füge demnächst noch eine Spalte ein, die die „Art der UDI“ angibt.
Danke für den Hinweis!
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Johner,
müssen den legacy devices auch bis zum 26.05.20 die UDI zugewiesen werden und in bestimmten in Abschnitt 7. aufgeführten Dokumenten enthalten sein? In dem von der Europäischen Kommission veröffentlichten Dokument „Unique Device Identification (UDI) System – FAQs“ steht, dass die UDI-Pflichten nicht für legacy devices gelten. Nur, dass sie in der EUDAMED (ohne Basis- und UDI-DI) registriert werden müssen. Demnach gehe ich davon aus, dass bspw. in den relevanten PMS- oder Logistikdokumenten eine UDI für legacy devices nicht enthalten sein muss. Sehe ich das richtig?
Viele Grüße
Peter Koszolka
Das sehe ich auch so, lieber Herr Koszolka!
Sehr geehrter Herr Johner,
ich habe eine Frage bzgl. Anhang VI Teil C Punkt 6.5 Software für Produkte (Device Software).
Ich würde dies so interpretieren, dass damit die Software gemeint ist, die für die Produkte verwendet wird. Beispiel: aktives MP mit Messfunktion, Software liefert Messalgorithmus.
In Ihrem Blockeintrag steht beim Punkt 4. UDI für Software, dass dies nur für Stand-alone Software gilt. Denn diese speziellen Regeln, die dort aufgeführt werden, decken sich ja mit dem Inhalt von Punkt 6.5.
Das ist ehrlich gesagt etwas verwirrend.
Liege ich mit meiner Interpretation falsch und der genannte Punkt gilt nur für reine Stand-alone Software?
Viele Grüße
Laura
Sehr geehrte Laura,
die MDR meint standalone Software, weil die Software als Teil eines MPs gar keine eigene UDI hat.
Mit „Device Software“ ist „Medizinprodukte-Software“, also „Software as a Medical Device“, wie es das IMDRF nennt, gemeint.
Hilft das? Falls nicht, einfach nachhaken.
Beste Grüße, Christian Johner
Sehr geehrer Herr Prof. Johner,
als Hersteller von Klasse 1 Produkten ist uns die Vorgehensweise zur Erlangung der UDI-DI bzw. UDI-PI soweit bekannt, aber woher ist die Basis-UDI-DI zu bekommen? Der erste Teil wird wohl auch die unternehmensspezifische (GS1)Basis-Nr. sein, am Ende mit 2 Prüfziffern, aber wird der mittlere Teil, der die Produktguppenspezifischen Informationen enthält, durch den Hersteller selbst vergeben? Nach welchen Kriterien?
Unsere Recherche endet immer an diesem offenen Punkt.
Herzliche Grüße
Jörg Dewert
Sehr geehrter Herr Dewert,
danke für Ihre Frage, die sicher viele Firmen umtreibt:
Die Basis-UDI entspricht bei GS1 der GMN (Global Model Number). Die Spezifikation ist hier zu finden:
https://ec.europa.eu/docsroom/documents/38563/attachments/1/translations/en/renditions/native
Nach dem Company Prefix (erste Ziffern) dürfen Sie den mittleren Teil frei vergeben (Zahlen, Buchstaben und Sonderzeichen). Dieser sollte entsprechend der Basis-UDI Vorgaben ein Produktmodell oder Produktfamilie abdecken.
Herzliche Grüße, Christian Johner
Hallo Herr Dr. Johner,
Die Definition der UDI-DI erscheint mir klar und wirft keine Fragen auf.
Die UDI-PI hingegen schon. Als Hersteller von Produkten der Klasse 1, wo kann ich nachlesen welche Informationen in den PI Code eingetragen werden müssen?
In vielen Publikationen wird nur geschrieben was man in den PI Code eintragen kann! Nicht was man muss.
Sehr geehrter Herr Strinzinger,
die PI müssen und können Sie frei vergeben. Entscheidend ist nur, dass diese wirklich eindeutig ist.
Ich hoffe, Ihre Frage richtig verstanden zu haben. Wenn nicht, haken Sie gerne nach.
Viele Grüße, Christian Johner
Hallo zusammen,
in Bezug auf Kommentar 76/77 habe ich eine Frage zur Einstufung der Software.
Wird die Software dazu genutzt ein MP klasse IIb zu steuern ist die SW Teil des MP und somit der selben klasse zuzuordenen, richtig? (Anhang III, Kapitel II, 3.3).
Wenn ich den Kommentar 77 richtig verstanden habe ist damit keine Zuteilung einer UDI nötig, da SW Teil eines MP. Aber woher weiß ich das es bei Teil C 6.5 um standalone Software geht?
eine weitere Frage wäre wie mit Geräten umzugehen ist, die zur STK ins Haus kommt. Müssen die Geräte nachträglich mit UDI konformen Etiketten versehen werden?
Sehr geehrter Herr Schweizer,
eine Software wird dann der gleichen Klasse wie das von ihr gesteuerte Medizinprodukt zugewiesen, wenn es eine standalone Software ist. Falls die Software Teil des Medizinprodukts ist, das sie steuert, wird nicht sie, sondern das Medizinprodukt klassifiziert.
Zwar vergleichbar aber dennoch völlig unabhängig davon ist die Frage mit der UDI: Nur standalone SW bekommt eine eigene UDI. Keine SW, die Teil eines Medizinprodukts ist.
Viele Grüße, Christian Johner
Hallo Herr Johner,
in der Tabelle unter Punkt 7 „Dokumente, die die UDI enthalten müssen“ sind bei den letzten beiden Punkten die Felder für „Artikel, Verweis“ leider leer.
Uns als Händler interessiert hierbei, ob (und wenn ja, auf welcher Grundlage) wir unsere Lieferanten „verpflichten“ können, die UDI-DI und UDI-PI auf ihre Lieferscheine und Rechnungen zu drucken, und ob wir das selbe für unsere Kunden gewähleisten müssen.
Viele Grüße
Achim Hürster
Sehr geehrter Herr Hürster,
Sie können sich am ehesten auf Artikel 14 der MDR beziehen.
Allerdings sollte eher der Hersteller daran Interesse haben, das Produkt im Markt nachverfolgen zu können. Denn genau das will die MDR von ihm.
Eine Pflicht, die UDI-PI auf dem Lieferschein anzugeben, gibt es nicht. Eine andere Art der Identifikation, über die auf die UDI-PI zurückgeschlossen werden kann, dürfte auch die regulatorischen Vorgaben erfüllen.
Beste Grüße, Christian Johner
Ich bin Softwareentwickler und muss „einfach nur“ die UDI-DI und UDI-PI im Splashscreen und Info-Dialog unserer Standalone Software anzeigen.
Da ich kaum taugliche, konkrete Hinweise gefunden habe, wie die UDI-DI und UDI-PI in meinem konkreten Fall aussehen muss, möchte ich meine Erfahrungen hier teilen.
1. Die einzig hilfreichen Informationen stecken in diesem Dokument auf den Seiten 59 und 60: http://www.imdrf.org/docs/imdrf/final/consultations/imdrf-cons-udi-system-n48-180712.pdf (das Dokument ist übrigens auch sehr hilfreich für Medizinprodukte, die nicht Standalone-Software sind)
2. Wir haben uns vor längerer Zeit bereits bei GS1 eine GS1-8 Prefix bzw. GS1 Company Prefix gekauft. Somit haben wir einen weltweit eindeutigen 8-stelligen Zahlen-Code, womit wir nun 1000 weltweit eindeutige EANs bilden können. GS1 verweist in https://www.gs1.org/sites/default/files/docs/healthcare/position-papers/gs1_udi_guide_final_20170324.pdf S. 3 darauf, dass die UDI-DI bei GS1 durch deren GTIN implementiert ist, was nichts anderes als die EAN ist. „The GS1 Global Trade Item Number (GTIN) enables this aspect of the UDI“
3. Für jede UDI-DI verbrauchen wir nun so eine EAN
4. Die UDI-PI ist einfach nur die Versionsnummer, die wir auch bisher dem Kunde anzeigen in unserem Info-Dialog
Über ein kurzes Feedback zu diesem Vorgehen würde ich mich freuen! Vielleicht kann dann auch Kapitel https://www.johner-institut.de/blog/regulatory-affairs/unique-device-identification-udi/#software dieses Blogs um ein hilfreiches Bsp. ergänzt werden.
Sehr geehrter Herr,
danke für Ihre Nachricht. Ich bin nicht ganz sicher, was die Frage ist. Ihr Vorgehen hört sich jedenfalls logisch an. Ob Sie die Version (genau genommen die „Minor-Version“) in die PI direkt kodieren oder die PI als Referenz auf diese Minor-Version haben, entscheiden Sie. Der Info-Dialog ist gut. Dass Sie mit jeder Major-Version eine EAN verbraten ist richtig, wobei sie nicht verpflichtet sind, diese EAN zu vergeben. Es besteht nur die Pflicht für die UDI. Natürlich bietet sich das an.
Wie gesagt, Ihr Vorgehen klingt schlüssig.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielleicht können Sie ein wenig Licht ins Dunkel zum Thema UDI bei Hersteller-Händler-Beziehungen bringen.
Wie muss ich mir die Kennzeichnung mit UDI (HIBC, Datamatrix) und die Registrierung durch den Hersteller in Eudamed vorstellen, wenn ein Hersteller das gleiche Material an verschiedene Händler (vorher bereits PLMs für das Produkt) verkaufen möchte, die dieses Produkt alle unter eigenem Handelsnamen und eigener Artikelnummer an die Anwender vertreiben wollen?
Dass der Hersteller auf dem Etikett bei allen Händlern angegeben werden muss, ist klar. Aber ist es so, dass das gleiche Produkt (mit gleicher Primärverpackung) pro Händler zu registrieren ist und es so zu unterschiedlichen UDI-DIs kommt?
In Annex VI, PART B Nr. 10 der MDR steht „if applicable, name or trade name“. Wie kann man das verstehen? Die Angabe des (Handels-)Namens ist „freiwillig“? Ist dies vielleicht der „Türöffner“, um ein Produkt mit verschiedenen Handelsnamen nur unter einer UDI-DI zu registrieren, ohne dass sofort augenscheinlich wird, welche Händler das gleiche Produkt anbieten? Dann gäbe es trotzdem einen Unterschied zwischen der ausgewiesenen Artikelnummer auf der Kennzeichnung (Artikelnummer des Händlers) und der Artikelnummer im HIBC-Code und der entsprechenden Klarschrift (Artikelnummer des Herstellers) auf dem Etikett. Wäre das trotzdem möglich?
Grundsätzliche Annahme bei meinen Fragen ist, dass die vollständige Konfektionierung, also einschließlich Etikettierung mit den unterschiedlichen Handelsnamen, beim Hersteller erfolgt.
Herzliche Grüße
K. Lippert
Sehr geehrte Frau Lippert,
es wird in der EUDAMED-Datenbank möglich sein, ein und dieselbe UDI-DI für mehrere Handelsnamen zu verwenden. Im UDI Data Dictonary der Kommission finden Sie die entsprechende Spezifikation: Die Beziehung UDI-DI zu Handelsname ist eine 1-n Beziehung, d.h. es sind mehrere Handelsnamen zu einer UDI erlaubt. Ob dies der Hersteller so umsetzt, bleibt ihm überlassen. Wenn Sie allerdings (bereits registrierte) Handelsnamen ändern, dann wäre die Rückverfolgbarkeit nicht mehr gegeben und eine neue UDI-DI müsste vergeben werden.
Zur Artikelnummer: Es kann sicherlich eine weitere händlerspezifische Artikelnummer ergänzt werden auf der Kennzeichnung. Die Original-Herstellerreferenz (z.B. REF-Nummer) sollte allerdings weiterhin erkenntlich sein.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Professor Johner,
meine Frage ergänzt ggf. die Feststellungen unter Punkt 65/66.
Wir haben 2 Medizinprodukte welche ausschließlich als Zubehör in eine System eingebaut werden (Röntgengenerator).
Wie ist der Begriff Produkt und evtl. System nach MDR hier in Verbindung mit Zubehör zu verstehen. Ist ein Produkt nur „komplett“ wenn alle zur Verwendung notwendigen Teile (also z.B. Kabel, Adapter, Netzteile, Schienen, Halter) als eine Produktvariante mit einer UDI-DI definiert werden? Oder ist das bereits ein System? Benötigen alle Zubehörteile eine eigene UDI-DI was dann zu einem System führt? Oder doch nicht, weil es kein Zubehör zum Zubehör gibt und spielt dies bei der UDI-Vergabe gar keine Rolle?
Vielen Dank.
Martin Barnett
Sehr geehrter Herr Barnett,
Wenn Ihre Generatoren tatsächlich Zubehör und keine Komponenten und keine Ersatzteile sind, dann gelten für dieses Zubehör die gleichen Regeln wie für „normale“ Medizinprodukte.
Wenn Sie verschiedene „Produktvarianten“ aus Kabel, Adapter usw. haben, dann würde man diese „Zusammenstellung“ eher als ein konfigurierbares Produkt mit einer UDI sehen als ein System aus vielen Produkten. Ich würde Ihnen das empfehlen, weil Sie sonst mit ziemlich vielen UDIs hantieren müssen.
Kabel, Schienen usw. sind wahrscheinlich keine „Zubehör“ im Sinne der MDR. Es sind entweder Teiles eines Systems oder besser Teile eines konfigurierbaren Produkts. Das Konzept des „konfigurierbaren Produkts“ verwendet die MDR speziell im Kontext der UDI.
Beste Grüße, Christian Johner
Hallo Herr Johner,
müsste es unter Punkt 5 c) „Wo die Basis-UDI-DI verwendet wird“ nicht heißen:
„In der technischen Dokumentation gemäß Anhang II“ – nicht wie aufgeführt Anhang I?
Mit freundlichen Grüßen
C. Distel
Liebe Frau Distel,
Sie haben vollkommen Recht, es ist natürlich der Anhang II. Der Fehler ist bereits korrigiert. Vielen Dank für Ihren Hinweis!
Sehr geehrter Herr Johner und Herr Salvatore,
ich habe noch nicht ganz verstanden wie sich die UDI-PI bilden lässt.
Kann oder muss sie eine der Komponenten Seriennummer, Losnummer, Software-Identifikation, Herstellungs- und/oder Verfallsdatum enthalten? Kann ich folglich die Seriennummer für alle meine Produkte als PI nutzen, auch wenn diese schon auf der Kennzeichnung angegeben ist?
Mit freundlichen Grüßen
A. Salmen
Guten Tag,
die Informationen, welche die UDI-PI enthalten muss, ergibt sich aus der Kennzeichnung. Falls Ihre Kennzeichnung eine Seriennummer enthält, muss diese auch Teil der UDI-PI sein. Wenn Ihre Kennzeichnung auch eine Chargennummer enthält, müsste diese ebenfalls in der UDI-PI kodiert werden. Gleiches gilt für das Haltbarkeitsdatum. Das Herstellungsdatum ist nur zu kodieren, falls es das einzige Attribut wäre.
Sehr geehrter Herr Johner und Herr Salvatore,
ich habe eine Frage zur UDI und GTIN-Vergabe. Eine UDI wird laut MDR ausschließlich vom Hersteller vergeben. Bedeutet dies, dass auch der GTIN (=UDI-DI) für ein Medizinprodukt ausschließlich vom Hersteller vergeben werden darf?
Oder kann ich als Importeur und Distributor einem Produkt, das ich unter meinem Markennamen verkaufe, einen „meiner“ GTINs geben, und wenn der Hersteller dieses Produkt z.b. nächstes Jahr gemäß MDR zertifizieren lassen möchte, darf er dann die UDI mit „meinem“ GTIN bilden? Der GTIN führt doch über die GS1-Basis Nummer zu einer bestimmten Firma (diejenige, die eben diesen GTIN gekauft hat).
Darf nun ein anderer GTIN als der des Herstellers auf dem Produkt sein und dadurch auch Teil der UDI sein, die vom Hersteller vergeben wird?
Oder darf ein Medizinproduktehersteller UDIs ausschließlich mit seinen eigenen GTINs vergeben? Völlig unabhängig davon, ob der Hersteller das Produkt unter seinem eigenen Markennamen Inverkehrbringt, oder durch einen Importeur und Distributor unter dessen Markennamen?
Mit freundlichen Grüßen,
Andrea Feiler
Sehr geehrte Frau Feiler,
vielen Dank für Ihre spannende Frage. Die UDI-Vergabe darf ausschließlich vom Hersteller erfolgen. Sicherlich darf dieser den Prozess der UDI-Vergabe und -Anbringung auslagern, z.B. an einen Händler. Die Gesamtverantwortung bleibt allerdings beim Hersteller.
Interessant ist die Frage, ob in diesem Fall nur GTINs mit dem company prefix des Herstellers verwendet werden dürfen. Die MDR und die bisherigen Guidance-Dokumente schreiben dazu leider nichts Konkretes vor. Vermutlich würde dies allerdings den GS1-Allocation Rules widersprechen (https://www.gs1.org/docs/gsmp/healthcare/GS1_Healthcare_GTIN_Allocation_Rules.pdf). Diese schreiben unter anderem vor:
„responsibility for GTIN allocation remains with the original brand owner“ oder „Distributed by: Products for which an agreement exists between the Brand Owner and the party identified on the label as the Distributor. The Brand Owner remains responsible for GTIN assignment…“ Demnach würde ich empfehlen, die GTINs des Herstellers zu nutzen. Die Vergabe und Anbringung darf dann durch den Händler erfolgen als ausgelagerter Prozess.
Herzliche Grüße
Luca Salvatore
In Anhang 6 Teil C 3.1 steht: „Eine UDI wird dem Produkt selbst oder seiner Verpackung zugeteilt. Höhere Verpackungsebenen verfügen über eine eigene UDI.“
Mir ist hier nicht klar, wann ich auf eine Umverpackung die UDI des Produkts aufbringe und wann eine eigene UDI verwendet wird. Auf der Folie um Implantat zum Beispiel wird die UDI des Implantates aufgebracht. Wenn ich das Implantat in der Folie in einen Einzelkarton tue wahrscheinlich auch. Wenn ich mehrere Implantate dann gemeinsam in einen Karton tue, muss dann eine karton-spezifische UDI verwendet werden, außer es handelt sich um einen Versandkarton?
Sehr geehrte Frau Toll,
Ihre Antwort ist korrekt. Sie geben unterschiedliche UDIs an, sobald Sie mehrere Produkte in einer Verpackungsebene zusammenfassen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Johner und Herr Salvatore,
ich habe jetzt bereits zweimal aus unterschiedlichen Quellen gehört, dass die Basis-UDI-DI nicht zu groß gewählt werden sollte, da diese gegebenenfalls für Sicherheitskorrekturmaßnahmen (wie bspw. Rückrufe) herangezogen werden könnte und nicht nur eine bestimmte Chargen zu UDI-DI´s. Bei meinen Recherchen habe ich leider dazu gar nichts gefunden.
Wissen Sie ob das der Fall ist und man deshalb zur Vorsicht die Basis-UDI-DI Gruppen wirklich kleiner halten sollte?
Viele Grüße
Maria
Sehr geerhte Frau Tschirner,
es gibt bereits konkrete Guidance-Dokumente der britischen Behörde MHRA zum Thema Sicherheitskorrekturmaßnahmen unter der MDR (https://www.gs1uk.org/sites/default/files/061119_Field_Safety_Corrective_Actions_document.pdf). Dort wird empfohlen, welche Informationen dringende Sicherheitsmeldungen des Herstellers (Field Safety Notices) enthalten müssen.Es sind GS1- und HIBCC-spezifische Formulare vorhanden, die genutzt werden können, um die von der Sicherheitskorrekturmaßnahme betroffenen UDIs zu listen (siehe z.B. auch https://www.gs1uk.org/sites/default/files/061119_Field_Safety_Corrective_Actions_document.pdf). Dort werden konkrete UDIs genannt und keine Basis-UDI-DIs. Ich denke auch nicht, dass eine Maßnahme auf alle Produkte/Varianten, die einer Basis-UDI-DI zugeordnet sind, anzuwenden ist. Die Bestimmung des Umfangs der Maßnahme/des Rückrufs ist zunächst Aufgabe des Herstellers.
Herzliche Grüße
Luca Salvatore
Im MDCG-Papier 2018-1 wird darauf hingewiesen, dass die Leitlinie nicht für wiederaufbereitete Medizinprodukte gilt.
Wie bildet man das nun in der TD für diese Medizinprodukte ab? Lässt man die Angabe einfach weg oder begründet man, warum es keine Nummer gibt.
Herzlichen Dank und viele Grüße
Heike Hauptmann
Sehr geehrte Frau Hauptmann,
die Anforderungen an die Basis-UDI-DI und die Anforderungen, wann neue UDI-DIs vergeben werden müssen, sind in der MDR angegeben. Die MDCG hat nun eine Reihe von Guidance-Dokumenten für bestimmte Aspekte des UDI-Systems veröffentlicht, u.a. die von Ihnen Genannte. Diese geht allerdings nicht auf die Besonderheiten von Software, Behandlungseinheiten und wiederaufbereiteten Produkten ein. Dennoch müssen Sie die UDI-Anforderungen auch für diese Produkttypen erfüllen. Somit können Sie nicht einfach die Basis-UDI-DI weglassen. Diese muss in jedem Fall angegeben werden.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
ich frage mich hinsichtlich der Platzierung der UDI-Carrier auf die Produkte und deren Timelines (MDR §27(4) und §123 (3)f) wie mit Lagerware und Ware im Umlauf umgegangen werden soll.
Ist meine Annahme richtig, dass ab dem betreffenden Stichtag nur noch Ware mit der UDI in den Verkehr gebracht werden darf. Lagerware wäre damit verfallen. Ware im Marktumlauf wäre aber weiter zu kaufen (oder ist Sie zurückzuziehen?).
Vielen Dank für Ihre Antwort
Sehr geehrter Herr Lang,
Ihre Annahme ist korrekt. Produkte die nach dem entsprechenden Stichtag (unter der MDR!) ohne UDI in Verkehr gebracht werden, wären nicht mehr MDR konform. Ware im Marktumlauf ist bereits in Verkehr gebracht und könnte auch ohne UDI-Kennzeichnung noch bis zur Inbetriebnahme bereitgestellt werden.
Herzliche Grüße
Luca Salvatore
Hallo zusammen,
gibt es Erfahrungen zur Interpretation von MDR, Anhang VI, Teil C, 4.10. b) ? Hier heißt es das wiederverwendbare Produkte kein UDI Träger brauchen, wenn es „technisch nicht möglich ist“.
Mir fällt es schwer zu glauben, dass im Wortsinn „technisch nicht machbar“ gemeint ist, denn „technisch machbar“ ist vieles. Aber ist es auch ökonomisch sinnvoll? Konkret könnte es bedeuten, dass ich meine Firma schließen muss, weil ich für die Umsetzung ein state-of-the-art Beschriftungslaser gekauft (dieser ist technsich in der Lage auf kleiner Fläche ein UDI-konformen Datamatrix Code aufzulasern), einen Mitarbeiter eingestellt (der das Gerät bedienen kann) und das Material gewechselt habe (damit es überhaupt laserbar ist). Auf halbem weg ist mir aber das Geld ausgeangen und die Firma ist insolvent.
Das Produkt desshalb einzustellen erscheint mir aber auch eine sehr dramatische Maßnahme zu sein.
vielen Dank und Grüße,
Christian
Lieber Herr Schweizer,
man kann davon ausgehen, dass diese Anforderung streng interpretiert werden wird. Dies zeigt z.B. ein Statement der FDA zum Thema „Technical Feasibility“:
„We expect that such situations [Anmerkung: Technically not feasible] will be rare. FDA does not consider exception requests that are based
on reasons other than technological infeasibility (including, but not limited to, financial burden, a claimed low rate of adverse events, or a claim that the product is somehow unique such that adverse events do not occur).“ Sie möchte demnach keine finanziellen Gründe gelten lassen. Sicherlich gibt es die Möglichkeit, anstatt teure Maschinen für das DPM anzuschaffen, diesen Prozess an geeignete Dienstleister auszulagern. Einige Beispiele, wo es Probleme mit der Machbarkeit gibt, finden Sie im IMDRF-Dokument ab Seite 50:
http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-190321-udi-sag.pdf
Herzliche Grüße
Luca Salvatore
Vielen Dank auch für Ihren Hinweis zu den zurückgezogenen Normen. Wir haben die Liste entsprechend aktualisiert.
Sehr geehrter Herr Salvatore,
als Hersteller einer recht robusten Orthese, die durchaus über viele Jahre hinweg täglich aufs neue angewendet werden kann bin ich mir zunächst einmal sicher, daß es sich hierbei keinesfalls um ein chirurgisches Instrument – und somit auch nicht um ein „wiederverwendbares chirurgisches Instrument“ handelt.
Gibt es für meine Orthese oder für einen als Medizinprodukt deklarierten Krückstock, in Bezug auf die regulatorischen Anforderungen der MDR bei der Anmeldung (einschließlich UDI-Beschaffung) vor dem 26.05.2021 noch eine weitere andere Meldungs- relevante Begrifflichkeit wie „zur Wiederverwendung geeignet“ [z.B. Dieser Blog unter 2. e], die für die von einer sterilen oder chirurgischen Zweckbestimmung losgelöst ist?
Falls ja,
wo/wie erläutert ?
in welchem Anmeldungs-Schritt relevant?
Sehr geehrter Herr Klobe,
vielen Dank für Ihre Frage. Im Data Dictonary zur EUDAMED (https://ec.europa.eu/docsroom/documents/38543) finden Sie alle zu meldende Informationen. Für Ihr Produkt sind mir keine speziell Eingabefelder bekannt. Ein Feld „zur Wiederverwendung geeignet“ existiert nicht, allerdings können Sie optional bei wiederverwendbaren Produkten (in EUDAMED: labeled as single use = „no“) die Anzahl der erlaubten Wiederverwendungen angeben oder als nicht anwendbar angeben.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
Ich glaube, der Groschen fällt gerade. Kann ich Ihre Worte dahingehend verstehen, daß das Feld 17 aus UDIWG 2018 1 und 2 ausreichend optional ist, daß
„Maximum number of reuses“ mit der Erklärung „If the device is reusable, an indication of how many times the device can be re-used according to ist label and instructions of use“
im Falle eines Krückstocks als Medizinprodukt nicht zwingend beziffert werden muss?
Sehr geehrter Herr Klobe,
ganz genau. In diesem Fall müssen Sie keine genaue Anzahl der möglichen Anwendungen/Wiederverwendungen angeben.
Herzliche Grüße
Luca Salvatore
Vielen Dank für Ihre ausführliche Information zu dem umfassenden Thema!
In Ihrem Artikel und Ihrem Webinar wird darauf hingewiesen, dass die maschinenlesbare Codierung (AIDC) der UDI zu bevorzugen ist, bei Problemen beide Formate auf ein Produkt aufzubringen. Dazu verweisen sie auch auf MDR Anhang VI Teil C Artikel 4.7. Da steht allerdings: „Gibt es erhebliche Probleme, beide Formate — AIDC und HRI — auf der Kennzeichnung unterzubringen, so ist nur das AIDC-Format auf der Kennzeichnung zu verwenden. Bei Produkten, die außerhalb von Gesundheitseinrichtungen verwendet werden sollen, wie etwa Produkte für die häusliche Pflege, ist allerdings das HRI-Format auf der Kennzeichnung zu verwenden, auch wenn dies dazu führt, dass für das AIDC-Format keine Fläche zur Verfügung steht. “
D.h. sie unterschlagen den zweiten Teil der Anforderung, oder? Es wird nicht immer wird das AIDC-Format bevorzugt.
Sehr geehrte Frau Blümke,
ganz genau. Im Normalfall ist bei Platzmangel das AIDC-Format zu wählen. Im Sonderfall, bei Produkten die außerhalb von Gesundheitseinrichtungen eingesetzt werden, ist dies genau umgekehrt. Hier soll das HRI-Format bevorzugt werden.
Herzliche Grüße
Luca Salvatore
Guten Abend,
ich habe einige Fragen;
1. Die Angabe ob single use oder nicht, wird das auf der Einheit der Basic UDI oder UDI-DI in EUDAMED eingegeben?
2. Gibt es eine maximale Länge/Größe für die UDI-DI bei GS1 zB?
3. UDI-PI, ist es Pflicht bestimmte Werte (zB LOT oder EXP) anzugeben oder kann man es sich selbst aussuchen?
4. Größere Versandpakete und Paletten bekommen keine UDI, ist das richtig?
Sehr geehrte Frau Frank,
vielen Dank für Ihre Fragen:
zu 1.) Das Merkmal „single use“ wird in EUDAMED auf Ebene der UDI-DI Registrierung angegeben.
zu 2.) Ja, die gibt es, abhängig vom gewählten Barcode (z.B. GS1 128, Datamatrix, etc.). Die Informationen finden Sie in der GS1-Spezifikation (https://www.gs1.org/docs/barcodes/GS1_General_Specifications.pdf). Dort steht beispielsweise zum GS1-128 Barcode: „The physical length, including Quiet Zones, cannot exceed 165.10 millimetres“
zu 3.) Die Antwort dazu finden Sie in der MDR in Anhang VI, 3.5:
„Wird auf der Kennzeichnung eine Losnummer, eine Seriennummer, eine Software-Identifikation oder ein Verfallsdatum angegeben, so ist diese bzw. dieses Teil der UDI-PI. Befindet sich auf der Kennzeichnung auch das Herstellungsdatum, so muss dieses nicht in die UDI-PI aufgenommen werden. Befindet sich auf der Kennzeichnung nur das Herstellungsdatum, so ist dieses als UDI-PI zu verwenden.“
Wenn also Losnummer und Haltbarkeitsdatum auf der Kennzeichnung angegeben werden, müssen diese auch in der UDI-PI kodiert werden.
Zu 4.) Typischerweise nicht, es sei denn, es handelt sich dabei um eine definierte Verpackungsebene mit einer definierten Anzahl an Produkten. Schauen Sie dazu mal in die GS1 Healthcare Allocation Rules (https://www.gs1.org/standards/gs1-healthcare-gtin-allocation-rules-standard/current-standard#2-GTIN-Allocation-Rules+2-1-New-product-introduction): „Higher levels of packaging, such as a case or pallet, can be identified with a GTIN when it is identifying a trade item“. Paletten und Versandpakete mit variablen Inhalten bekommen typischerweise keine eigene GTIN zugeordnet.
Herzliche Grüße
Luca Salvatore
Sehr geehrte Damen und Herren,
ich habe eine Frage bezüglich der Kennzeichnung bei der Produktverpackung mit UDI.
1. Muss neben dem UDI-Code zwingend das UDI-Symbol zu sehen sein?
2. Wenn neben dem UDI-Code das HIBC-Symbol zu sehen ist, muss das HIBC-Symbol durch das UDI-Symbol ersetzt werden? Meiner Meinung nach muss dieser nicht ersetzt werden, da der HIBC-Code dem UDI-Code entspricht.
Wie sehen Sie es?
Vielen Dank im Voraus.
Sehr geehrte Frau Alici,
vielen Dank für Ihre Fragen zur UDI, die ich sehr gerne im Folgenden beantworte:
Zu 1.) Die Angabe des UDI-Symbols ist generell optional. Es sollte verwendet werden, falls mehrere Datenträger auf der Kennzeichnung vorhanden sind, damit der UDI-Code eindeutig bestimmt werden kann (siehe auch ISO 15233-1)
Zu 2.) Das wäre sicherlich auch eine Option. Wie gesagt, gemäß ISO 15223-1 ist die Angabe des UDI-Symbols optional.
Herzliche Grüße
Luca Salvatore
Sehr geehrte Damen und Herren,
zunächst einmal vielen herzlichen Dank für die Ausarbeitung der relevanten Informationen rund um UDI. Eine zentrale Frage bleibt für mich aktuell noch offen. Diese bezieht sich auf die Fristen und die damit zusammenhänge aktive Bearbeitung dieses Themas.
Müssen wir als Hersteller von Produkten, u.a. Medizinprodukte (Reinigungsmittel), bis Mai 2021 aktiv eine Meldung (mehrere Meldungen) unserer Produkte vornehmen. Momentan erfolgt die Argumentation innerhalb unseres Unternehmens auf Basis der Kennzeichnungspflichten. Für Kategorie II Produkte wäre dies Mai 2023. Wie ich jedoch hier lesen kann, müssen entsprechende Meldungen aber bereits im Mai 2021 vorliegen (Basis UDI-DI).
Vielen Dank für Ihre Rückmeldung.
Freundlichen Gruß
F. Heinen
Sehr geehrter Herr Heinen,
die MDR fordert die Registrierung von Medizinprodukten (unabhängig ob Konformität mit der MDD oder der MDR erklärt wird) ab dem 26.05.2021, vorausgesetzt, das Registrierungsmodul ist bis dahin funktionsfähig und freigeschaltet. Zusätzlich gibt es aber eine Übergangsfrist von 18 Monaten. D.h. Sie haben bis November 2022 Zeit, die Registrierung abzuschließen. Für Produkte, welche Sie unter der MDD in VErkehr bringen, müssen keine UDIs vergeben oder registriert werden. In diesem Fällen wird automatisch eine Nummer von EUDAMED generiert. Diese muss aber nicht auf Ihren Produkten angebracht werden.
Herzliche Grüße
Luca Salvatore
Guten Tag,
Ich beziehe mich auf die Frage von „Antje | Freitag, 8. Januar 2021 um 16:04 Uhr“.
Wenn z.B. bei einer Implantatschraube eine AIDC UDI-Kennzeichnung aufgrund der Wölbung nicht möglich ist, obwohl diese zu bevorzugen ist, kann ich die HRI UDI-Kennzeichnung wählen, da diese technisch umsetzbar ist?
mfg
Sehr geehrter Herr Grill,
ganz genau. Bevor gar keine UDI-Kennzeichnung angebracht wird, sollte in diesem Fall zumindest, wenn technisch machbar, die HRI-Variante gewählt werden.
Herzliche Grüße
Luca Salvatore
Sehr geehrte Damen und Herren,
bei der Planung von UDI sind meinem Team noch einige Fragen aufgekommen.
Wir stellen stoffliche Medizinprodukte her und haben sehr viele Distributoren außerhalb der EU. Nun zu unserer Frage, muss der UDI Carrier nur auf Produkte, die innerhalb der EU vertrieben werden oder auch auf Produkte, die außerhalb der EU vertrieben werden? Denn in die Eudamed sollen ja so oder so alle Produkte registriert werden.
Ist es möglich sowohl Barcode als auch Datamatrix gleichzeitig auf die Faltschachtel zu drucken?
Wie sieht es mit der Unit of Use DI aus, muss diese zwangsläufig ebenfalls nach zB GS1 erstellt werden mit den gleichen Konditionen, wie die UDI-DI erstellt wird, also Companyprefix-Nummer-Prüfziffer? Oder können hier Firmeninterne beliebige „Artikelnummern“ verwendet werden?
Danke und viele Grüße
V. Frank
Sehr geehrte Frau Frank,
in EUDAMED müssen nur Produkte registriert werden, die auch in der EU bzw. unter der MDR in Verkehr gebracht werden. Dementsprechend müssen Sie auch nur solche Produkte mit der UDI versehen. Beachten Sie aber, dass andere Länder wie die USA, ebenso Anforderungen an eine UDI-Markierung stellen.
Generell müssen Sie sowohl die maschinenlesbare Form/HRI als auch den Klartext auf der Verpackung anbringen. Eine Unit of Use DI benötigen Sie nur, wenn Sie einzelne Produkte nicht kennzeichnen (z.B. mehrere Handschuhe in einem Karton). Hier ist mir nicht bekannt, dass man von den generellen UDI-Anforderungen der gewählten Zuteilungsstelle abweichen darf.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore
als Hersteller von lediglich einer einzigen aber ganz speziellen Produktfamilie von Orthesen ( mit ganz eindeutig nur eine Basis-UDI-DI) fertige ich 5 Produktvarianten und hoffte mit 5 UDI-DI auszukommen.
Meine Orthesen selbst sind bisher – abgesehen von einem CE Symbol und einer Seriennummer – unbeschriftet; sie werden weltweit gezielt angefragt und danach erst mit einer Gebrauchsanleitung und einem Label auf der Produktverpackung versehen in den Versand gebracht.
Nun erfuhr ich, daß die UDI-Zuteilungsstelle GS1 für identische Gegenstände („Produkte“), jedoch mit von einander abweichenden Sprachen in der Gebrauchsanleitung auch jeweils von einander abweichende UDI-DI einfordert. (Unter allgemein kommerziell-logistischen Gründen kann ich dies meist sogar nachvollziehen.)
Meine Frage lautet jedoch : Ist dies (für jede abweichende Sprache in der Gebrauchsanleitung) eine abweichende UDI-DI) auch auch schon von der MDR zwingend vorgeschrieben?
Falls NEIN, kennen Sie eine UDI-Zuteilungsstelle, welche für identische Gegenstände („Produkte“) auch dann noch eine identische UDI-DI zulässt, wenn die Gebrauchsanleitungen in von einander abweichenden Sprachen dazu gepackt werden?
Hintergrund: Als Kleinunternehmer (Micro Enterprise) möchte möchte ich in Hinblick auf die künftige UDI-Kennzeichnungspflicht möglichst lange offen halten können, mit welcher Sprache ausgestattet welche Orthese einmal verkauft wird.
Mit freundlichen Grüßen
Eckart Klobe
Sehr geehrter Herr Klobe,
in der Tat verlangt GS1 in ihren Healthcare Allocation Rules eigene UDI-DIs pro „trade item“ und für jede Sprachvariante. Zum „trade item“ zählen auch Begleitdokumente nach GS1: „If a manual or leaflet is included in the product package, it is considered to be part of the trade item identified with a GTIN.“ Diese Forderung geht über jene der MDR hinaus. Allerdings fordert die MDR die Einhaltung der Regeln der jeweiligen Zuteilungsstelle. Somit sehe ich in Ihrem Fall keinen Ausweg von der Vergabe für sprachenspezifische UDI-DIs. Bei HIBCC sind mir solche Regeln nicht bekannt. Ich würde Ihnen empfehlen, dazu direkt bei der Zuteilungsstelle anzufragen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
vielen Dank für die schöne Übersicht zu diesem Thema! Uns ist nun bei der Umsetzung die Frage aufgekommen, welche Zuteilungsstelle man wann wählen sollte. Da wir ein kleiner Hersteller von nur 5 verschiedenen Produkten sind, müssen wir die Kosten gut im Blick behalten. Die Preise zwischen GS1 und IFA GmbH weichen erheblich voneinander ab. Sie erwähnen eigentlich immer nur GTIN von GS1, aber würde nicht auch die PPN von IFA möglich sein, wenn beide vom MDR bzw. der Europäischen Kommission als Zuteilungsstellen benannt sind? Sind Ihnen irgendwelche Unterschiede bekannt, warum man GTIN statt PPN wählen sollte?
MfG
Stefanie Neidhardt
Sehr geehrte Frau Neidhardt,
Sie haben generell die Freiheit die Zuteilungsstelle selbst zu wählen und somit auch IFA. IFA basiert auf der PZN/PPN-Nummer und wird eher im Umfeld der Apotheken eingesetzt, GTIN finden wir in vielen Bereichen des Einzelhandels. Ein weiterer Unterschied ist die Kodierung ausschließlich über die Datamatrix bei IFA. Bei GS1 gibt es weitere Formate, die möglich sind. Alternativ gibt es auch noch HIBCC, die ein direktes Kodierungssystem anbieten. Wenn Ihre Kunden keine konkreten Anforderungen stellen and die Zuteilungsstelle / Kodierungsstandard, dann haben Sie sicherlich die freie Wahl.
Herzliche Grüße
Luca Salvatore
Muss neben dem UDI Code das Symbolzeichen ‚UDI‘ der ISO 15223 verwendet werden? Oder reicht es, wenn der UDI Code auf der Produkt und Verpackungskennzeichnung angebracht wurde?
Guten Tag,
die Antwort auf Ihre Frage finden Sie weiter oben in den Kommentaren (Antwort vom 22.01.2021).
Herzliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
herzlichen Dank für den Hinweis. Ich hatte mich zwar quer durch den Chat gelesen, aber nicht vollständig.
MfG
Hello Mr. Salvatore,
We manufacture cables to be used with our variety of products.
As per classification, they come under class I self certified class and are listed as accessory to medical device.
My question is, do we need to assign a Basic UDI-DI for each cables (different specification of use).
We have UDI-DI in place on its packaging level and also UDI-PI is also present.
Please let me know
Thank you.
Paul
Dear Paul,
you may only combine the cables under the same Basic UDI-DI of the essential design and manufacturing characteristics are similar. Without knowing the specifications of these cables, it is not possible to provide a more detailed answer. You may use the MedTech Europe’s Guidance on the Basic-UDI-DI assignment (https://www.medtecheurope.org/wp-content/uploads/2020/06/200602_MTE-Basic-UDI-DI-guidance-v1.1_final.pdf).
Best regards
Luca Salvatore
Hello Mr. Salvatore,
Thank you for your response.
I have another question to ask.
Do you have any reference to a actual template for MDR Technical file for Class I devices.
Thank you.
Paul
Dear Skaria,
we offer a set of templates for the technical documentation according to MDR. These are included in our Auditgarant Platform: https://www.johner-institut.de/auditgarant/
Best regards
Luca Salvatore
Guten Tag,
bis wann sind Basis UDI-DIs zu vergeben?
Wir haben Klasse I und IIa Medizinprodukte, die derzeit unter der MDD zugelassen sind. Ich dachte, für Klasse I Produkte, müssen UDI-DIs schon mit Mai 2021 vergeben werden?
Ein Kollege meinte, dass diese erst vergeben werden müssen, wenn EUDAMED funktionsfähig ist, daher bin ich jetzt verunsichert.
Vielen Dank.
Mit freundlichen Grüßen,
Martina
Guten Tag,
Basis UDI-DIs sind nur für Produkte, die gemäß der MDR CE-gekennzeichnet werden, notwendig ab dem 26.05.2021. Für MDD-Produkte müssen Sie keine Basis UDI-DIs vergeben.
Herzliche Grüße
Luca Salvatore
Guten Tag,
ein Paar Fragen zur IVDRs.
Müssen die Liste aller vergebenen UDIs als Teil der technischen Dokumentation für jedes Medizinprodukt pflegen oder nur basic UDI-DI. Ich könnte das nicht in IVDR finden. Eine Komponente, die identisch in verschiedenen IVDR Kits ist, muss auch identische UDI haben? Wenn ein IVDR Kit (besteht aus 4 Komponenten) das heißt unterschiedliche UDIs, welche UDI kommt dann auf dem KIT? Ich meine welche UDI-PI?
Vielen Dank im Voraus!
R. Fayyad
Liebe(r) R. Fayyad,
da ein Kit gemäß der IVD-Definition in Artikel 2 selbst ein IVD sein kann, brauchen die vier Komponenten keine eigene UDI zu tragen. Das Kit ist in diesem Fall das Produkt. Sie sollten dann zwar die Einzelkomponenten rückverfolgen können. Eine eigene Basis-UDI benötigen diese aber nur, wenn Sie die vier Komponenten auch einzeln in Verkehr bringen. Welche UDI-PI im letzteren Fall beim Kit auf das Label muss ist tatsächlich eine knifflige Frage. Meine Empfehlung wäre hierfür die kritischste Komponente in Bezug auf die Haltbarkeit zu wählen und das Vorgehen so in der technischen Dokumentation und Ihren Prozessen festzulegen.
Herzliche Grüße,
Sebastian Grömminger
Guten Tag,
ich habe einige Fragen zur UDI, die momentane Implementierung dieses Systems in unsere Prozesse hat doch noch einige Fragen aufgeworfen.
Es ist ja so, dass wir als legaler Hersteller natürlich ab MDR für die Aufbringung des UDI Carriers zuständig sein werden, sobald wir Produkte haben, die wir nach MDR registrieren können. Zur Zeit ist es bei uns so, dass der Distributor ihre eigene GTIN in Form des EAN13 Barcodes auf die Produkte aufbringt. Wissen Sie, ob es möglich ist, dass dieser Barcode vom Distributor auf der Schachtel bleibt und wir nur die Datamatrix als UDI Carrier aufbringen oder muss sowohl EAN13 Barcode als auch Datamatrix (in UDI Format und unser Nummernkontingent) von uns als Hersteller aufgebracht werden, vor Allem wenn der Distributor die gleiche Zuteilungsstelle wie wir verwendet? Es wäre natürlich technisch leichter, wenn wir einfach sagen würden, der Barcode macht unser Distributor drauf mit ihrer eigenen Nummer und um die Datamatrix mit unserer spezifischen Nummer kümmern wir uns.
Und habe ich das richtig verstanden, dass wir eine UDI-DI für verschiedene Produkte verwenden können, also für ein und das selbe Produkt, aber andere Trade names, andere Sprachen etc? Ich habe es bisher immer so verstanden, dass zB Produkt A in 3 verschiedenen Ländern unter 3 verschiedenen Trade names ebenfalls 3 verschiedene UDI-DIs erhält, liege ich da falsch?
Vielen Dank im Voraus
V. Frank
Sehr geehrte Frau Frank,
die Aufbringung des EAN-Codes ist für die Einhaltung der MDR nicht relevant. Verpflichtend ist, dass Sie für die UDI-Vergabe verantwortlich sind. Somit ist Ihr beschriebenes Vorgehen möglich. Die UDI sollte allerdings als solche erkennbar sein (z.B. durch Aufbringung des UDI-Symbols aus der ISO 15223-1).
Zu Ihrer 2. Frage: Dies ist in der Tat eine GS1 spezifische Regel, die so nicht von der MDR gefordert ist. Dennoch sind Sie verpflichtet, die Regeln der Zuteilungsstelle einzuhalten. Somit sind in den genannten Fällen verschiedene UDI-DIs zu vergeben.
Herzliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
danke für die schnelle Antwort. Wenn ich die Allgemeinen Spezifikationen von GS1 aber richtig verstehe, dann ist es laut Kapitel 4.14.1 nicht möglich 2 verschiedene Carrier mit unterschiedlichen GTINs (UDI-DIs) zu verwenden. Ich verstehe das so, dass dann beide Carrier die gleiche GTIN aufweisen müssten, auch wenn nur einer dieser Carrier den hauptsächlichen UDI Carrier darstellt. Oder ich habe einen Denkfehler in dem Fall.
Viele Grüße
V. Frank
Hallo Frau Frank,
vielen Dank für das Nachhaken. Sie haben natürlich recht. Es ist zwar möglich, mehrere Barcodes anzubringen (wobei hier die UDI identifizierbar sein sollte), diese müssen aber dieselbe GTIN enthalten. In Ihrem Fall würde der Händler eine eigene GTIN vergeben und Anbringen, was somit gemäß GS1-Spezifikation nicht erlaubt wäre.
Entschuldigen Sie die Verwirrung.
Herzliche Grüße
Luca Salvatore
Lieber Herr Professor Johner,
vielen Dank im Voraus für diese unbeschreiblich hilfreichen Texte und Kommentare, die mich wirklich weiter bringen.
Nun habe ich noch eine Frage und finde keine deutliche Antwort.
Wir stellen zum einen I er MPs und zum anderen IIa MPs her.
Muss ich die UDI (UDI-DI+UDI-PI) ab dem 25.05.2021 auf die Produkte und auf die Verpackung aufbringen oder erst ab 2023?
Vielen Dank schonmal für Ihre Bemühungen.
Viele Grüße und einen schönen Nachmittag.
T. Herzog
Sehr geehrte Frau Herzog,
die UDI-Pflicht gilt generell nur für MDR-Produkte, d.h. Produkte, welche Sie unter der MDR in Verkehr bringen. Für die Aufbringung auf den Produkten/Verpackungsebenen gibt es Übergangsfristen. Für Klasse IIa-Produkte ist dies der 26.05.2023, für Klasse I Produkte der 26.05.2025.
Herzliche Grüße
Luca Salvatore
Dear Mr. Salvatore,
if my products are falled into 2 different categories, does this mean that i must generate 2 different basic udi-di, must not i? Because i think the same basic udi-di can be given at the level of medical product groups or the variants, could not it?
Many thanks in advance and my best regards, Rania
Dear Rania,
the Basic UDI-DI is not related to device categories or generic device groups. There are specific grouping criterias defined in the MDR:
– Same manufacturer
– Same risk class
– Same intended purpose
– Same essential design and manufacturing characteristics
You may refer to MedTech Europe’s guidance document on assigning Basic UDI-DIs: https://www.medtecheurope.org/wp-content/uploads/2020/06/200602_MTE-Basic-UDI-DI-guidance-v1.1_final.pdf
Best regards
Luca Salvatore
Dear Luca,
Many thanks for your kind reply and the interesting link. Actually, i did understand from your youtube presentation, that a main question is to decide at which level of the MP hierarchy, could the same basic udi-di be given. So now you mean it does not matter if i give 2 kits that belong to 2 different categories but they have the.
– Same manufacturer
– Same risk class
– Same intended purpose
– Same essential design and manufacturing characteristics
Thank you and my best regards, Rania
Dear Rania,
that’s correct. However, if 2 kits have the same intended purpose they may probably belong to the same device category.
Best regards
Luca Salvatore
Many thanks Luca! Actually, have the same intended purpose but because of the IVR code, they do not belong to the same category. It is really complex, I know! Anyhow, many thanks! I do very much appreciate!
Yours truly,
Rania
Guten Tag,
da mir das UDI Helpdesk leider nicht weiterhelfen konnte, schreibe ich hierhin. Vielleicht haben Sie weitere Infos für mich.
Es ist ja so, dass wir als Hersteller für die Vergabe der Basic UDI und UDI-DI zuständig sind, allerdings ist es so, dass unsere Distributoren bereits für unsere Produkte GTIN Nummern in Form eines EAN13 Barcodes auf Faltschachteln aufgedruckt haben. Das heißt, dass dies alles geändert werden müsst, es müssten unsere Nummern als UDI auf die Faltschachteln, alle Infos ebenfalls in den Apotheken müssten geändert werden und dies ist sehr umständlich, wie sie es sich vorstellen könnten. Laut Artikel 16 müsste es doch möglich sein, dass wir unsere Artikel mit einer von uns vergebenen Basic UDI bei EUDAMED registrieren, aber die UDI-DI stammt in dem Fall von unserem Distributor, sodass kein Change Management vonstatten gehen würde. Können Sie mir sagen, ob dies möglich wäre, dass unser Distributor die UDI-DI vergibt und wir es unter unserer Basic UDI-DI registrieren?
Oder bleibt es dabei, dass wir die UDI-DI vergeben?
Ich bedanke mich im voraus
Mit freundlichen Grüßen
Viktoria Frank
Guten Tag Frau Frank,
generell bleibt die Pflicht zur UDI-Vergabe gemäß MDR immer bei Ihnen als Hersteller.
GS1 hat zusätzliche Regelungen beim sogenannten Co-Branding und Own-Brand-Labeling (siehe https://www.gs1.org/docs/gsmp/healthcare/GS1_Healthcare_GTIN_Allocation_Rules.pdf, Kapitel 2.6). Demnach wäre der Co-Brand Owner für die GTIN-Vergabe zuständig, was aber im Widerspruch zur MDR steht. Sicherlich könnten Sie die UDI-Vergabe und UDI-Kennzeichnung an einen Händler auslagern, die Verantwortung bleibt aber immer bei Ihnen als Hersteller. Zur Absicherung würde ich Ihnen empfehlen, sich mit GS1 in Verbindung zu setzen.
Herzliche Grüße
Luca Salvatore
Guten Tag,
wenn wir mehrere Medizinprodukte in einem „Set“ zusammenstellen und dieses Set eine eigene Artikelnummer hat, müssen wir dann auf das Set für jedes in dem Set befindliche Medizinprodukt ein Label anbringen mit der entsprechenden UDI, Artikelnummer und LOT? Oder reicht es dem Set eine eigene LOT zu generieren?
Besten Dank!
Sehr geehrter Herr Fehr,
wenn es sich bei Ihrem „Set“ um eine Behandlungseinheit gemäß Artikel 22 der MDR handelt, wovon ich ausgehe, dann benötigt jedes enthaltene Medizinprodukt sowie das „Set“ selbst eine eigene Kennzeichnung einschließlich UDI.
Herzliche Grüße
Luca Salvatore
Guten Tag.
Eine Frage zur Vergabe der Basis UDI DI. Kann man einem Produkt, das einmal mit reduzierter Zweckbestimmung in einem Zielareal eines Organs verwendet wird und einmal mit größerer Zweckbestimmung am ganzen Organ verwendet wird, wobei die Form abweicht (Beispiel kantige und runde Klinge) die gleiche Basis UDI DI zuordnen?
Guten Tag,
ohne das Produkt und die genaue Zweckbestimmung zu kennen, ist eine Aussage schwer zu treffen. Generell scheint es mir, dass beide Produkte eine gemeinsame übergeordnete Zweckbestimmung haben, sich allerdings die Indikationen unterscheiden. Dies und die Form der Klinge würde ich nicht als Ausschlusskriterium ansehen, um eine gemeinsame Basis UDI-DI vergeben zu können. Ein weiteres Indiz, welches für eine gemeinsame Basis UDI-DI sprechen könnte, wäre die Beschreibung beider Produkte in einer gemeinsamen Technischen Dokumentation.
Herzliche Grüße
Luca Salvatore
Sehr geehrte Frau H.,
diese Frage ist ohne weitere Informationen nicht eindeutig zu beantworten. Dazu müsste ich wissen, ob die Auslegung und Herstellung des Produkts im Wesentlichen gleich ist. Falls das der Fall ist und falls man die Zweckbestimmungen unter eine gemeinsame subsumieren kann, ist solch eine Basis-UDI-DI denkbar.
Was dagegen spricht ist, dass die beiden Produkte wahrscheinlich unterschiedliche Risiken haben und damit über keine gemeinsame Risikomanagementakte verfügen können.
Mehr kann ich bei meinem Kenntnisstand nicht sagen und hoffe, dass trotzdem ein nützlicher Gedanke dabei war.
Viele Grüße, Christian Johner
Guten Tag,
nachdem ich alle Anfragen mit Interesse studiert habe, bin ich noch nicht zu einer Lösung unserer Fragen gekommen und möchte diese hiermit stellen:
Als Hersteller von 2 Schlauchklemmen aus Kunststoff, die seit Jahrzehnten vertrieben werden, wollen wir jetzt die Beantragung der UDI’s durchführen.
Für beide Produkte jeweils eine Basis UDI-DI, die dann gleichzeitig der UDI-DI entspricht, wenn ich das richtig sehe.
Derzeit wird auch dem Label der Verpackungseinheit – 50 Stk. – die Lot-Nummer aufgedruckt, aus der sich das Herstelldatum bzw. Charge ableiten lassen. Gleichzeitig enthält jedes Teil die für Kunststoffteile typische Datumsuhr, von der auch auf die Lotnummer bzw. Charge geschlossen werden kann.
Die Produkte sind Klasse 1 unsteril. Ein Verfallsdatum bzw. Begrenzung der Einsatzdauer ist nicht vermerkt, aber eine Klemme ist ein Einmalteil und zu der zweiten Klemme gibt es eine Aufbereitungsanweisung.
Laut Übergangsfrist haben wir das Aufbringen der Kennzeichnung bei Klasse 1 bis zum 26.05.2025 umzusetzen.
Die UDI-DI auf die Produkte neben dem Datumsstempel zu setzen sollte umsetzbar sein, jedoch eine UDI-PI in der für jedes Los/Charge das Herstelldatum neu anzugeben ist, ist technisch sehr schwierig. Reicht da nicht die UDI-DI und der Datumsstempel aus?
Lt. Anhang 1 Abschnitt 23.1.b ist es zulässig, dass einige oder alle Informationen zum Produkt, wenn es nicht praktikabel oder angemessen ist, auf der Verpackung jeder Einheit angebracht werden kann.
Da die UDI-PI ja den Zweck der Rückverfolgbarkeit des Produktes im Reklamationsfall dienen soll, kann ich aber dies zum Einen über das Label der Verpackungseinheit nachvollziehen oder beim einzelnen Produkt über die UDI-DI und den Datumsstempel im Kunststoffteil.
Können wir das als Hersteller so praktizieren?
Ich sehe Ihrer Antwort mit Interesse entgegen.
Mit freundlichen Grüßen
Karl-Heinz Sommerlade
Guten Tag Herr Sommerlade,
die Basis UDI-DI entspricht nicht der UDI-DI. Diese müssen separat generiert werden. Die Basis UDI-DI wird auch nicht auf dem Produkt und der Kennzeichnung angebracht.
Für das Aufbringen der UDIs haben Sie im Falle von Klasse I-Produkten in der Tat bis zum 26.05.2025 Zeit.
Für Einmal-Produkte der Klasse I ist generell keine direkt Markierung notwendig gemäß Anhang VI:
„4.3. Bei Produkten für den einmaligen Gebrauch der Klassen I und IIa, die einzeln verpackt und gekennzeichnet werden, ist zwar das Anbringen des UDI-Trägers auf der Verpackung nicht erforderlich, doch ist er auf einer höheren Verpackungsebene anzubringen, z. B. einem Karton, der mehrere einzeln verpackte Produkte enthält. Wenn allerdings der Gesundheitsdienstleister voraussichtlich keinen Zugang zur höheren Verpackungsebene hat, wie z. B. bei der häuslichen Pflege, wird die UDI auf der Verpackung des Einzelprodukts angebracht.“
Für die wiederverwendbare Klemme wäre allerdings eine direkte Kennzeichnung mit einer vollständigen UDI notwendig.Der Datumsstempel erfüllt diese Anforderung nicht. Generell ist die Codierung des Herstellungsdatums in der UDI-PI nicht zwingend vorgeschrieben. Die Codierung der LOT-Nummer wäre ausreichend. Nur falls technisch nicht machbar, könnte auf die direkte Kennzeichnung mit einer UDI verzichtet werden.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Johner, sehr geehrter Herr Salvatore,
ich habe einige Fragen zum Thema UDI, vielleicht können Sie mir weiterhelfen. Ich versuche, die Fragen klar und knapp zu stellen. Hoffentlich gelingt es mir, da die Zusammenhänge etwas komplex sind.
Frage 1)
Es geht um konfigurierbare Produkte. Wir haben ein Zubehör der Klasse I, das in Zahnärztlichem Bereich Verwendung findet. Es handelt sich um ein aktives Produkt, welches in zwei Varianten angeboten wird. Diese zwei Varianten haben die identische Zweckbestimmung, ein „sehr ähnliches“ Design und sind beide der Klasse I. Einige Komponente sind unterschiedlich: Die eine Variante hat eine Komponente aus Stahl (die andere Variante aus Kunststoff). Die erste Variante verfügt über eine automatische Spülung, die andere Variante nicht. Das Risikoprofil, der Einsatz und die sonstigen Funktionen sind gleich. Ich würde an beide Varianten die gleiche Basis UDI-DI als Produktfamilie, jedoch unterschiedliche UDI-DI als Produktvarianten zuteilen. Wie sehen Sie das?
Frage 2)
Die beiden o. g. Varianten können mit „Bausätzen“ an die verschiedenen Bedürfnisse des Anwenders angepasst werden. Diese Bausätze enthalten zum Beispiel ein Lichtschutz , einen längeren Stromanschluss, einen Schallschutz. Zweckbestimmung, Risikoprofil, wesentliche Funktion des Produktes sind identisch. Diese Modifikationen können nachträglich vom Anwender bestellt werden. Wir sehen alle diese Bausätze als Komponenten einer Konfiguration. Jede Produktvarianten würde demzufolge eine eigene UDI-DI als konfigurierbares Produkt erhalten. Würde Sie auch so vorgehen?
Frage 3)
Ich sehe einen Widerspruch in den MDR-Vorgaben in Bezug auf konfigurierbare Produkte.
Nach Anhang VI Teil C 3.6 gilt: „Jeder Komponente, die als Produkt gilt und für sich genommen verfügbar ist, wird eine gesondert UDI zugewiesen, es sei denn, die Komponenten sind Teil eines konfigurierbaren Produkts, das mit einer eigenen UDI gekennzeichnet ist.“
Nach Anhang VI Teil C 6.4.5 (konfigurierbare Produkte) gilt: „Jede Komponente, die als Produkt gilt und allein kommerziell verfügbar ist, bekommt eine gesonderte UDI zugewiesen“
Sehen Sie auch einen Widerspruch?
Frage 4)
In einem Dokument eines Verbandes ist Folgendes zu lesen: „Verpflichtend auf Grundlage der gesetzlichen Regelung ist bei Klasse-I-Produkten eigentlich nur die Angabe des Device Identifiers (UDI-DI), d.h. die eindeutige Codierung des Artikels.“
Abgesehen von den unterschiedlichen Fristen je nach Risikoklasse ist aus meiner Sicht die Angabe der PI auch für Produkte der Klasse I verpflichtend.
Sehen Sie es auch so?
Besten Dank und herzliche Grüße
Alberto Di Benedetto
Sehr geehrter Herr Di Benedetto,
vielen Dank für Ihre spannenden Fragen. Der Umfang übersteigt leider dem, was wir kostenlos anbieten können. Ich antworte Ihnen dennoch gerne kurz zu jeder Frage:
1. Wenn die Unterschiede (z.B. Material) der Varianten keine anderen Fragestellungen aufwerfen bezüglich Sicherheit oder Leistung, d.h. auch keine anderen/zusätzlichen Risiken einführt, sollte eine Gruppierung unter einer Basis UDI-DI möglich sein.
2. Ich würde das Produkt als konfigurierbares Produkt definieren. Dazu zählen auch die zusätzlichen Bausätze. Somit bekommt das konfigurierbare Produkt eine UDI-DI zugewiesen bzw. jeder Variante (aber nicht jede Konfiguration).
3. Das sehe ich auch als Widerspruch. Wenn die Bausätze bei Ihnen als eigenes Medizinprodukt oder med. Zubehör gelten, würden diesen eine eigene UDI zugewiesen werden. Dies dürfte vermutlich nicht der Fall sein.
4. Diese Sonderregelung kenne ich nur von der FDA für Klasse I-Produkte. Unter der MDR muss immer eine vollständige UDI vergeben werden.
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore,
vielen Dank für Ihre Antwort auf meine Fragen. Ich stimme Ihnen inhaltlich zu. Sorry für die viele Fragen und der damit verbundene Aufwand.
Herzliche Grüße
Alberto Di Benedetto
Sehr geehrter Herr Johner,
müssen Hersteller, deren MDD Zertifikat noch aufrecht ist, ebenfalls bereits das UDI-System einführen? Oder haben diese bis zum Auslaufen der Zertifikate noch Zeit?
LG Astrid Unterkreuter
Sehr geehrte Frau Unterkreuter,
die UDI-Anforderungen gelten ausschließlich für Produkte für die gemäß MDR Konformität erklärt wird. Für MDD-Produkte mit gültigem Zertifikat muss kein UDI-System eingeführt werden.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Johner, sehr geehrter Herr Salvatore,
Ich habe eine Frage bezüglich der in Artikel 27 Absatz (7) gemachten Anforderung: „Der Hersteller führt eine auf dem neuesten Stand zu haltende Liste aller von ihm vergebenen UDI als Teil der technischen Dokumentation gemäß Anhang II.“
Genauer geht es um die „Liste aller von ihm vergebenen UDI“: Bezieht sich UDI in diesem Fall auf die UDI, so wie sie in Anhang VI definiert ist, sprich die Kombination aus UDI-DI und UDI-PI? Müssen wir als Hersteller also eine explizite Liste über all unsere freigegebenen Chargen führen?
Oder wäre es eventuell ausreichend eine Liste aller vergebenen UDI-DIs zu führen? Über diese, und die Chargenverfolgung wäre es ja so auch jeder Zeit machbar, die UDI-PI der entsprechenden UDIs zu bestimmen, da sich diese bei unseren Produkten aus der Chargen-Lot und dem Ablaufdatum zusammensetzt.
Freundliche Grüße
Fabian Schulze Bisping
Sehr geehrter Herr Schulze Bisping,
da in dem von Ihnen zitierten Abschnitt von UDI die Rede ist, ist davon auszugehen, dass der Gesetzgeber vollständige UDIs inkl. -DI und -PI gemeint hat.
Im Idealfall können Sie sich solch eine Liste z.B. aus einem ERP-System exportieren lassen.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Johner, sehr geehrter Herr Salvatore,
ich habe eine Frage zum Thema Basic UDI-DI. Gemäß des Dokumentes von MedTech Europe (Guidance for assigning Basic UDI-DI, v1.1 2 June 2020), auf Seite 11 stand, dass man eine gleiche TD (Teschnische Dokumentation) für mehrere Basic UDI-DI erstellen kann, stimmt wirklich das? Ich dachte, eine TD für die Produkte, die nur eine gleiche Basic UDI-DI haben? Oder? Noch kurze Frage dazu, warum wird es teuere bei benannten Stelle, wenn man alle Produkte unter einer gleichen Basic UDI-DI erstellen als unter mehreren Basic UDI-DIs?
Vielen Dank im Voraus!
Mit freundlichen Grüßen
Rony
Guten Tag,
es ist möglich, in einer TD mehrere Produkte mit unterschiedlichen Basic UDI-DIs zu dokumentieren. Dies ist z.B. der Fall, wenn ein Hauptprodukt und Zubehör in einer TD behandelt werden und das Zubehör einer eigenen Basis UDI-DI zugewiesen wird.
Die Kosten der benannten Stellen hängen eher mit der Anzahl der zu prüfenden TDs zusammen.
Freundliche Grüße
Luca Salvatore
Vielen Dank Herr Salvatore für die Antwort!
Aber heisst das denn, dass man nur in diesem Fall (Hauptprodukt und Zubehör) eine TD für mehrere Produkte mit unterschiedlichen Basic UDI-DIs dokumentieren darf, ODER auch in den Fällen, wobei man unterschiedlichen Hauptprodukte (IVD-Kits) mit unterschiedlichen Basic UDI-DI hat?
Vielen Dank im Voraus!
MfG
Rony
Guten Tag,
Im Idealfall haben Sie ein Produkt/Kit mit einer Basis UDI-DI in einer TD. Es gibt aber kein Verbot, mehrere Kits mit unterschiedlichen Basis UDI-DIs in einer TD zu dokumentieren. Der entgegengesetzte Fall, d.h. mehrere Produkte mit derselben Basis UDI-DI über mehrere TDs verteilt, ist allerdings nicht möglich.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
zum Production Identifier hätte ich noch eine Frage. Sie beschreiben den PI als:
„UDI-PI: Der Produktions-Identifier kennzeichnet jede einzelne Instanz eines Produkts bzw. eines Batches. Jeder Defibrillator hat somit eine eigene UDI-PI.“
Hieraus ergibt sich, dass jedes einzelne Produkt einer Charge einen eigenen PI bekommt. Das hieße dann, wenn z.B. in einer Charge 10.000 Packungen Heftpflaster hergestellt werden, jede Packung einen eigenen PI bekommt und man am Ende 10.000 verschiedene UDIs hat. Denn die einzelne Instanz eines Batches ist meinem Verständnis nach die einzelne Packung Heftpflaster.
Auch bestätigte Herr Professor Johner in einem Kommentar darüber, dass die UDI eine Art „Reisepass“ für das einzelne Produkt ist und eine eindeutige Identifizierung dessen erlaubt. Nach dieser Logik bräuchte wieder jede einzelne Packung Pflaster einen eigenen PI.
Die MDR enthält in Anhang VI Teil C zur UDI den Satz: <>
Zugegebenermaßen verstehe ich diesen Satz nicht zu 100%,, aber es klingt so, als dürfte im o.g. Beispiel eine UDI für die gesamte Charge verwendet werden (alle einzelnen Packungen dieser Charge haben dieselbe UDI).
Wie würden Sie das beschriebene Szenario einschätzen?
Ein UDI-PI für die gesamte Charge oder ein UDI-PI pro einzelne Packung der Charge?
Vielen Dank schonmal und viele Grüße
Michael Becer
Sehr geehrter Herr Becer,
mit dem Zitat ist gemeint, dass z.B. eine Seriennummer nicht zwingend in der UDI-PI codiert werden muss, wenn ansonsten keine Seriennummern z.B. für einzelnen Packungen vergeben werden. In Ihrem Fall würde also eine UDI-PI für eine Charge ausreichen, d.h. Sie würden die Chargennummer in der UDI-PI codieren und keine zusätzliche Seriennummer für einzelne Produkte oder Packungen vergeben.
Herzliche Grüße
Luca Salvatore
Sorry, hier ging irgendwie das Zitat in den Anführungszeichen verloren. Die MDR schreibt:
„Der Begriff „einmalig“ bedeutet nicht, dass einzelne Produktionseinheiten serialisiert werden“
Viele Grüße
Michael Becer
Vielen Dank, das war sehr hilfreich!
Viele Grüße
Michael Becer
Guten Tag,
wir sind Private Label Hersteller und lassen unser Produkt durch einen chinesischen Hersteller produzieren – dieser steht auch als Produzent auf der Verpackung. Wir haben allerdings die EAN zur Verfügung gestellt. In der MDR steht an einigen stellen, dass der Hersteller (also nicht wir!!) für die Bereitstellung und viele weitere Themen im Zusammenhang mit der Basis-UDI verantwortlich ist. Dadurch dass wir die EAN bereit gestellt haben gestaltet sich das nun aber schwierig / unmöglich. U.a. stellt sich gerade die Frage für die Erstellung der Konformitätserklärung, kann dort die von uns (nicht Hersteller!!) generierte Basis-UDI angegeben werden? Wir interpretieren die MDR so, dass dies nicht möglich ist – hierdurch befinden wir uns aber in einem Teufelskreislauf.
Gibt es hierfür eine Lösung?
Viele Grüße,
Bas
Guten Tag,
gemäß den GS1 Healthcare Allocation Rules (https://www.gs1.org/docs/gsmp/healthcare/GS1_Healthcare_GTIN_Allocation_Rules.pdf), Kapitel 2.6, sind Sie als PLM für die GTIN-Vergabe verantwortlich:
„Products wherein an agreement exists between the Original Manufacturer and the party identified on the label as the Brand Owner. The Brand Owner takes responsibility for GTIN assignment and therefore retains the alignment of brand to GTIN allocation.“
Dies ist in der Tat etwas widersprüchlich zur MDR, da gemäß MDR der Hersteller für die UDI-Vergabe und -Anbringung verantwortlich ist. Sie sollten vertraglich regeln, dass Ihre GTINs/EANs für Ihren Brand genutzt werden sollen.
Bei der Basis UDI-DI sehe ich die Verantwortung für die Vergabe allerdings eindeutig beim Hersteller.
Freundliche Grüße
Luca Salvatore
Danke für die Antwort Luca.
Es kann doch nicht sein, dass jetzt jede Marke (es wird ja nicht nur uns betreffen) seine EAN umändert, damit dies mit diesen neuen UDI Bestimmungen konform ist? Das ist ja meine Wissens auch gar nicht der Grundgedanke der UDI (Nachvollziehbarkeit, etc).
Ich weiss, dass es in der MDR so strikt vorgeschrieben ist, aber was ist denn eure Meinung bei Johner dazu? Gibt es hier keine praktikable Lösung für? Gibt es keine Präsedenzfälle?
Kann man eventuell die Basis-UDI-DI vom Hersteller generieren lassen und dann als Markeneigner selbst die UDI-DI (= EANs) bereit stellen und mit der Basis-UDI-DI des Herstellers verknüpfen? Wo eigentlich verknüpfen, in der Eudamed, oder? Falls man dann als Markeneigner den Hersteller wechselt (dass kann bei Private Label Brands ja häufiger vorkommen), dann wechselt man einfach die Verknüpfung der Basis-UDI-DI zur UDI-DI. Wäre das praktikabel?
Guten Tag,
gemäß der GS1-Regeln müsste ja die EAN/GTIN des Brand Owners verwendet werden, was in Ihrem Fall ja gewünscht ist. Ich würde versuchen so vorzugehen, wie Sie es beschreiben, d.h. Ihre (PLM) EANs nutzen und der Hersteller muss diese mit seiner Basis UDI-DI verknüpfen in der Dokumentation und in EUDAMED. Das würde ich als praktikabels Vorgehen ansehen. Ich würde aber empfehlen, dies mit GS1 abzusprechen.
Herzliche Grüße
Luca Salvatore
Guten Tag,
gibt es so ein Templet für PA (Prozessanweisung) zu dem Thema UDI und EUDAMED?
Vielen Dank!
MfG
Rony
Lieber Rony,
wollen Sie dazu vielleicht einmal unsere Kollegen kontaktieren? Das Erstgespräch ist natürlich kostenlos. So wäre es einfacher herauszufinden, ob und wie genau Ihnen das Johner Institut bei der Frage helfen kann. Einen Termin dazu können Sie hier buchen: https://meetings.hubspot.com/florian-wettlaufer/kostenloses-erstgesprach-johner-institut
Herzliche Grüße
Anja Segschneider | Redaktion
Sehr geehrter Herr Professor Johner,
ich habe eine Frage bezüglich Art. 27, Abs. 7:
Der Hersteller führt eine auf dem neuesten Stand zu haltende Liste aller von ihm vergebenen UDI als Teil der technischen Dokumentation gemäß Anhang II.
Ist hier mit UDI die gesamte UDI -bestehend aus UDI-DI UND UDI-PI – gemeint?
Oder (hoffentlich)nur die UDI-DI?
Wenn eine Liste der kompletten UDI geführt werden muss, dann müsste diese immer wieder aktualisiert werden-aufgrund der beweglichen Daten der UDI-PI,richtig?
Oder bin ich hier auf dem falschen Weg?
Vielen Dank für Ihre Antwort im Voraus!
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
Ihre Hoffnung ist gerechtfertigt! Sie sind auf dem richtigen Weg: Es reicht für die TD die Liste der UDI-DIs.
Man will einfach wissen, für welche UDI-DIs eine TD gedacht ist. Die TD darf ja auf der Ebene der Basis-UDI-DI erstellt werden und gilt dann für mehrere dazu gehörige UDI-DIs.
Allerdings benötigen Sie einen Prozess, der die Vergabe der UDI-PIs regelt und nachvollziehen lässt, welches Gerät welche UDI-PI bekommen hat. Das sind aber Aufzeichnung im Sinne der ISO 13485 und keine Vorgabedokumente.
Viele Grüße, christian Johner
Sehr geehrte Damen und Herren,
dieses Kommentar steht im Wiederspruch zu den gemachten Aussagen vom:
Luca Salvatore (30. Juli 2021 um 13:09 Uhr)
„da in dem von Ihnen zitierten Abschnitt von UDI die Rede ist, ist davon auszugehen, dass der Gesetzgeber vollständige UDIs inkl. -DI und -PI gemeint hat.“
und
Benedikt Tölle (9. Mai 2022 um 15:45 Uhr)
„Wir interpretieren den Abschnitt wie Sie: Da in dem von Ihnen zitierten Abschnitt (Artikel 27 (7)) von UDI die Rede ist, ist davon auszugehen, dass der Gesetzgeber vollständige UDIs inkl. -DI und -PI gemeint hat.“
Was ist nun richtig?
Mit freundlichen Grüßen
Steffen K
Sehr geehrter Steffen K,
das MDCG-Guidance 2021-19 sagt dazu: „As part of the technical documentation, the manufacturer shall keep up-to-date a list of all
UDIs (UDI-DIs and UDI-PIs) that it has assigned (Article 27(7) MDR, Article 24(7) IVDR).“
Demnach wären also die vollständig vergebenen UDIs der Technischen Dokumentation hinzuzufügen. In der Praxis ist es üblicherweise ausreichend, nur die UDI-DIs (neben der Basis UDI-DI) direkt in der Technischen Dokumentation zu führen und zumindest zu referenzieren, wo die vollständigen UDIs inkl. UDI-PIs bei Ihnen abliegen (z.B. in einem ERP-System). Das sollte für die Benannte Stelle im Rahmen der Begutachtung der Technischen Dokumentation ausreichend sein.
Herzliche Grüße
Luca Salvatore
Hallo Herr Johner,
vielen Dank für die Bestätigung meiner Hoffnung 🙂 Ich bin sehr erleichtert.
Viele Grüße
Christine Graß
Guten Tag,
zum Punkt 7 (Dokumente, die die UDI enthalten müssen), wurde in der Tabelle gemeint, alle UDI-Elemente (Basic UDI-DI, UDI-DI und UDI-PI)?
Vielen Dank im Voraus!
MfG
Rony
Guten Tag,
das können Sie aus der Tabelle leider nicht rauslesen. In den meisten der genannten Fälle ist es ausschließlich die Basis UDI-DI. In manchen Fällen aber auch die UDIs (z.B. bei den FSCAs).
Freundliche Grüße
Luca Salvatore
Was bedeutet Sehr geehrter Herr Professor Johner,
ich habe mal wieder eine Frage zur UDI….
Es geht um die Angabe der UDI in der Kennzeichnung.
Der UDI-Träger muss bei der Kennzeichnung angegeben werden: Anhang I,Kap. III, 23.2 h.
Der UDI-Träger wird auf der Kennzeichnung oder auf dem Produkt selbst angegeben : Anhang VI,Teil C, 4.1
– Steht dieser Satz nicht im Widerspruch zu 23.1 b? Hier steht doch, dass die Angaben zur Kennzeichnung auf dem Produkt selbst angebracht werden sollen.
Wenn ich nun das Produkt mit den geforderten Angaben unauslöschlich kennzeichne, indem ich es mit einem eingenähten Textiletikett versehe, muss dann gemäß Anhang VI, Kap. II, 4.7 die AIDC- UND HRI Form des UDI auf das Textillabel gebracht werden?
Wenn dieses aus Platzgründen nicht möglich ist, steht in 4.7, dass die HRI-Form bevorzugt werden soll, wenn das Produkt außerhalb von Gesundheitseinrichtungen verwendet werden soll.
– Zählen zu diesen Produkten all die Produkte, die in der häuslichen Umgebung von Patienten verwendet werden?
– Was bedeutet es dann für die Kennzeichnung? Nur HRI-Format, nur AIDC-Format oder beide Formate des UDI auf das Textillabel?
Ich hoffe, dass ich Ihnen meinen Gedankengang verdeutlichen konnte.
Vielen Dank im Voraus für Ihre Hilfe!!
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
die Angabe in Anhang VI,Teil C, 4.1 ist in der Tat etwas verwirrend. Die Kennzeichnung sollte natürlich wenn möglich immer auf dem Produkt angegeben werden.
Zu Ihrer zweiten Frage: Ja, im Regelfall sind beide Formate AIDC und HRI anzugeben. Bei Platzmangel würde bei einem Einsatz außerhalb von Gesundheitseinrichtungen das HRI-Format zu nutzen sein. Dazu zählen typischerweise „home use“ Produkte.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
vielen Dank für Ihre zeitnahe Antwort.
Dazu habe ich noch eine Nachfrage:
Was sind in diesem Fall „home use“ Produkte?
Mit Produkten, die in der häuslichen Umgebung verwendet werden, meinte ich, z.B. Orthesen und Bandagen. Diese werden ja vom Endanwender (=Patienten) überwiegend außerhalb von Gesundheitseinrichtungen getragen.
Demnach müsste für diese Produkte vorzugsweise das HRI-Format genutzt werden?
Und wieder vielen Dank im Voraus !
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
Ihre Produkte fallen sicherlich unter diesen Bereich. Somit wäre das HRI-Format zu bevorzugen.
Freundliche Grüße
Luca Salvatore
Sehr geehrte Damen und Herrn,
bei welchen Änderungen ist eine neue GTIN erforderlich? Wir wollen zusätzlich zum MHD noch das Produktionsdatum in den UDI aufnehmen und am Etikett aufdrucken. Muss deshalb eine neue GTIN vergeben werden? Und muss das Produktionsdatum in der GUDID auch gepflegt werden?
Danke im Voraus für die Hilfe!
Schöne Grüße,
Karl Heinz
Guten Tag,
wir werden Ihnen so bald wie möglich antworten!
Herzliche Grüße
Anja Segschneider | Redaktion
Guten Tag,
bitte entschuldigen Sie vielmals die ungewöhnlich lange Wartezeit!
Meine Kollegin Maria Laitenberger war so freundlich, mir die folgende Antwort zu geben:
Normalerweise muss nur ein neuer DI (*D*evice *I*dentifier oder GTIN) vergeben werden, wenn es ein neues Modell oder eine neue Versionsnummer gibt. Aber die einfachste Variante, um herauszufinden, ob ein neuer DI/GTIN nötig ist, ist ein Blick auf die GUDID Data Elements Reference Table der FDA zu werfen https://www.fda.gov/media/120974/download, ode https://www.fda.gov/medical-devices/global-unique-device-identification-database-gudid/gudid-guidance
Ich hoffe, das hat Ihre Frage beantwortet!
Herzliche Grüße
Anja Segschneider | Redaktion
Guten Abend,
bei der Kennzeichnung des UDI als solchen, sieht man öfters die Beschriftung mit HIBC und zusätzlich UDI. Ist es erforderlich, dass die Vergabestelle auch angegeben wird? Ist das eventuell eine Anforderung der US-FDA?
Oder ist das UDI-Symbol alleine ausreichend? Müsste man z.B. bei der Vergabestelle GS1 den UDI mit „GS1 UDI“ kennzeichnen, wenn mehrere (Bar)codes verwendet werden?
Beste Grüße,
Martina
Sehr geehrte Frau M.,
sowohl in der EU als auch in de USA sind mir keine Anforderungen bekannt, die eine Angabe der Zuteilungsstellen auf der Kennzeichnung erfordert.
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrte Damen und Herren,
wir sind ein Hersteller von IVD-Kits für Diagnostik und diskutieren intensiv den Umgang mit genrischen Komponenten im UDI-System.
In verschiedenen Produkten zum Nachweis von Erregern befinden sich als Komponente identische Pufferlösungen (z.B. ein Puffer A der sowohl in Produkt 1 für Erreger X, als auch in Produkt 2 für Erreger Y als Komponete enthalten ist). Bekommen dieser Puffer A dann eine eigene UDI-DI (immer die gleiche) und werden dann der Basis-UDI des jeweiligen Produkt zugeordnet? Das würde dann bedeuten, dass diese UDI-DI für den Puffer A dann pro Produkt einer anderen Basis-UDI zugeordnet würde. Das ist jedoch, soweit ich das System verstanden habe, nicht möglich. Oder würden dann in Technischer Dokumentation und Konformitätserklärung beide basic UDIs (die des Puffers und die des Kits) auftauchen können?
Oder bekommen diese diese generische Pufferlösung gar keine eigene UDI-DI? Oder muss der Puffer A pro Produkt eine individuelle UDI-DI erhalten, die dann wiederum der der jeweiligen basis-UDI des Kits zugeordent wird (wobei die Zweckbestimmung des Puffers im Kit sicher eine andere ist, als die Zweckbestimmung des Gesamtkits).
Wie verhält es sich dann, wenn der Puffer A zusätzlich noch als IVD (als Klasse A Produkt nach IVDR) in Verkehr gebracht werden möchte?
Vielen Dank für Ihre Einschätzung.
Harald Guldan
Lieber Herr Guldan, vielen Dank für Ihre spannende Frage zu Komponenten eines IVD-Reagenzien-Kits. Sie beschreiben zwei unterschiedliche Fälle:
Fall 1: Der Puffer ist eine Komponente eines bzw. mehrerer Reagenzien-Kits. Dann ist das jeweilige Kit das Produkt, das eine eigene UDI-DI erhält. Der Puffer ist dann eine Komponente und muss keine UDI tragen, sondern kann über Ihre internen Rückverfolgbarkeits-Merkmale zugeordnet werden. Das jeweilige Kit hätte dann auch eine eigene Basis-UDI. Ein weiteres Kit könnten Sie nur derselben Basis-UDI zuordnen, wenn es die gleiche Zweckbestimmung hat, also zum Beispiel das gleiche Kit nur in größerer Ausführung für mehr Durchsatz.
Fall 2: Der Puffer wird als Produkt in Verkehr gebracht. Dann erhält der Puffer eine eigene UDI-DI. Wenn er nur in Verbindung mit Ihren Kits verwendet werden soll, dann ist er ggf. Zubehör mit eigener Zweckbestimmung. Gerade bei der Zuordnung zu verschiedenen Kits mit jeweils unterschiedlicher Zweckbestimmung braucht der Puffer somit eine eigene Basis-UDI-DI.
Ich hoffe ich konnte Ihre Frage damit beantworten. Wenn noch etwas offen geblieben ist, kontaktieren Sie uns gerne.
Mit besten Grüßen,
Sebastian Grömminger
Guten Tag,
Wir sind Hersteller von digitalen Thermometern. Leider haben wir weiterhin offene Fragen zur UDI, durch welche wir nicht weiterkommen. Deswegen wäre ich Ihnen sehr dankbar, wenn Sie uns weiterhelfen könnten.
Um es etwas einfach zu machen, beziehen sich alle meine Fragen auf einen digitalen Thermometer (als Beispiel Modellnummer: DT-1000).
1. Kann die Modellnummer (DT-1000) in der UDI-DI integriert werden?
2. Genügt als UDI-PI die Seriennummer oder müssen weitere Angaben wie zum Beispiel die Lotnummer und Herstelldatum in der UDI-PI beinhaltet sein?
3. Jedes Thermometer befindet sich in einer einzelnen Verpackung (GB) mit einer Bedienungsaleitung (IM). Die Thermometer werden in Kartonschachteln (MC) an die Apotheken geliefert. In jeder Kartonschachtel befinden sich je 100 Einzelverpackungen (GB) mit digitalen Thermometern. Die digitalen Thermometer werden in der Apotheke nur einzeln verkauft.
Nun zu meiner Frage: Müssen das digitale Thermometer, die einzelne Verpackung (GB) und die Kartonschachtel (MC) je eine eigene UDI haben? Falls nicht, wo braucht es keine UDI?
4. Nehmen wir an, auf der Bedienungsanleitung kommt ein neuer wichtiger Warnhinweis dazu und es muss eine neue UDI-DI vergeben werden.
Muss auf allen Verpackungsebenen eine neue UDI-DI vergeben werden oder nur auf einer? Falls nur auf einer, auf welcher?
Vielen Dank im Voraus!
Freundliche Grüße,
Mario Ammann
Lieber Herr Ammann,
da das eine ganze Menge an konkreten Punkten sind, die sich sehr stark auf Ihr individuelles Produkt beziehen, schlage ich die Kontaktaufnahme zu unseren Beratern vor ([email protected]). So können wir Ihnen besser helfen, als über die Kommentarfunktion eines öffentlichen Beitrags.
Herzliche Grüße
Christian Rosenzweig
Guten Tag,
ich habe eine Frage zur UDI-Kennzeichnung von konfigurierbaren Produkten. Es heißt, die Konfiguration ist eine vom Hersteller festgelegte Kombination von Baueinheiten, die als Produkt zusammenwirken, um eine Zweckbestimmung zu erfüllen. Nach meinem Verständnis erhält jedes, ggf. individuell, konfiguriertes Produkt eine UDI-PI, die auch diese Konfiguration widerspiegelt. Was muss beachtet werden, wenn sich die Konfiguration eines Produktes ändert, z.B. nach einigen Jahren der Kunde ein Bauteil, welches nicht einzeln CE-gekennzeichnet ist, ergänzen möchte? Kann/muss dann die UDI-PI angepasst werden? Ist dies überhaupt möglich?
Vielen Dank im Voraus!
Mit freundlichen Grüßen
Carina Gasch
Liebe Frau Gasch,
danke Ihnen für die spannende Frage!
Die MDR macht für die Änderungen bei konfigurierbaren Produkten – neben den allgemeine Anforderungen – keine speziell Vorgaben.
Jedoch finden sich in den Regelungen der Zuteilungsstellen Vorgaben dazu. Bei der GS1 finden Sie bspw. folgende Ausführungen in Kapitel 2.2.2 der GTIN Allocation Rules :
– A change in form, fit, or function to a mandatory component affecting intended use, requires a change to the GTIN and/or GTIN plus serial number of the entire configurable medical device. Mandatory components are those that are required to deliver the functionality of the device. Changes to or the removal of, a mandatory component, impacting device form, fit, or function, require a GTIN change.
– Changes to optional components impacting device form, fit, or function, require a GTIN change. Similarly, removal of optional components from the set of available components requires a GTIN change.
Es kommt also darauf an, ob die Änderungen an ihrem konfigurierbaren Produkt „form, fit or function“ beeinflussen. Falls ja ist sogar eine Änderung der UDI-DI/GTIN notwendig bei der GS1.
Mit meinen allerbesten Grüßen
Benedikt Tölle
Sehr geehrter Herr Tölle,
vielen Dank für Ihre Antwort und den Hinweis auf die GS1 Ausführungen!
Ich hätte noch eine grundsätzliche Frage zur UDI-PI von konfigurierbaren Produkten:
Wenn einzelne Komponenten eines konfigurierbaren Produktes, alle nicht einzeln CE-gekennzeichnet, bereits Identifikationsnummern (Serien-, LOT-Nr.) aufweisen. Reicht es dann aus, der UDI-PI die Seriennummer der „Hauptkomponente“ zuzuweisen (gem. MDR Anhang VI Abschnitt 6.4.4.)? Der Hersteller würde dann in seiner Dokumentation festhalten, welche weitere Komponenten inklusive Identifikations-Nummern zu jeder einzelnen Konfiguration gehören, um die Rückverfolgbarkeit zu gewährleisten. Oder muss ggf. noch woanders die Zusammensetzung der einzelnen spezifischen Konfiguration dokumentiert werden?
Mit freundlichen Grüßen
Carina Gasch
Liebe Frau Gasch,
für die Erstellung einer UDI-PI eines konfigurierbaren Produktes macht die MDR keine speziellen Vorgaben, außer, dass Sie die UDI-PI für jede ausgelieferte konkrete Konfiguration vergeben. Die Seriennummer, der „Hauptkomponente“ (also die Komponenten, bei dem die geringste Wahrscheinlichkeit besteht, dass es während der Lebensdauer des Systems ausgetauscht wird) ist eine Möglichkeit. Eine andere wäre das Datum des „Zusammenbaus“/Herstelldatum zu wählen.
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrter Herr Tölle,
ich hätte da eine Frage undzwar ob bei Änderung der Gesetzgebung, also bei Umstellung von MDD auf MDR eine neue UDI-DI-Vergabe erforderlich ist? Unsere jetzigen Produkte die nach MDD laufen haben bereits eine UDI. Müssen diese Produkte beim Umstieg auf MDR eine neue UDI bekommen? Die Gesetzgebung ist meines Wissens ein Trigger-Feld für eine neue „GTIN“ Vergabe. Dann müssten die Produkte unter MDD eine andere GTIN haben als dieselben Produkte unter MDR, jedoch sind wir uns in diesem Fall nicht sicher. Wissen Sie etwas hierzu? Ich bedanke mich herzlichst.
Mit freundlichen Grüßen,
Alen Sen
Sehr geehrter Herr Sen,
danke Ihnen für ihre spannende Frage!
Sie sollten für das MDR Device eine neue UDI-DI vergeben. Die UDI soll ein Produkt eindeutig identifizieren, was die MDCG auch im Dokument 2018-1 Rev. 4 noch einmal verdeutlicht hat:
A new UDI—DI shall be required whenever there is a change that could lead to misidentification of the device and/or ambiguity in its traceability.Ihr Legacy Device wird sich zu Ihrem MDR-Device in jedem Fall unterscheiden und bspw. versch. Technische Dokumentationen haben, weswegen Sie dem MDR-Device zur eindeutigen Identifizierung eine eigene UDI-DI geben sollten.
Damit Sie mich nicht falsch verstehen: Das heißt aber nicht, dass Sie ihre bereits in Verkehr gebrachten Legacy Devices, nach der Inverkehrbringung noch mal „umlabeln“ müssen. Es geht um die Produkte, die Sie unter der MDR dann „erstmalig“ in Verkehr bringen.
Mit allerbesten Grüßen
Benedikt Tölle
Guten Tag,
ich habe eine Frage zur UDI, in dem Artikel 27 heißt es:
„Der Hersteller führt eine auf dem neuesten Stand zu haltende Liste aller von ihm vergebenen UDI als Teil der technischen Dokumentation gemäß Anhang II. “
Bedeutet für mich, dass die gesamte UDI gemeint ist, also auch jede UDI-PI, was bei uns die SN ist.
Kann es sein, dass es tatsächlich so von der Kommission gefordert ist, jede einzelne vergebene SN in der technische Doku aufzuführen?
Ich sehe leider den Sinn nicht, denn die UDI-PI, ist ja im System enthalten, nur nicht in der technischen Doku.
MfG
Dominik Theunert
Lieber Herr Theunert,
danke Ihnen für Ihre Nachfrage!
Wir interpretieren den Abschnitt wie Sie: Da in dem von Ihnen zitierten Abschnitt (Artikel 27 (7)) von UDI die Rede ist, ist davon auszugehen, dass der Gesetzgeber vollständige UDIs inkl. -DI und -PI gemeint hat.
Im Idealfall können Sie sich solch eine Liste z.B. aus einem ERP-System exportieren lassen.
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrter Herr Tölle,
ich stolpere über die Definition des UDI-Trägers:
Teil C: Das UDI-System
1. Begriffsbestimmungen
UDI-Träger
Der UDI-Träger ist das Mittel, mit dem die UDI durch die AIDC und gegebenenfalls in ihrer HRI wiedergegeben wird.
UDI-Träger sind u. a. lineare 1D-Strichcodes, 2D-Matrix-Strichcodes und RFID.
4. UDI-Träger
4.1. Der UDI-Träger (AIDC- und HRI-Darstellung der UDI) wird auf der Kennzeichnung oder auf dem Produkt selbst sowie auf allen höheren Ebenen der Produktverpackung angebracht. Versandcontainer gelten nicht als höhere Verpackungsebene.
Ist als UDI-Träger nur die AIDC-Form zu verstehen, oder auch die HRI-Form?
Ich verstehe 4.1 so, dass AIDC- und HRI-Darstellung der UDI jeweils UDI-Träger sind. Ist das so korrekt?
Vielen Dank für Ihre Hife!
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
sie haben bereits die korrekten Stellen in der MDR gefunden.
Wenn Sie also die AIDC und HRI als 1D-Barcode (AIDC) mit darunter stehenden Nummer (HRI) als „Label/Sticker“ auf ihr Produkt kleben, ist dieses „Label“ ihr Mittel mit der Sie die AIDC und HRI wiedergeben und somit ihr UDI-Träger.
Mit allerbesten Grüßen
Benedikt Tölle
Liebes Team vom Johner-Institut,
ich habe eine Frage zur Vergabe der Basis UDI-DI. Sie schreiben in Ihrem Artikel, dass gemäß MDCG Produkte einer gemeinsamen Basis UDI zugeordnet werden, wenn
– sie über die gleiche Zweckbestimmung verfügen,
– wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und
– wenn sich auch die Herstellung, also die Produktion im Wesentlichen nicht unterscheiden.
Wie eng ist „im Wesentlichen gleich“ auszulegen?
Wenn wir beispielsweise einen Antikörper als IVD in Folgenden Varianten anbieten:
– Konzentrat
– gebrauchsfertig in Puffer 1
– gebrauchsfertig in Puffer 2
Dann gäbe es zwischen Konzentrat und gebrauchsfertig natürlich den Unterschied in der Herstellung, dass der Verdünnungsschritt entfällt. Und zwischen den beiden gebrauchsfertigen Varianten gibt es leichte Unterschiede in der Pufferzusammensetzung. All das wirkt sich jedoch nicht wesentlich auf die Leistung des Produkts aus, sondern ist lediglich eine Frage der Vorliebe des Anwenders.
Würde ich für die drei Varianten in dem Beispiel nun eine, zwei (Konzentrat vs. gebrauchsfertig) oder drei Basis UDI(s) vergeben?
Herzlichen Dank für Ihre tolle Arbeit! Viele Grüße,
Stefanie Schreyer
Liebe Frau Schreyer,
zunächst einmal Danke für Ihre Wertschätzung!
Leider gibt es keine mir bekannten Vorgaben, wie eng „im wesentlichen“ auszulegen ist. Das ist die Veratnwortung von Ihnen als Hersteller, eine schlüssige (verschriftliche) Begründung zu finden, warum Produktvarianten „im wesentlichen“ gleich sind. Anhaltspunkte, die eher für einen wesentlichen Unterschied sprechen:
– Wesentliche Leistungsmerkmale (IEC 60601-1) des Produkts sind betroffen
– Sicherheitsbezogene Merkmale (ISO 14971) sind beeinflusst
– Wirkweise oder physikalisches Prinzip des Produktes beeinflusst
– Produktrisiko ändert sich bspw. weil durch Unterschiede neue/höhere Risiken entstehen
Sehen sie dies Liste bitte als Hilfe und nicht voll ausschöpfende Liste an.
Ihren Fall „aus der Ferne“ zu bewerten ist mir leider nicht möglich, aber die o.g. Anhaltspunkte sollten Ihnen bei der Argumentation helfen.
Mit allerbesten Grüßen
Benedikt Tölle
Guten Tag,
wir sind Hersteller von Klasse II Medizinprodukten und stofflichen Medizinprodukten. Regelmäßig werden unterschiedliche Produkte als Gebinde dem Handel zur Verfügung gestellt. Die Herstellung dieser Gebinde ist ausgelagert und bekommen eine eigene Bestellnummer und Lot-Code.
Frage: Müssen wir diese Gebinde als „Behandlungseinheiten“ ansehen und dementsprechend eigenständige UDIs vergeben? Wohl gemerkt bilden die enthaltenen Produkte keine Einheit im medizinischen Sinne, es handelt sich eher um ein Produktsortiment aus eigenständigen MDs.
Vielen Dank
Freundliche Grüße aus München.
Liebe Frau Bauer,
als erstes müssen Sie sich die Frage beantworten, ob es sich bei Ihren Zusammenstellungen, um eine Behandlungseinheit im Sinne der MDR handelt. Hier hilft ihnen unser Fachartikel zum Thema Systeme und Behandlungseinheiten
weiter.
Falls Ihre Zusammenstellungen dann Behandlungseinheiten sind, beachten Sie bei der UDI die Anforderungen an Systeme und Behandlungseinheiten aus Anhang VI Abschnitt 6.3. der MDR. Dieser Abschnitt verpflichtet die Hersteller auch für Systeme und Behandlungseinheiten eine eigene vollständige UDI zu vergeben. Zusätzlich müssen Sie entweder auf den einzelnen enthaltenen Produkten oder auf den Verpackungen dieser Produkte UDIs aufbringen.
Mit allerbesten Grüßen
Benedikt Tölle
Hallo Johner-Team,
wir möchten gerne das Konzept eines „configurable device“ für unser Medizinprodukt anwenden. Dieses besteht aus mehreren Modulen, die nur zusammen den intended use erfüllen. Wir würden diese Module als Komponenten bezeichnen und nur für jede „group of configurations“ eine UDI vergeben.
Wir möchten jedoch z.B. für den Servicefall jedes dieser Module einzeln in Verkehr bringen, ohne dass diese Module eine eigene UDI tragen. Medical Devices inkl. Zubehör müssten dies ja in diesem Falle.
Jetzt sind wir uns unsicher, was eigentlich eine Komponente sein kann? Ist dies immer entweder ein Medizinprodukt, ein Zubehör (das zur Erfüllung des intended use eines Medizinproduktes unbedingt notwendig ist) oder ein Spare Part oder gibt es noch eine weitere Möglichkeit?
Vielen Dank für Ihren Support.
Viele Grüße
Anna
Liebe Anna,
was Komponenten Ihres Medizinprodukts sind, bestimmten Sie in Ihrer technischen Dokumentation. Bei einem konfigurierbaren Produkt können Komponenten auch für sich genommen Produkte sein, müssen es aber nicht. Da Zubehör zu einem Medizinprodukt wie ein Medizinprodukt selbst zu behandeln ist, könnte auch ein Zubehör eine Komponenten eines konfigurierbaren Produktes sein.
Wenn eines Ihrer Komponenten auch ein eigenständiges Medizinprodukt oder Zubehör ist, dürfen Sie dieses auch „alleine“ in Verkehr bringen also dies bspw. einzeln verkaufen, da dieses MP/Zubehör selbst ja eine Konformitätserklärung hat.
Mit allerbesten Grüßen
Benedikt Tölle
Liebes Johner-Team,
Ich habe eine eine frage zu der UDI-DI Vergabe.
wie stellen IVD Produkte der Gruppe B, C, D her.
in einer Verkaufseinheit befindet sich 50 Flaschen mit dem selben Produkt , selbe Charge und Verfallsdatum.
Meine Frage:
1.muss die UDI auf allen einzelnen Flasche oder nur auf die Verpackung der Verkaufseinheit (50 Flaschen). wenn ja muss dann jede einzelnen Flasche in der Verkaufseinheit eine eigene UDI bekommen oder bekommen alle Flaschen in dieser Verkaufseinheit dieselbe UDI.
2. haben innerhalb einer Produktionscharge alle Verkaufseinheiten eine eigene UDI oder hat die gesamte Produktionscharge eine gemeinsame UDI.
Liebe Frau Keller,
danke für Ihre Fragen:
zu 1.: Ich gehe davon aus, dass die Flaschen ein Einmalprodukt sind. Zunächst sagt die IVDR in Anhang VI, Teil C:
Bei Produkten für den einmaligen Gebrauch gilt allerdings auch:
Zu 2.: Leider ist mir nicht ganz klar, was für Sie der Unterschied ist zwischen einer Produktionscharge und einer Verkaufseinheit einer Prodkutionscharge. Die Produkte einer Charge werden aber die gleiche UDI-DI haben und je nachdem wie Sie die UDI-PI definiert haben, auch die gleiche UDI-PI (bspw. wenn Sie das Verfallsdatum in der UDI-PI kodieren). Beachten Sie für die Vergabe der UDI-PI die Vorgaben der IVDR:
Mit allerbesten Grüßen
Benedikt Tölle
Hallo Herr Johner,
die Anzahl der Kommentare zu diesem Thema ist mittlerweile sehr groß und ich habe sie jetzt nicht alle gelesen. Meine Frage dazu ist: Wenn sich der Legal Manufacturer eines Medizinproduktes innerhalb der Firmenorganisation ändert, benötigen die MD dann eine neue Basis UDI-DI?
Viele Grüße
G.Schumacher
Liebe Frau Schumacher,
eine Übernahme der Basis-UDI-DI von einem Hersteller zu einem nächsten Hersteller wird schon an der EUDAMED scheitern. Dort muss sich Hersteller 2 als neuer Wirtschaftsakteur registrieren und auch eine neue Basis-UDI-DI angeben.
Mit allerbesten Grüßen
Benedikt Tölle
Liebes Johner-Team,
gilt die UDI Kennzeichnungspflicht auch für Legacy Produkte (Klasse IIa – EG-Zertifikat gültig bis 26.05.2024) ab dem 26.Mai 2023? Oder ausschließlich für Produkte, die nach der MDR in Verkehr gebracht werden?
Ich lese es so, dass die UDI Anforderung schon ab Mai 2023 für alle (=MDD und MDR Produkte) Klasse IIa gilt. Jedoch wird bei uns intern der Artikel 123 der MDR sehr unterschiedlich interpretiert. Daher wäre ich für eine zusätzliche Meinung sehr dankbar!
Schöne Grüße,
M. Marker
Liebe Frau Marker,
die Antwort finden Sie in unserem Fachartikel zum Thema Legacy Produkte im Kapitel 1. e) oder in der MDCG Guidance 2021-15. Der Artikel 27 ist für Legacy Devices nicht anwendbar und somit benötigen Sie auch keine UDI für diese Produkte. Sie können aber eine UDI vergeben.
Mit allerbesten Grüßen
Benedikt Tölle
Guten Tag Herr Tölle,
ich habe hierzu noch eine weiterführende Verständnisfrage:
Wenn ein Legacy Produkt der Klasse IIa auf die MDR umgestellt wird, ist es dann auch nicht erforderlich einen UDI aufzudrucken?
Schöne Grüße, Susanne
Liebe Frau Arnhold,
für das Legacy-Device ist es nicht erforderlich, für das MDR-Produkt müssen Sie eine UDI vergeben.
Mit allerbesten Grüßen
Benedikt Tölle
Liebes Johner-Team,
ich wollte fragen, ob Leitfaden zum UDI-Modul auf Deutsch gibt?
Danke und schöne Grüss
Rony
Lieber Herr Rony,
der Leitfaden ist meines Wissens nach nur auf Englisch verfügbar.
Mit allerbesten Grüßen
Benedikt Tölle
Übernahme vorhandene Basic UDI-DI oder neue generieren?
Sehr geehrter Herr Tölle,
bei meinem Anliegen geht es um die Basic UDI-DI.
Wir übernehmen gruppenintern die CE-Verantwortung für ein Produkt, für das wir bisher Importeur waren. Das CE stammt ursprünglich aus den USA. Für das CE des Produkts wurde von den amerikanischen Kollegen bereits eine Basis UDI-DI vergeben. Mir stellt sich die Frage, ob wir diese vorhandene BUDI übernehmen können bzw. müssen, oder ob wir eine neue generieren müssen/können?
Ist diese Entscheidung davon abhängig, ob die BUDI mit den dazugehörigen UDI-DIs bereits an die EUDAMED gemeldet wurde? Oder ist das irrelevant?
Vielen dank für Ihre Unterstützung.
Mit freundlichen Grüßen
C.Graß
Liebe Frau Graß,
eine Übernahme der Basis-UDI-DI von einem Hersteller zu einem nächsten Hersteller wird an der EUDAMED scheitern. Dort muss sich Hersteller Nr. 2 als neuer Wirtschaftsakteur registrieren und auch eine neue Basis-UDI-DI angeben.
Mit allerbesten Grüßen & Ihnen ein frohes neues Jahr!
Benedikt Tölle
Sehr geehrter Herr Tölle,
vielen Dank für die Hilfe.
Ich hatte die Antwort bereits einem Beitrag im November entnommen-Sie waren schneller 🙂
Viele Grüße
Christine Graß
Sehr geehrter Herr Tölle,
ich wollte kurz fragen, was wurde gemeint im Anhang VI, Teil C, Punkt 5.3 (Die Hersteller erstellen geeignete Methoden/Verfahren für die Validierung der bereitgestellten Daten)? Ich bitte um eine Erklärung.
Danke im Voraus
Schöne Grüße
Rony
Sehr geehrter Rony,
danke für Ihre Frage!
Diese Anforderung ist dem Abschnitt „5. Allgemeine Grundsätze der UDI-Datenbank“ zugeordnet. Der Punkt 5.3 gibt an, dass in der EUDAMED Methoden/Verfahren verfügbar sein müssen, um bereitgestellte Daten zu validieren. Das macht die EUDAMED bspw., wenn Sie eine Basis-UDI-DI der GS1 eingeben. Dann checkt die EUDAMED die Syntax und sollte diese nicht mit den Vorgaben der GS1 übereinstimmen, akzeptiert die EUDAMED die Basis-UDI-DI nicht.
Diese Anforderung ist also nicht direkt an Hersteller gerichtet.
Mit allerbesten Grüßen
Benedikt Tölle
Ich stimme zu Herr Tölle, dass es keine Aufgabe der Hersteller. Leider aber in der deutschen IVDR-Version der Verordnungen wurde der Satz als Hersteller Aufgabe übersetzt. Ich gehe aber davon aus, dass die Englische Version stimmt. Oder? Danke im Voraus!
Lieber Herr Younis,
das scheint mir ein Übersetzungsfehler in der deutschen Version der IVDR zu sein. Sowohl in der englischen IVDR und MDR Version, als auch in der deutschen MDR Version ist der Satz ohne „Hersteller“ zu finden. Deswegen gehe ich stark davon aus, dass es sich um einen Übersetzungsfehler handelt. Das ist uns bisher auch noch nicht aufgefallen, danke Ihnen für den Hinweis!
Mit allerbesten Grüßen
Benedikt Tölle
Lieber Herr Tölle,
ich habe eine Frage zur UDI-PI. Wir verkaufen große Medizingeräte und nutzen nur manchmal eine Umverpackung in Form einer Holzkiste für den Transport. Dies aber auch nicht immer. Da diese Kiste eigentlich nur zum Schutz beim Transport gedacht ist und lediglich ein kleines Label angebracht ist mit Produktname – Empfänger und Seriennummer weiß ich nun nicht genau ob dies als „Verpackung“ zu zählen ist. Ist es da notwendig eine UDI-PI anzugeben? Oder zählt die Holzkiste hier als „Container“ der von UDI befreit ist?
Eine kurze Erklärung wäre sehr nett.
Vielen Dank Yvonne
Liebe Frau Quardockus,
die MDR definiert einen Versandcontainer wie folgt
Für einen solchen Versandcontainer definiert Artikel 27
Ob die Holzkiste nun ein Versandcontainer ist oder nicht hängt davon ab, ob die Rückverfolgbarkeit über einen für Logistiksysteme spezifischen Kontrollprozess ermöglicht wird. Ist dies der Fall, können Sie die Holzkiste als Versandcontainer deklarieren.
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrter Herr Johner,
Sehr geehrter Herr Salvatore,
Sehr geehrter Herr Tölle,
muss man beim UDI-Code mit dem GS1-System die Nummern (Application Identifier) in Klammern, also (01) für GTIN, (17) für Verfallsdatum, (21) für Seriennummer usw. abbilden oder kann man auf diese Nummern in Klammern verzichten?
Wäre dann eine Abtrennung der einzelnen Code-Teile mit „-“ zulässig?
Wann brauche ich den Application Identifier? Also die 8013 bei der Basis UDI? Die 01 usw. bei allen anderen Codes?
Die 8013 wird dabei nicht in der Eudamed angegeben. Muss ich diese Nummer aber in der Dokumentation angeben?
Vielen Dank.
Mit freundlichen Grüßen
Florian
Lieber Herr Sedlmeir,
danke für Ihre Fragen! Leider können wir im Rahmen der Kommentarfunktion eines Fachartikels nicht auf alle Fragen im Detail eingehen, das würde unserer Ressourcen sprengen, die uns für dieses kostenfreie Angebot zur Verfügung stehen.
Die Antworten auf Ihre Fragen finde Sie alle in den General Specifications der GS1 im Kapitel 3 und den Vergaberegeln für das Gesundheitswesen
Kurz zu Ihrer ersten Frage: Im GS1 System sind die Application Identifier verpflichtend. Welche Angaben (bspw. Seriennummer oder Herstelldatum) in der UDI-DI enthalten sein müssen, regelt aber die MDR in Anhang VI, Teil C, 3.5.
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrter Herr Johner & Co,
vielen Dank für die vielen Hinweise, die man hier im Forum sowie durch die Kommentare erhält! Diese hier waren soweit ich es sehe noch nicht dabei:
Wird für eine Standalone-Software überhaupt ein Barcode der UDI benötigt? Wir verwenden keinerlei Datenträger, sondern installieren via Download. Ich würde hoffen, dass dann die Angabe der UDI als Zahl (HRI) im Info-Screen ausreicht? Das würde bedeuten, ich brauche überhaupt keinen Anbieter für Barcodes?
Und: Muss auf die IFU eine UDI oder Basis-UDI-DI oder gar nichts?
Herzlichen Dank im Voraus!
Manuela Hartmann
Liebe Frau Hartmann,
danke Ihnen für ihre positive Rückmeldung, das freut uns riesig, dass unser Angebot Ihnen weiterhilft!
Die Anforderungen an Standalone-Software finden Sie im Abschnitt 6.5 des Anhang VI, Teil C der MDR
Reine standalone Software muss die UDI tragen, aber nur im menschenlesbaren Format HRI und nicht im maschinenlesbaren Format AIDC. Die UDI sollten Hersteller z.B. auf dem Start- oder Splash-Screen, in der Hilfe oder bei „About“ angeben. Einen Barcode benötigen Sie somit nicht.
Ohne die Anforderungen der MDR an die IFU noch einmal genau geprüft zu haben, muss weder die UID noch die Basis-UDI-DI in der IFU angegeben werden. Nur für implantierbarer Produkte gibt es die Anforderung, dass auch die in Artikel 18 angegeben Informationen in der IfU zu finden sein sollten. Sinnvoll diese anzugeben ist es aber schon, da immer klar sein muss, welche Version einer IfU zu welcher Produktversion/-variaten gehört.
Mit allerbesten Grüßen
Benedikt Tölle
Herzlichen Dank für die schnelle Antwort!
sehr geehrte Johner Team,
Eine Frage zur Basis UDI-DI.
wenn sich beide Produkte nur in der Menge, Inhalt (1x 250 ml und 1x 500ml) unterscheiden und somit auch gleich die Verpackungsgröße unterscheidet , darf man dann auch dieselbe Basis UDI verwenden.
(wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und)
Liebe Frau Ben,
Liebe Anna,
Produkte dürfen laut MDCG 2018-1 v4 dann eine gemeinsame Basis-UDI-DI haben, wenn
– sie über die gleiche Zweckbestimmung verfügen,
– wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und
– wenn sich auch die Herstellung, also die Produktion im Wesentlichen nicht unterscheiden.
Wenn all diese Bedingungen erfüllt sind, erlaubt die MDR, Produkte mit eigener UDI-DI zu einer Gruppe mit einer Basis-UDI-DI zusammenzufassen.
In Ihrem Fall können Sie also die zwei Produktvarianten unter einer Basis-UDI-DI zusammenfassen.
Mit allerbesten Grüßen
Benedikt Tölle
sehr geehrte Johner Team,
Eine Frage zur Basis UDI-DI.
wenn sich beide Produkte nur in der Menge, Inhalt (1x 250 ml und 1x 500ml) unterscheiden und somit auch gleich die Verpackungsgröße unterscheidet , darf man dann auch dieselbe Basis UDI verwenden.
(wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und)
Liebe Anna,
Produkte dürfen laut MDCG 2018-1 v4 dann eine gemeinsame Basis-UDI-DI haben, wenn
– sie über die gleiche Zweckbestimmung verfügen,
– wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und
– wenn sich auch die Herstellung, also die Produktion im Wesentlichen nicht unterscheiden.
Wenn all diese Bedingungen erfüllt sind, erlaubt die MDR, Produkte mit eigener UDI-DI zu einer Gruppe mit einer Basis-UDI-DI zusammenzufassen.
In Ihrem Fall können Sie also die zwei Produktvarianten unter einer Basis-UDI-DI zusammenfassen.
Mit allerbesten Grüßen
Benedikt Tölle
I know article and questions were in German so far, but i don’t trust my German grammatic to write a public question, thus please apologies for asking in English. Reply in German is welcomed though, so that others can benefit of the question.
In case of standalone SaMD, If we have developed several SW applications, each:
– is a standalone SaMD on its own,
– with own independent intended use and indications for use,
– some cloud based deployment and some on premise, but
– all with same MD risk class (IIb) and
– that were group under same EMDN Code with a high level Intended Purpose Statement.
And by being grouped under same EMDN, these will be covered by same Technical Documentation sampling Report.
Therefore our question, although the differences in product intended use or indications, as well as eventually the deployment technologies, if all were grouped under same EMDN, would make sense to group them all also under one single Basic-UDI ?
Or sharing EMDN code is not a criteria and despite sharing same EMDN, would rather be required or recommended by any reason to build groups by product families, creating product families on other level of shared similarities? what could be those criteria to consider that could require a split in several families?
Thanks a lot in advance.
L.I.
Thanks a lot in advance.
Liebe Frau Ipsen,
danke Ihnen für Ihre Frage und ich antworte gerne auf deutsch, danke, dass Sie dies angeboten haben!
Die EMDN Codes sind für die Basis-UDI-DI nicht relevant. Produkte dürfen laut MDCG 2018-1 v4 dann eine gemeinsame Basis-UDI-DI haben, wenn
– sie über die gleiche Zweckbestimmung verfügen,
– wenn deren Auslegung (d. h. das Design) im Wesentlichen gleich ist und
– wenn sich auch die Herstellung, also die Produktion im Wesentlichen nicht unterscheiden.
Wenn all diese Bedingungen erfüllt sind, erlaubt die MDR, Produkte mit eigener UDI-DI zu einer Gruppe mit einer Basis-UDI-DI zusammenzufassen.
Nun zu ihrer Frage:
The differences in product intended use or indications, as well as eventually the deployment technologies, if all were grouped under same EMDN, would make sense to group them all also under one single Basic-UDI ?
Sie können diese Produkte nicht unter einer Basis-UDI-DI zusammenfassen, da Sie sich im Intended Use oder der Indikation unterscheiden.
Mit allerbesten Grüßen
Benedikt Tölle
Hallo liebes Johner-Team,
gibt es eine Abverkaufsfrist für Medizinprodukte, die nach der Übergangsfrist für das Aufbringen des UDI-Trägers, noch ungekennzeichnet auf dem Markt sind?
Für ein MDR in Klasse I-Produkt muss der UDI-Träger bis 26.05.2025 aufgebracht sein. Wie verhält es sich dann für die Produkte die nach diesem Datum noch ungekennzeichnet auf dem Markt sind? Müssen die Hersteller diese dann sofort vom Markt zurückziehen oder bzw. sicher stellen, dass bis 26.05.2025 keine ungekennzeichneten Klasse I-Produkte sich auf dem Markt befinden? Oder haben diese Produkte noch eine bestimmte Abverkaufszeit?
Herzlichen Dank im Voraus
Mit freundlichen Grüßen
I. Milke
Liebe Frau Milke,
wie sie richtig sagen, müssen Produkte der MDR Klasse I, die ab dem 26.05.2025 in Verkehr gebracht werden, einen UDI-Träger haben. Produkt die vor diesem Termin ohne Träger in Verkehr gebracht wurden, sind rechtskonform in Verkehr gebracht und können auf dem Markt verbleiben. Die Rückverfolgbarkeit müssen Sie selbstverständlich auch bei den Produkten ohne UDI-Träger aufrecht erhalten.
Falls Ihnen die genaue Definition von in Verkehr bringen und v.a. die Abgrenzung zur Inbetriebnahme nicht zu 100% klar ist, schauen Sie gerne in unseren Fachartikel Inverkehrbringung: Definition und regulatorische Anforderungen
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrtes Johner-Team,
in obiger Übersicht in Abschnitt 7 ist dargestellt, dass die UDI auch in Reparatur-, Aufbereitungs- und Wartungsdokumenten enthalten sein muss.
Woraus leitet sich diese Anforderung ab? Gilt dies auch für Instandhaltungen, die der Betreiber selbst durchführt (z.B. STK-Protokolle, Serviceberichte)?
Herzlichen Dank schon einmal für Ihre Antwort und viele Grüße
Thomas Thienel
Lieber Herr Thienel,
vielen Dank für Ihre spannende Frage!
Die UDI ist die zentrale Identifikationsnummer für Medizinprodukte. Sie müssen sicherstellen, dass klar ist, welche Dokumente zu welchen Produkt gehören, hier bietet sich die UDI an und Benannte Stellen fordern diese auch auf den genannten Produkten (hier stammt die Liste von der Berlin Cert). Indirekt ableiten lässt sich dies aus Artikel 25, Artikel 27 und (für aufbereitete Produkte) Artikel 17.
Ich sehe diese Rückverfolgbarkeit auch auf Instandhaltungstätigkeiten bei den Betreibern als sinnvoll an.
Zusammengefasst: Die Dokumente müssen eindeutig einem Produkt zuzuordnen sein, die UDI muss regulatorisch nicht zwingend dafür genutzt werden, bietet sich dafür aber an, da sie genau dafür gemacht wurde.
Mit allerbesten Grüßen
Benedikt Tölle
Liebes Team vom Johner-Institut,
ich habe eine Frage zur Instandsetzung wieder verwendbarer Medizinprodukte, die eine UDI tragen, genauer gesagt zum Anhang VI, Teil C, Ziffer 4.10 der MDR. Demnach muss der UDI-Träger „dauerhaft angebracht und nach jedem Verfahren, das zur Vorbereitung des Produkts für die nachfolgende Verwendung durchgeführt wird, während der gesamten erwarteten Lebensdauer des Produkts lesbar sein.“
Es wird in der Praxis beobachtet, dass der UDI-Code oftmals so stark abgenutzt oder gar beschädigt ist, dass er nicht lesbar und somit auch nicht einlesbar ist.
Wie ist mit der Reparatur von solchen Medizinprodukten umzugehen?
1. Kann eine Instandsetzung erfolgen und der UDI-Code weggelassen werden, da es Pflicht des Herstellers bzw. Betreibers ist, die Aufrechterhaltung des Codes sicherzustellen?
2. Eine Instandsetzung kann in diesen Fällen nicht erfolgen (obwohl technisch möglich), da der UDI-Code der Medizinprodukte nach der Reparatur nicht dauerhaft lesbar ist.
Vielen Dank für für Ihre Antwort, Melanie Mayer
Liebe Frau Mayer,
vielen Dank für Ihre spannende Frage!
Wenn die UDI nicht mehr lesbar ist schlage ich folgendes Vorgehen vor: Melden Sie die nicht Lesbarkeit der UDI an den Hersteller. Dieser muss dann entsprechend handeln und Ihnen auch Anweisungen geben, wie Sie mit dem Produkt weiter zu verfahren haben.
Mit allerbesten Grüßen
Benedikt Tölle
Sehr geehrter Herr Tölle,
ich habe eine kurze Frage zum Thema UDI und technische Dokumentation. Muss man in der TD beide die UDI-DI und UDI-PI schreiben? Wenn ja wie kann man in Realität schaffen? Da UDI-PI ist keine statische info.
Vielen Dank!
Rony
Lieber Herr Rony,
vielen Dank für Ihre spannende Frage!
mit UDI meint die MDR und IVDR die UDI-DI und UDI-PI. D.h. Sie müssen in Ihrer technische Dokumentation beide Teile der UDI ablegen.
Aktualisieren Sie die UDI nicht fortlaufend sondern in bestimmten Intervallen (bspw. alle 3 Monate), indem Sie die vergebenen UDI-PIs aus Ihrem Warenwirtschaftssytem ziehen.
Mit allerbesten Grüßen
Benedikt Tölle
Hallo liebes Team vom Johner Institut,
ich habe eine Frage zur Basic UDI-DI. Wir sind Importeur von Klasse I Medizinprodukten und fragen uns, ob wir auf die Basis UDI-DI sowie die UDI-DIs unserer Hersteller beziehen können. Wir sind nicht der Hersteller im Sinne der MDR wenn ich das richtig verstehe. Als Importeur sind wir auch bereits in EUDAMED registriert und ich kann dort keine Basis UDI-DI hinterlegen.
Vielen Dank im Voraus.
Beste Grüße an den Bodensee.
Lieber Herr Andreas,
vielen Dank für Ihre spannende Frage!
Die Basis-UDI-DI und UDI-DIs können Sie über drei Möglichkeiten herausfinden:
– Fragen Sie den Hersteller direkt an
– Schauen die in der EUDAMED nach
– Schauen Sie auf die Konformitätserklärung der Produkte
Mit allerbesten Grüßen
Benedikt Tölle
Hallo –
Im Rahmen einer Registrierung wurden wir aufgefordert GTINs zu übermitteln. Allerdings wurden wir nach GTIN 12 bzw. 13 Codes gefragt – Nach langem Hin und Her haben wir herausgefunden, dass man eigentlich EAN/UPN codes für die Endabgabestelle (z.B. Apotheke) gemeint hat. Daher meine Frage: In wie weit besteht die Verpflichtung, dass Medizinprodukte in Anlehnung an Handelswaren mit dem EAN/UPN gekennzeichnet werden müssen nebst der regulatorischen Anforderung einer UDI Kennzeichnung. Oder woher kommt dieses requirement?
Lieber Herr Müllner,
vielen Dank für Ihre spannende Frage!
Mir ist keine Anforderung an eine EAN/UPN bekannt und ich höre dies zum ersten Mal. Meines Wissen hat die GTIN die EAN ersetzt.
Bei welche Registrierung wurde von wem diese Anforderung gestellt? Haben Sie diese Person bereits direkt gefragt, woher die Bitte nach der EAN/UPN kommt?
Mit allerbesten Grüßen
Benedikt Tölle
Guten Tag Herr Tölle,
Die Anforderung wurde im Rahmen einer lokalen Türkischen Distrutorenregistrierung gestellt. Nach einer etwas mühsamen Recherche haben wir herausgefunden, dass Handelswaren grundsätzlich mit Gtin-12/13 Codes (vormals EAN/UPC_A) gekennzeichnet werden sollen,damit die Abrechnungs/Logistik Systeme die Waren verarbeiten können. Einige Systeme sind nicht in der Lage eine UDI Datamatrix korrekt zu lesen und die Daten korrekt zu entschlüsseln. Daher ist eine Doppelkennzeichnung sinnvoll.
Beste Grüße
Peter Müllner
Lieber Herr Müllner,
danke für das recherchieren und dass Sie uns dies Information mitteilen! Das war mir so bisher nicht bekannt.
Mit allerbesten Grüßen
Benedikt Tölle
Hallo liebes Johner-Team,
wir stellen unter anderem ein Medizinprodukt Klasse IIa (MDR: IIb) her und arbeiten derzeit intensiv an der Einreichung der Technischen Dokumentation für die MDR. Das Medizinprodukt besteht aus einem Wirkstoff in einer Cellulose-Kapsel die verblistert ist.
Zur Frage UDI:
1) Müssen wir die UDI zum Zeitpunkt der MDR Beantragung schon auf den Produkten vorweisen? Es gibt einige widersprüchliche Informationen. In erster Linie die noch nicht voll funktionsfähige Eudamed.
2) Muss man auf dem Blister (Primärverpackung) die UDI anbringen oder erst eine Ebene darüber (Sekundärverpackung)? Die MDR und auch MDCG-Dokumente sind dahingehend für uns nicht ganz eindeutig.
Ich freue mich über Ihre Rückmeldung.
Viele Grüße
Christiane
Liebe Frau Schmidt,
vielen Dank für Ihre spannende Fragen!
1. Die Übergangsfrist bei der UDI gilt nur für das Anbringen der UDI-Träger und ist unabhängig von der EUDAMED. MDR Produkte der Klasse IIb müssen den UDI Träger seit dem 26.05.2023 tragen.
2. Gemäß MDR Anhang VI, Teil C 4.3 gilt: bei Einmalgebrauchsprodukten können Sie den UDI-Träger auf Produkt oder Einzelverpackung auftragen. Das ist Ihnen als Hersteller überlassen. Da bei Tabletten das Aufbringen des UDI Trägers keinen Sinn ergibt, müssen Sie die Blisterverpackung mit einer UDI-DI versehen.
Mit allerbesten Grüßen
Benedikt Tölle
Liebes Johner-Team,
ein Hersteller kennzeichnet ein Medizinprodukt direkt auf dem Produkt mit einer LOT-Nummer, die sich aus der Produktionswoche herleitet. Auf der Verpackung mit UDI-Träger gibt der Hersteller jedoch in der UDI-PI eine komplett andere LOT-Nummer an, die den Verpackungstag angibt.
Ist das zulässig? oder gibt es eine Anforderung, nach der die LOT-Nummern übereinstimmen müssen?
Herzlichen Dank im Voraus,
beste Grüße,
Thomas Ertner
Lieber Herr Ertner,
vielen Dank für Ihre Frage! Mein Kollege Benedikt Tölle wird sich diesbezüglich demnächst per E-Mail an Sie wenden.
Herzliche Grüße
Tea Bodrusic
Sehr geehrter Herr Tölle,
ich bin jetzt mal ganz forsch und hänge mich an die Frage dran: Wenn ich stoffliche Medizinprodukte habe, z.B. Ultraschallkontaktgel und 10 Flaschen in einen Karton verpacke, handelt es sich bei der Versandeinheit dann auch um einen Versandcontainer? Über das Warenwirtschaftssystem ist der Versand/Transport an den Händler (und weiter an den Endkunden) ja nachvollziehbar und somit die geforderte Rückverfolgbarkeit gegeben, die dann über den UDI ja „nur“ innerhalb der Endanwender-Organisation sichergestellt werden müsste. Ob das dann allerdings den Aufwand für den UDI rechtfertigt, wage ich zu bezweifeln, und komme ich daher zur Schlußfolgerung, dass der UDI Einzelprodukte und sämtliche Verpackungsvarianten umfasst (ggf. gibt es ja die Einzelgebinde in verschiedenen Gebindegrößen und Stückzahlen je Karton sowie ggf. noch mit Zubehör und folglich wären dementsprechend viele UDIs auf Verpackungsebene erforderlich). Leider habe ich aber keine dahingehende, konkretere und offizielle Angabe gefunden und besteht hier intern große Uneinigkeit, ob es nur der Kennzeichnung der Einzelgebinde oder der Versandeinheit bedarf, wobei letzteres voraussetzt, dass eine Versandeinheit genau definiert ist und man daher diesbezüglich dementsprechend unflexibel ist/wird.
Ich bin gespannt auf Ihre Meinung und sage schon mal herzlichen Dank für Ihre Einschätzung!
Viele Grüße,
Nick Rößler
Lieber Herr Rößler,
vielen Dank für Ihre spannende Frage!
Wenn Sie die Ultraschallkontaktgele im 10er Pack in Ihrem Sortimente anbieten, ist es aus meiner Sicht eine Verpackungseben. Wenn Sie die Ultraschallkontaktgele nur einzeln anbieten und auf Bestellung des Kunden mal 3, dann 5 und manchmal 10 Gele zusammenverpacken, ist es eher ein Versandcontainer.
Mit allerbesten Grüßen
Benedikt Tölle
Hallo,
gibt es einen Beitrag wie genau die Fristen der Anbringung eines UDI-Codes auf dem Produkt sind?
Welche Frist gilt für die verschiedenen Klassen und für wiederverwendbare Produkte?
Vielen lieben Dank für Ihre Unterstützung!
Hallo Frau Coßmann,
die Fristen zur UDI-Anbringung finden Sie im Kapitel 2 d) in folgendem Beitrag:
https://www.johner-institut.de/blog/regulatory-affairs/uebergangsfristen-mdr/
Freundliche Grüße
Luca Salvatore
Hallo liebes Johner Team,
Ich habe eine Frage zum scannen des UDI-Codes bei Medizinprodukten. Der menschenlesbare Teil wird mit Klammern angezeigt Beispiel (01)04046153092604(17)290331(10)289-154-2416. Sollte der maschinenlesbare Teil (scannen) mit Klammern oder ohne Klammern angezeigt werden? Gibt es dazu eine Richtlinie oder Verordnung?
Herzliche Grüße A.Brandt
Lieber Herr Brandt,
vielen Dank für Ihre interessante Frage.
Die Klammern der Application Identifier dienen nur der besseren Lesbarkeit in der HRI. In der AIDC werden die Klammern nicht codiert und beim Scannen auch nicht angezeigt.
Dies ist in der GS1 Spezifikation definiert: https://www.gs1-germany.de/fileadmin/gs1/fachpublikationen/gs1-germany-allgemeine-gs1-spezifikationen-mit-nationalem-Anhang-v24.pdf
Herzliche Grüße,
Katharina Keutgen
Liebe Frau Keutgen,
gerne möchte ich mich auf diesem Weg für Ihre geleistete Arbeit bedanken.
Ihre Antwort ist für mich sehr hilfreich gewesen. Dankeschön!
Ich wünsche Ihnen noch eine angenehme Woche.
Herzliche Grüße A.Brandt
Hallo liebes Team,
ich hätte nochmal eine etwas spezifischere Frage. Wir führen ein Medizinprodukt (OP-Tray), das aus einem Boden, einem Deckel und Einsätzen besteht. Zusammengesetzt ist es ein Medizinprodukt. Jede Komponente für sich aber nicht. Wo genau bringe ich die UDI-DI nun auf? Auf jedem einzelnen Teil oder auf einem mit einem Hinweis, dass dies z.B. Komponente 1/3 ist?
Vielen Dank für Ihre Hilfe.
Liebe Grüße,
Mona Coßmann
Liebe Frau Coßmann,
herzlichen Dank für Ihre Frage.
Anhang VI, 4.13 der MDR definiert:
„Bei einzelnen Endprodukten, die aus mehreren Teilen bestehen, die vor ihrer ersten Anwendung zusammengefügt werden müssen, reicht es aus, wenn der UDI-Träger auf lediglich einem Teil jedes Produkts angebracht wird.“
Wenn das Produkt zwischen Anwendungen auch wieder in seine Einzelkomponenten zerlegt werden kann/soll, kann es dennoch erforderlich sein, diese dem eigentlichen Produkt eindeutig zuordenbar zu machen. Dies geschieht aber in der Regel nicht über die UDI, sondern über die Angabe des Herstellers und z.B. der Artikelnummer oder -bezeichnung.
Herzliche Grüße,
Katharina Keutgen
Hallo liebes Johner Team,
muss Zubehör eine eigene UDI-DI bekommen, auch wenn dieses selbst kein Medizinprodukt ist? Und ist es möglich das Zubehör bei der EUDAMED zu registrieren?
Danke und viele Grüße
Yvonne Schoote
Liebe Frau Schoote,
vielen Dank für Ihre Frage!
Wenn es sich um Zubehör gemäß Artikel 2, MDR handelt, das schon per Definition keinen eigenen medizinischen Zweck hat, aber die Zweckbestimmung eines Medizinprodukts unterstützt oder ermöglicht, fällt das Zubehör in den Geltungsbereich der MDR. Somit benötigt es eine eigene UDI und muss in der Eudamed eigenständig registriert werden.
Dies ist nicht erforderlich, wenn es sich um eine Komponente eines Medizinprodukts handelt, die gemeinsam mit diesem ein Konformitätsbewertungsverfahren durchläuft.
Herzliche Grüße,
Katharina Keutgen
Hallo Frau Keutgen,
ich hab die selbe Frage wie Fraus Schoote, möchte das aber noch etwas spezifizieren. Wenn es bei dem Zubehörteil für unser aktives Medizinprodukt der Klasse 1 um einen optional als Zubehör bestellbaren, handelsüblichen 2-Punkt-Sicherheitsgurt (sog. Beckengurt) geht, welchen wir selbst fertig als „Katalogware“ beziehen und welcher grundsätzlich auch (bzw. „vor allem“) als Sicherheitsgurt in Baumaschinen (z.B. Bagger) oder Landmaschinen (z.B. Traktor) zur Anwendung kommt, braucht dieses Zubehörteil wirklich eine eigene UDI-DI. Das komm mir ziemlich überzogen vor. Oder sollten wir das besser als Konfiguration einordnen? Denn grundsätzlich wird der Beckengurt ausschließlich von uns oder von geschultem Fachpersonal der Sanitätshäuser angebaut.
Vielen Dank im Voraus für Ihre Mühen und schöne Grüße,
Daniel Mohr
Lieber Herr Mohr,
vielen Dank für Ihre Nachfrage.
Wenn der Sicherheitsgurt als Zubehör im Sinne von Artikel 2, MDR einzuordnen ist, dann muss er auch die Anforderungen an das UDI-System vollumfänglich erfüllen und benötigt eine eigene UDI-DI.
Die Konfigurationen gemäß Anhang VI sind ein möglicher Weg, die Anzahl der UDI-DIs zu reduzieren – allerdings müssen die möglichen Konfigurationen einer Konfigurationsgruppe dann in einer Technischen Dokumentation des konfigurierbaren Produkts beschrieben werden. Sollte der Sicherheitsgurt als Bestandteil eines konfigurierbaren Produkts jedoch weiterhin einzeln kommerziell verfügbar sein, müsste er dennoch eine eigene UDI-DI erhalten.
Melden Sie sich gern bei mir, wenn wir uns Ihre Konstellation einmal gemeinsam anschauen sollen.
Herzliche Grüße,
Katharina Keutgen
Hallo liebes Johner Team,
muss ich pro Basic-UDI eine eigene Technische Dokumentation anlegen?
Wir haben rezepturidentische Produkte, die wir unter verschiedenen Handelsnamen vertreiben und haben pro Handelsnamen einen Basic-UDI vergeben. Ich würde aber gerne z.B. nur eine Klinische Bewertung pro Rezeptur schreiben und darin auf alle der Rezeptur zugewiesenen Basic-UDI´s verweisen. Ist das möglich?
Herzlichen Dank im Voraus
Viele Grüße
Julia
Liebe Julia,
vielen Dank für deine interessante Frage.
Es ist grundsätzlich möglich, mehrere Basis UDI-DIs in einer Technischen Dokumentation zu führen, wenn eine klare Zuweisung der jeweils relevanten Informationen erfolgt.
In Ihrem Fall sollten Sie allerdings noch einmal prüfen, ob überhaupt unterschiedliche Basis UDI-DIs erforderlich sind, wenn sich die Produkte nur im Handelsnamen unterscheiden.
Sollten Sie bei dieser Einschätzung Hilfe benötigen, melden Sie sich gern bei uns.
Herzliche Grüße,
Katharina Keutgen
Liebes Team.
Wir sind ein Hersteller eines Software-Medizinprodukts. Benutzer können dieses Software-Medizinprodukt sowohl auf mobilen Geräten als auch auf der Website verwenden. In diesem Fall, sollen wir unterschiedliche UDI-DIs für die mobile und die Website-Version zuweisen? Oder können wir nur eine zuweisen? Wenn es ein Batch-Update für die mobile Version gibt, wird dann nur die UDI-PI für den mobilen Teil betroffen sein? Wenn ja, ist die UDI unterschiedlichen zwischen mobilen Geräten und Website-Version obwohl sie beziehen sich auf das gleiche Software-Medizinprodukt (Funktionsweise)
wie kann man die UDI in diesem Fall besser verwalten?
Viele Grüße.
Jason Li
Sehr geehrter Herr Li,
wenn die eine (1) Software eindeutig identifizierbar ist und (!) in unveränderter Art auf beiden Plattformen betrieben werden kann, reicht es, wenn Sie eine UDI vergeben.
Die UDI-PI sollten sie nicht für verschiedene Varianten der Software verwenden, sondern für verschiedene Versionen der Software, also minor Updates.
Wenn Sie ein minor Update fahren, führt das zu einer neuen UDI-PI. Falls es sich um ein (1) identifizierbares Software-System handelt, das auf zwei Plattformen läuft, dann hat dieses Software-System die neu PI auf beiden Plattformen. Schließlich ist es das gleiche System.
Ich kann Ihre Fragen nicht genauer beantworten, weil mir nicht ganz klar ist, was Ihre Software umfasst. Falls es letztlich zwei Software-Systeme sind, die sich z.B. durch die Programmiersprache oder durch die UI unterscheiden, dann führt das zumindest zu zwei UDI-DIs, bei ggf. gleicher Basis-UDI-DI.
Beste Grüße, Christian Johner
Liebe Frau Keutgen,
eine UDI Frage ist bei mir noch aufgetaucht.
Wird bei den Zahlen in der vom Menschen lesbaren Form (Human Readable Interpretation (HRI))
eine Reihenfolge vorgeschrieben, wie aufgeführt?
(01) GTIN
(10) Chargennummer
(11) Produktionsdatum
(17) Verfallsdatum / Haltbarkeit
Oder ist es auch möglich die Zahlen in den Klammern durcheinander aufzuführen
Hier ein Beispiel:
(01)
(11)
(17)
(10)
Ich würde mich freuen, wenn Sie mir bei dieser Frage behilflich sein könnten.
Ein sonniges Wochenende wünsche ich Ihnen
Herzliche Grüße André Brandt
Lieber Herr Brandt,
herzlichen Dank für Ihre interessante Frage.
Die MDR macht hierzu keine Vorgabe, innerhalb des GS1 Systems wird die Frage direkt in der Spezifikation im Abschnitt 3.1 beantwortet:
„In most cases predefined length element strings SHOULD be followed by non-predefined element strings. The sequence of predefined and non-predefined element strings should be at the discretion of the creator of the element string“ (https://ref.gs1.org/standards/genspecs/)
Entsprechend sollten sie Attribute mit definierter Länge voranstellen und Attribute mit variabler Länge dahinter codieren. Vorgaben zum Format der einzelnen „Application Identifier“ finden Sie übrigens auch in der Spezifikation, genauer gesagt im Abschnitt 3.2 ff.
Herzliche Grüße,
Katharina Keutgen
Ist meine Annahme richtig, dass ab dem betreffenden Stichtag nur noch Ware mit der UDI in den Verkehr gebracht werden darf. Lagerware wäre damit verfallen. Wir stellen Medizinprodukte der der Klasse 1 her und dürften dann ab Mai 2027 keine ungekennzeichnete Ware (mit direkt Markierung) mehr verkaufen.
Liebe Frau Grötsch,
vielen Dank für Ihre spannende Frage.
Richtig ist, dass ab Mai 2025 auch Klasse I Produkte eine UDI auf ihrer Kennzeichnung tragen müssen und im Fall von wiederverwendbaren Produkten diese ab Mai 2027 auf dem Produkt selbst gekennzeichnet sein müssen (MDR, Artikel 123(3)f) und g)).
Ob die Lagerware damit verfällt, hängt von Ihrem Produkt und den Möglichkeiten der Nacharbeit ab. Eventuell ist es möglich, eine UDI nachträglich aufzubringen.
Herzliche Grüße,
Katharina Keutgen
Liebes Johner-Team,
können Sie uns beantworten, ob die FDA für die UDI-Kennzeichnung von Medizinprodukten, die Verwendung von 2D-DataMatrix Barcodes akzeptiert und wo dies in den Regularien zu finden ist?
Herzlichen Dank im Voraus und viele Grüße
Peter Koszolka
Lieber Herr Koszolka,
vielen Dank für Ihre Frage.
Die FDA selbst macht keine Vorgabe zum Format der AIDC. Im Guidance Dokument „Unique Device Identification System: Form and Content of the Unique Device Identifier (UDI)“ heißt es:
„Therefore, while the UDI Rule does not require the use of a specific form of AIDC or a specific AIDC technology to convey the UDI, the AIDC form of the UDI must be in a format that can be read by a bar code scanner or other AIDC technology (21 CFR 801.3 and 801.40(a)). […] Labelers should consult the guidelines of their FDA-accredited issuing agency to determine which forms of AIDC are supported by the issuing agency’s UDI system.“ (https://www.fda.gov/media/99084/download)
Es gelten also die Vorgaben der Zuteilungsstellen, die sie in den jeweiligen Spezifikationen finden. Grundsätzlich ist der DataMatrix Code ein gängiger Barcode im Gesundheitswesen und wird entsprechend auch von der FDA akzeptiert.
Herzliche Grüße,
Katharina Keutgen
Sehr geehrtes Johner-Team,
gehe ich recht in der Annahme, dass das Schaubild zu den Datenelementen einer BUDI (Abschnitt 5) nicht mehr aktuell ist, respektive ein einheitlicher EMDN-Code für alle „Members“ einer BUDI bzw. die Angabe eines Nomenklatur-Codes auf BUDI-Level nicht mehr gefordert ist ? Danke für eine Klarstellung.
Mit freundlichen Grüßen
Susan Schirmer
Liebe Frau Schirmer,
vielen Dank für Ihren wichtigen Hinweis.
Das hier referenzierte Dokument der EU UDI Work Group aus 2018 ist zwar immer noch in dieser Form im docsroom der Europäischen Kommission zu finden, allerdings ist die Zuordnung des EMDN Codes in der aktuellen Fassung des Eudamed Data Dictionary auf die Ebene der UDI-DI verschoben. Das aktuelle Data Dictionary finden Sie hier: https://webgate.ec.europa.eu/eudamed-help/en/documentation/technical-documentation.html
Dass dies entsprechend umgesetzt wird bestätigt auch ein Blick in den Eudamed Playground.
Den Artikel werden wir zeitnah aktualisieren.
Herzliche Grüße,
Katharina Keutgen
Sehr geehrtes Johner-Team,
ich bin gerade in Diskussion mit einem Medizinproduktehersteller, der an einen Händler verkauft. Folgende Situation bezüglich Verpackungsebenen (die ja dann auch die UDI betreffen) kann ich darstellen:
Hersteller verkauft an Großhändler 50 Säckchen eines bestimmten Medizinprodukts (in einem durchsichtigen Nylon-Säckchen befinden sich 20 Einzelprodukte). Diese 50 Säckchen werden wiederum zusammengefasst in einem großen durchsichtigen Nylonsäckchen. Nehmen wir an, das Label wäre nur auf dem äußersten Säckchen angebracht (wo dann QTY: 50×20 angegeben ist), aber nicht auf den Einzelsäckchen – darf das sein? Meine Meinung ist es notwendig, dass die kleinstmögliche Einheit gelabelt sein muss (schon allein wegen der UDI – das wäre für mich hier das Säckchen mit 20 Stück). Der Hersteller meinte aber zum Großhändler, dass seine „Verpackungseinheit“ sowieso 50×20 beim Verkauf ist und diese auch nicht kleiner bestellt werden kann und daher auch keine Labels auf den einzelnen Säckchen notwendig sind. Falls von Belangen: Es handelt sich hier noch um ein Legacy Device.
Ich bin gespannt auf Ihre Antwort!
Mit lieben Grüßen,
Kathrin
Liebe Kathrin,
vielen Dank für die spannende Frage!
Zur Anbringung der Kennzeichnung an sich schreibt die MDR in Anhang I, 23.1 folgendes vor: „Medium, Format, Inhalt, Lesbarkeit und Anbringungsstelle der Kennzeichnung und der Gebrauchsanweisung eignen sich für das jeweilige Produkt, seine Zweckbestimmung und die technischen Kenntnisse, die Erfahrung, Ausbildung oder Schulung der vorgesehenen Anwender.“ Der Hersteller entscheidet muss also ausgehend von der Zweckbestimmung und Anwendung des Produkts risikobasiert beurteilen, auf welcher Ebene Kennzeichnungsangaben gemacht werden (unter Berücksichtigung von Absatz b), der besagt, dass die Kennzeichnungsangaben, wenn möglich, auf dem Produkt oder höheren Verpackungsebenen anzubringen sind).
Falls das Produkt also ausschließlich in der beschriebenen Versandeinheit 50×20 verschickt und der Hersteller davon ausgehen kann, dass die Produkte bis zum Gebrauch in der gekennzeichneten Verpackung verbleiben, ist es grundsätzlich möglich, nur den äußeren Beutel zu kennzeichnen. Die Tatsache allerdings, dass nicht 1000 Stk. in einem Beutel verpackt, sondern jeweils 20 Stk gemeinsam verpackt werden, legt die Vermutung nahe, dass diese zur weiteren Verwendung aus dem großen Beutel entfernt werden. Das spräche wiederum für eine Kennzeichnung auf den einzelnen Beuteln.
Hinsichtlich der UDI Kennzeichnung gilt, dass jede Verpackungsebene mit Ausnahme des Versandcontainers mit einer UDI zu versehen ist. Verpackungsebenen sind definiert als „die verschiedenen Ebenen der Produktverpackungen, die eine festgelegte Menge an Produkten enthalten, wie z. B. eine Schachtel oder Kiste.“ Damit bleibt in diesem Fall wenig Spielraum. Sowohl die kleinen als auch der große Sack enthalten eine festgelegte Menge an Produkten und benötigt damit eine UDI.
Wichtiger Hinweis: Für Legacy devices ist das UDI-System nicht verpflichtend!
Liebe Frau Keuchen, vielen Dank für Ihre rasche und ausführliche Antwort!
Sehr geehrtes Johner-Team,
wir haben hier eine Diskussion zwischen Logistik und Regulatory: Wir stellen einen Führungsdraht (Klasse IIb) her, welcher mit der UDI gekennzeichnet ist. Diesen Führungsdraht verkaufen wir ausschliesslich in einer 10er-Packung (kein Einzelverkauf, keine übergeordnete Verpackungseinheit).
Nun herrscht Uneinigkeit darüber, ob wir sowohl eine UDI für den einzelnen Draht als auch eine weitere UDI für die 10er-Packung benötigen, oder ob auf der 10er-Packung die gleiche UDI angebracht werden kann wie auf dem einzelnen Draht.
Was würden Sie uns hier raten?
Freundliche Grüsse
Daniel Hertig
Lieber Herr Hertig,
vielen Dank für Ihre interessante Frage.
Wenn es sich bei dem Führungsdraht nicht um ein wiederverwendbares Produkt handelt, muss keine UDI auf dem einzelnen Draht angebracht werden. Sie beginnen dann mit der UDI-Kennzeichnung auf der Ebene der 10er Einheit.
Allerdings müssen Sie dem einzelnen Draht eine sogenannte virtuelle „Unit-of-Use“-DI zuweisen. Diese wird nicht auf dem Produkt oder der Kennzeichnung angebracht und lediglich in der EUDAMED Datenbank registriert.
Sollten Sie hierzu Fragen haben, melden Sie sich gern noch einmal bei uns.
Herzliche Grüße,
Katharina Keutgen
Hallo liebes Johner-Team,
ich habe eine Frage zum Aufbau der UDI. Auf dem Produkt selbst würden wir gerne im UDI-Code die Rohstoffnummer des Produktes angeben, da wir so eine Rückverfolgbarkeit besser/einfacher sicherstellen können. Auf der Verpackung des Produktes würden wir dann gerne die UDI mit der REF-Nummer angeben. Ist das so möglich?
Vielen Dank fürs Beantworten meiner Frage.
Liebe Frau Cossmann,
vielen Dank für Ihre spannende Frage.
Die MDR schreibt vor, dass die UDI auf allen Ebenen der Produktverpackung einmalig sein muss. Zudem werden die Mindestanforderungen an die UDI-PI in Anhang VI, 3.5 definiert. Weitere optionale Angaben, wie z.B. die Artikelnummer, dürfen aber durchaus in die UDI-PI aufgenommen werden. Eine Vorgabe, nach der die freiwilligen Attribute in der UDI-PI auf jeder Verpackungsebene gleich sein müssen, ist uns nicht bekannt.
Herzliche Grüße,
Katharina Keutgen
Sehr geehrter Herr Johner, liebe Community,
in unserer Gruppe ist die Frage aufgekommen: ist es gesetzliche Pflicht oder GS-1 Pflicht, dass die UDI-DI gleich der GTIN ist (so wie wir es erfolgreich praktizieren), oder ist dies nur ein „good practice“?
Herzlichen Dank
Tjark Siedentop
Lieber Herr Siedentop,
vielen Dank für Ihre interessante Frage.
die MDR definiert, dass die UDI aus der UDI-DI und der UDI-PI besteht. Jede Zuteilungsstelle verwendet hierfür Ihre eigenen Begrifflichkeiten.
Der Begriff „GTIN“ stammt von der GS1 und wird im Medizinprodukte-Kontext für die UDI-DI verwendet. Die UDI-DI ist also für GS1-Kunden immer die GTIN.
Herzliche Grüße,
Katharina Keutgen
Liebe Frau Keutgen,
herzlichen Dank für Ihre Antwort, die unsere Haltung bestätigt und mir sehr weiter hilft.
Liebes Johner-Team,
weiter oben gab es einige Fragen zu Zubehör / Komponenten, ich würde es allerdings gerne nochmal in einem konkreten Beispiel verstehen:
Das Medizinprodukt ist ein Blutdruckmessgerät mit beigelegtem Steckernetzteil und Manschette.
Dieses Gesamtpaket hat eine Bestellnummer und damit eine UDI-DI und UDI-PI.
Wir sind Hersteller des Grundgerätes und legen dem Gesamtpaket das kommerzielle Steckernetzteil bei (das Teil der Konformitätsbewertung des Gerätes ist).
Weiterhin legen wir die Manschette bei, die wir als Katalogware mit eigener Konformitätserklärung zukaufen.
Sowohl Steckernetzteil als auch Manschette wird nicht einzeln als Zubehör verkauft, lediglich als Ersatzteil.
Reicht hier eine UDI-DI und UDI-PI für das Gesamtpaket?
Herzlichen Dank vorab,
Thomas Wirl
Lieber Herr Wirl,
vielen Dank für Ihre interessante Frage.
Ihrer Beschreibung nach scheint es sich bei Ihrem Verkaufsartikel um ein System nach Artikel 22 zu handeln, das aus dem Produkt 1 „Blutdruckmessgerät inkl. Netzteil“ sowie dem Produkt 2 „Manschette“ besteht.
Hier gilt, dass die einzelnen Produkte ihre eigene UDI tragen (MDR, Artikel 29(3)) und zusätzliche eine UDI für das System vergeben wird (MDR, Anhang VI, 3.7). In Ihrem Fall sind Sie als Hersteller von Produkt 1 verantwortlich für dessen UDI, sowie als Ersteller des System verantwortlich für die System-UDI. Da Sie die Manschette als CE-gekennzeichnetes Produkt zukaufen, muss diese (sofern es sich nicht um ein legacy device handelt) die UDI des legalen Herstellers tragen. Diese behält sie innerhalb Ihres Systems.
Herzliche Grüße,
Katharina Keutgen
Liebes Team des Johner-Instituts,
wir haben ein Produkt, dass aus zwei Boxen besteht, die unterschiedliche Lagertemperaturen haben.
Es gibt daher keine Verpackung, in der beide Boxen enthalten sind.
Werden beide Boxen mit der gleichen UDI-DI gelabelt?
Benötigen beide Boxen eine spezifische UDI-PI? Gibt es hierfür eine AI um die Boxen anhand dessen zu unterscheiden?
Lieber Herr Handt,
vielen Dank für Ihre Frage. Leider adressiert die MDR die Kennzeichnungsanforderungen für Produkte mit mehreren Packstücken nicht. Grundsätzlich gilt die UDI aber für die Gesamtheit des Produkts. Sie vergeben also keine eigene UDI (und auch keine eigene UDI-PI) für die separaten Boxen – davon ausgehend, dass es sich hierbei nicht um Komponenten handelt, die für sich genommen Medizinprodukte und einzeln kommerziell verfügbar sind.
Auf der Kennzeichnung beider Komponenten sollte erkennbar sein, dass weitere Komponenten zum Medizinprodukt gehören. Mein Vorschlag für beide Boxen: „Component 1 of 2 of medical device X with UDI Y“.
Sollten Sie weitere Fragen zur Kennzeichnung haben, melden Sie sich gern bei uns.
Herzliche Grüße,
Katharina Keutgen
Sehr geehrte Frau Keutgen,
Wann muss ein UDI-Träger auf einem Legacy-Produkt erscheinen? Gibt es eine Deadline? Wird es in einem MDCG-Dokument irgendwo erwähnt? Darf man eine UDI für ein Legacy-Produkt in EUDAMED eingeben, obwohl sie nicht auf dem Produkt, das auf dem Markt ist, vorhanden ist?
Vielen Dank im Voraus!
MfG, Younis
Lieber Younis,
vielen Dank für deine interessanten Fragen.
Die Anforderungen im Zusammenhang mit dem UDI-System gelten nicht für Legacy Devices. Dies ergibt sich aus den Übergangsbestimmungen in Artikel 120 der MDR und wird bestätigt durch das MDCG-Dokument 2021-25.
Für Legacy Devices, die noch keine UDI haben, wird für die Registrierung in der EUDAMED ein alternativer Identifier, die sogenannte EUDAMED ID verwendet. Details hierzu findest du auch im EUDAMED User Guide zu den Legacy Devices: https://webgate.ec.europa.eu/eudamed-help/en/files/Legacy%20Devices%20-%20user%20guide.pdf
Herzliche Grüße,
Katharina Keutgen
Sehr geehrte Damen und Herren,
im Zuge der Harmonisierung von UDI stellen wir uns für ein Medizinprodukt, welches eine PZN der Zuteilungsstelle IFA hat, die Frage, ob zwingend die NTIN des GS1 Standards für die Erzeugung des UDI DataMatrix Codes für den UDI-DI (01) Teil herangezogen werden muss.
Aktuell wird der PZN-Code 39 auf den Verpackungsebenen ausgegeben.
– Genügt es, nebst dem PZN-Code den UDI Data-Matrix Code mit der GTIN (01) zu implementieren und nicht die NTIN heranzuziehen?
Freundliche Grüße
Walter
Lieber Walter,
vielen Dank für Ihre Frage.
Die NTIN dient der Produktidentifikation von Arzneimitteln, nicht aber von Medizinprodukten.
Bei Medizinprodukten entscheiden Sie sich für eine Zuteilungsstelle und vergeben die UDI nach deren Vorgaben. Entscheiden Sie sich für die IFA, so können Sie die PZN in die UDI-DI (hier: PPN) integrieren.
Wählen Sie die GS1 als Zuteilungsstelle, so ist eine GTIN als UDI-DI zu verwenden. Die bestehende PZN kann dann weiterhin als solche auf der Kennzeichnung erscheinen.
Wichtig ist jedoch, dass die GTIN (UDI-DI) nicht ausreicht, sondern um die jeweilige UDI-PI ergänzt werden muss.
Sollten Sie Unterstützung bei der Vergabe Ihrer UDIs benötigen, melden Sie sich gern bei uns.
Herzliche Grüße,
Katharina Keutgen
Liebe Frau Keutgen,
vielen Dank für Ihre zügige Rückmeldung.
Das ASC Format wird von unserem QM und Software für die UDI Codegenerierung (UDI-DI und UDI-PI) nicht unterstützt. Es wird nur der GS1 Standard mit den entsprechenden Identifiern unterstützt. Es ist uns also nicht möglich, den Barcode mit den Datenstrings nach der IFA-Vorgabe zu codieren (lt. IFA, Beispiel: [)>069N1112345678421TABC12345D241231). Mehrere Code-Verifier können dieses Format zudem nicht lesen, da sie nur den GS1 DataMatrix Code unterstützen.
Daher nochmal die Nachfrage, welcher Teil als UDI-DI mit welchem Standard verwendet werden sollte.
– Kann dann die UDI-DI (PPN) mit dem GS1 Standard (14 Stellen für die UDI-DI) kombiniert werden oder sollte hier dann auf die GTIN mit dem GS1 Standard ausgewichen werden?
Freundliche Grüße
Dr. Walter
Lieber Dr. Walter,
zunächst einmal vielen Dank für Ihre Geduld.
Wenn Sie das ASC Format aus technischen Gründen nicht nutzen können, scheidet die Verwendung der IFA Standards für die UDI aus.
Entsprechend sollten Sie auf die vollständige UDI (UDI-DI + UDI-PI) im GS1 Format ausweichen.
Die UDI-DI wird im GS1 System als GTIN bezeichnet und durch eine UDI-PI ergänzt. Ihre PZN darf als solche dennoch weiterhin auf der Kennzeichnung erscheinen.
Sollte weiterhin Unsicherheit bestehen, melden Sie sich gern direkt bei uns, damit wir in einem persönlichen Gespräch auf Ihre Fragen eingehen können.
Herzliche Grüße,
Katharina Keutgen
Hallo liebes Johner Team
Wir verkaufen orthopädische Produkte ausschließlich über B2C E-commerce Kanäle und sind gerade dabei unsere Verpackungen für den Endkunden an die neuen Anforderungen anzupassen. Für die Abbildung der UDI-DI & UDI-PI Daten wollen wir das Format der GS1 DataMatrix nutzen (auf der Verpackung, am Produkt selbst ist kein Platz). Aktuell planen wir nur ein Minimum an vorgeschriebenen Daten in den Code zu laden (GTIN, LOT, EXP). Wir wissen, dass die Daten der Matrix (AIDC) auch ein HRI benötigen. Fraglich ist das korrekte Format in welchem die HRI Angaben angegeben werden dürfen. Wir sehen folgende Optionen:
1) DataMatrix ohne zugehörige GS1-codes (01,10,17) mit den zugehörigen Textangaben für GTIN, LOT, EXP. Dafür mit Darstellung derselben Informationen mittels der bewährten ISO Symbole und zugehörigen Angaben auf der selben Verpackungsseite + textliche Angabe der GTIN.
2) DataMatrix mit zugehörigen GS1 nummerischen Label-codes (01,10,17) für GTIN, LOT, EXP und deren Inhalt, aber ohne die dann redundante Erwähnung derselben Informationen mit den bewährten ISO Symbolen + Angaben an anderer Stelle.
3) Eine Art Mischform aus 1) & 2) wie beispielsweise hier: https://www.gs1.at/sites/default/files/2020-08/GS1-Datamatrix-Anwendungsbeispiel-Medikamente.jpg
Ich gehe mittlerweile davon aus, dass es hier keine konkrete Vorgabe gibt, sondern etwas Handlungsspielraum besteht. Stimmt meine Annahme?
Weiterhin frage ich mich ob entweder die Angabe „GTIN“ oder „UDI-DI“ als Textlabel korrekt ist.
Vielen Dank für Antwort und beste Grüße
Johannes
Lieber Johannes,
vielen Dank für die spannende Frage.
Die MDR schreibt zunächst vor, dass, sofern nicht aus Platzmangel unmöglich, beide Formate (AIDC + HRI) anzubringen sind.
Zudem definiert sie in Anhang VI, 2.1: „Die Anbringung der UDI ist eine zusätzliche Anforderung — sie ersetzt keine anderen Markierungs- oder Kennzeichnungsanforderungen gemäß Anhang I dieser Verordnung.“ Damit ist Ihre Option 2 ausgeschlossen.
Wie die AIDC und HRI darzustellen sind, regelt die jeweilige Zuteilungsstelle. Die Regeln für die GS1 HRI finden Sie im aktuellen GS1 Standard unter 4.14.1 (https://ref.gs1.org/standards/genspecs/).
Hier wird als bevorzugte Lösung die Angabe der HRI unter Verwendung der zugehörigen Application Identifier ((01), (17) etc.) definiert.
Es darf seitens der GS1 davon abgewichen und eine Mischform unter Verwendung von non-HRI Text wie in 4.14.1-3 und 4.14.1-4 beschrieben verwendet werden, falls andernfalls die HRI nicht gedruckt werden könnte. Da die MDR jedoch trotzdem die Verwendung international anerkannter Symbole auf der Kennzeichnung erwartet (Anhang I, 23.1.(h)), führt dies i.d.R. nicht zu einer Platzersparnis.
Falls Sie dies für Ihren konkreten Anwendungsfall besprechen möchten, melden Sie sich gern bei uns.
Herzliche Grüße,
Katharina Keutgen
Hallo liebes Johner Team,
Wir verkaufen als medizinisches Zubehör Saugleitungen für unsere EKG-Sauganlage. In unserer Gebrauchsanweisung empfehlen wir, die Elektroden (Saugdome und Elektrodenplatten) nach jeder Benutzung zu reinigen bzw. zu desinfizieren. Die Saugleitung als Ganzes muss jedoch nicht nach jeder Anwendung wiederaufbereitet werden.
Gilt die Saugleitung in diesem Fall dennoch als wiederverwendbares Produkt, das wiederaufbereitet wird, im Sinne der MDR, sodass der UDI-Träger dauerhaft direkt auf dem Produkt angebracht werden muss?
Oder kann argumentiert werden, dass nicht das gesamte Produkt wiederaufbereitet wird und daher die Anbringung des UDI-Trägers auf der Verpackung ausreichend ist?
Vielen Dank für Ihre Rückmeldung,
Mit freundlichen Grüssen
Philipp Bucher
Lieber Herr Bucher,
vielen Dank für Ihre Frage!
Die Antwort darauf ist abhängig von der Abgrenzung des Produkts. Wenn Saugleitung und Elektroden ein Produkt bilden, von dem eine Komponente nach der Anwendung aufbereitet werden muss, gilt die Anforderung der direkten Kennzeichnung für das Produkt in seiner Gesamtheit. Handelt es sich um zwei Produkte, von denen nur die Elektroden zwischen den Anwendung aufbereitet werden müssen, gilt die Anforderung für die Saugleitungen nicht. Hier dürfen Sie dann auf die erste Verpackungsebene ausweichen.
Falls wir uns diesen Fall gemeinsam anschauen sollen um die UDI-Anforderungen für Ihr Produkt zu definieren, melden Sie sich gern bei uns.
Herzliche Grüße,
Katharina Keutgen
Guten Tag Frau Keutgen,
Vielen Dank für Ihre Rückmeldung und die Informationen.
Freundliche Grüsse
Philipp Bucher
Liebes Johner-Team,
die Übergangsfrist für die Kennzeichnung von Klasse-I-Produkten mit UDI ist Ende Mai in Kraft getreten. Für wiederverwendbare Produkte ohne Verpackung, die nur in einem Versandkarton an den Kunden gehen, gilt jedoch eine Übergangsfrist bis 2027. Welche Übergangsfrist gilt dann? Muss das Produkt, obwohl es in der Direktkennzeichnung erst 2027 gekennzeichnet werden muss, jetzt schon gekennzeichnet werden, wenn nur Versandkartonagen vorhanden sind?
Vielen Dank im Voraus.
Liebe Frau Petzold,
es ist korrekt, dass ab Mai 2025 auch Klasse I Produkte eine UDI auf ihrer Kennzeichnung tragen müssen. Wenn das Produkt jedoch selbst keine kennzeichnungspflichtige Verpackung hat, und es sich um ein wiederverwendbares Produkt handelt, das direkt markiert werden muss, so gilt für die Direktmarkierung die Kennzeichnungspflicht der UDI ab Mai 2027 (MDR, Artikel 123(3)f) und g)).
Zusätzlicher Hinweis: Wenn ein Produkt immer im gleichen „Versandcontainer“ mit der festgelegten Stückzahl 1 verschickt wird, sollten Sie noch einmal neu bewerten, ob es sich dabei wirklich nur um einen Versandcontainer oder eine Produktverpackung handelt. Wir helfen Ihnen gern dabei.
Herzliche Grüße
Katharina Keutgen
Vielen lieben Dank, Ihre Antwort hilft mir sehr!
Liebes Johner-Team,
wir produzieren komplexe Test- und Trainingsgeräte für den Rehabereich. Derzeit noch als Legacy-Geräte und in der Umstellungsphase zur MDR.
Ich habe eine Frage zur Basis-UDI bei Produktänderungen:
Bei Produktänderungen erfordern Änderungen in Design und Herstellung die Vergabe einer neuen UDI-DI. Zum Beispiel ein neuer Zeichnungssatz mit geänderten mechanischen Einzelteilen oder geänderte Elektronikkomponenten, wobei Zweckbestimmung und Funktionalität des Produkts gleich bleiben.
Ergibt sich daraus auch zwingend eine neue Basis-UDI?
Viele Grüße und vielen Dank im Voraus.
Lieber Herr Dutschke,
vielen Dank für Ihre interessante Frage.
Nicht jede Änderung des Designs oder der Herstellung eines Produkts erfordert eine neue UDI-DI. Vielmehr müssen Sie sich entlang Ihres Änderungsmanagements die Frage stellen, ob eines der in MDR Anhang VI, Teil C, Abschnitt 3.9 genannten Attribute betroffen ist.
Wenn eine Änderung der UDI-DI erforderlich ist, bedeutet diese jedoch nicht zwangsläufig, dass sich die Basic-UDI-DI des Produkts ändert. Dies wäre der Fall, wenn sich die Zweckbestimmung oder die Risikoklasse ändert oder sich das Design und die Herstellung in einer Weise ändern, dass das Produkt nicht mehr als „im Wesentlichen gleich“ zu anderen unter dieser Basic-UDI-DI betrachtet werden kann.
Sollten Sie Unterstützung bei einer spezifischen Änderungsbewertung benötigen, melden Sie sich gern bei uns.
Herzliche Grüße,
Katharina Keutgen