
Die MDR legt in den Artikeln 120 bis 123 die Übergangsbestimmungen einschließlich der Übergangsfristen fest. Allerdings sind diese Übergangsbestimmungen und Übergangsfristen sehr komplex formuliert. Daher laufen die Hersteller Gefahr, sie falsch zu verstehen und regulatorische Anforderungen nicht zu erfüllen oder unnötige Aufwände zu betreiben.
Ein Ablaufdiagramm in Kapitel 2 dieses Artikels fasst die regulatorischen Anforderungen zusammen und schafft Klarheit. Eine Zusammenfassung steht auch als kostenloser Download zur Verfügung.
Dieser Beitrag berücksichtigt die im Februar 2023 vom EU-Parlament verabschiedete weitere Verschiebung der MDR-Übergangfristen sowie die Änderungsverordnung 2024/1860 zum „gradual roll-out“ von EUDAMED vom Juni 2024.
1. MDR-Übergangfristen: Einführung
Die Diskussion um die Übergangsfristen beschränkt sich meist auf die Frage, wie lange ein Produkt in den Verkehr gebracht werden darf. Dabei kennt die MDR eine Vielzahl von Übergangsfristen.
Hersteller sollten bei den Übergangsfristen unter anderem die folgenden Aspekte unterscheiden:
- Inverkehrbringung von Legacy-Produkten
- Bereitstellung und Inbetriebnahme von Legacy-Produkten
- OEM-PLM-Konstrukte
- „Verantwortliche Person“
- Post-Market-Surveillance
- Vigilanz
- QM-System
- UDI
- Klinische Prüfungen
Dieser Artikel verschafft einen Überblick über diese Übergangsfristen.
Unser Regulatory-Affairs-Experte Luca Salvatore schafft in dieser Podcast-Episode Klarheit, zeigt aber auch, welche Fragen noch nicht beantwortet sind.
Diese und weitere Podcast-Episoden finden Sie auch hier.

2. MDR-Übergangsfristen im Überblick
a) Zusammenfassung
| Aspekt | Übergangsfrist | Kommentar |
| Inverkehrbringung von Legacy-Produkten | siehe Ablaufdiagramm (Abb. 1) | Es geht um die erstmalige Bereitstellung |
| Weitere Bereitstellung und Inbetriebnahme von Legacy-Produkten | — | Die Einschränkungen („Sell-Off-Regelung“) sind mit der Änderungsverordnung weggefallen. |
| OEM-PLM-Konstrukte | Für diese Produkte gelten die gleichen Übergangsfristen. | Dem PLM muss die Technische Dokumentation vorliegen, um die Forderungen der MDR an die Post-Market-Surveillance sowie die Vigilanz erfüllen zu können. |
| „Verantwortliche Person“ / PRRC | — | Wird nicht benötigt für Hersteller von Legacy-Produkten. Lesen Sie hierzu den ausführlichen Absatz „Übergangsfristen“ im Artikel zur PRRC. |
| Post-Market-Surveillance | 26.05.2021 | Mit Einschränkungen bezüglich der EUDAMED; keine Einschränkung bei Überwachung und Berichten (z.B. PSUR) |
| Vigilanz | 26.05.2021 | Mit Einschränkungen bezüglich der EUDAMED |
| QM-System | 26.05.2021 (MDR-Produkte) 26.05.2024 (Legacy-Produkte) | Die Änderungsverordnung verlangt ein MDR-Artikel-10-konformes QM-System für Hersteller von Legacy-Produkten. |
| UDI | siehe Tabelle unten | |
| Klinische Prüfungen | 26.05.2021 | Eingeleitete Prüfungen dürfen fortgeführt werden, allerdings gelten neue Meldepflichten. |
Der Artikel 10 der MDR findet bei Produkten, die von der Übergangsfrist profitieren, keine Anwendung. Davon explizit ausgenommen sind die o.g. Anforderungen u.a. zur Post-Market-Surveillance und zur Vigilanz.
Die Zusammenfassung finden Sie in diesem Merkblatt zum Download:
Im August 2023 hat auch die EU auf ihrer Webseite ein Ablaufdiagrammpubliziert.
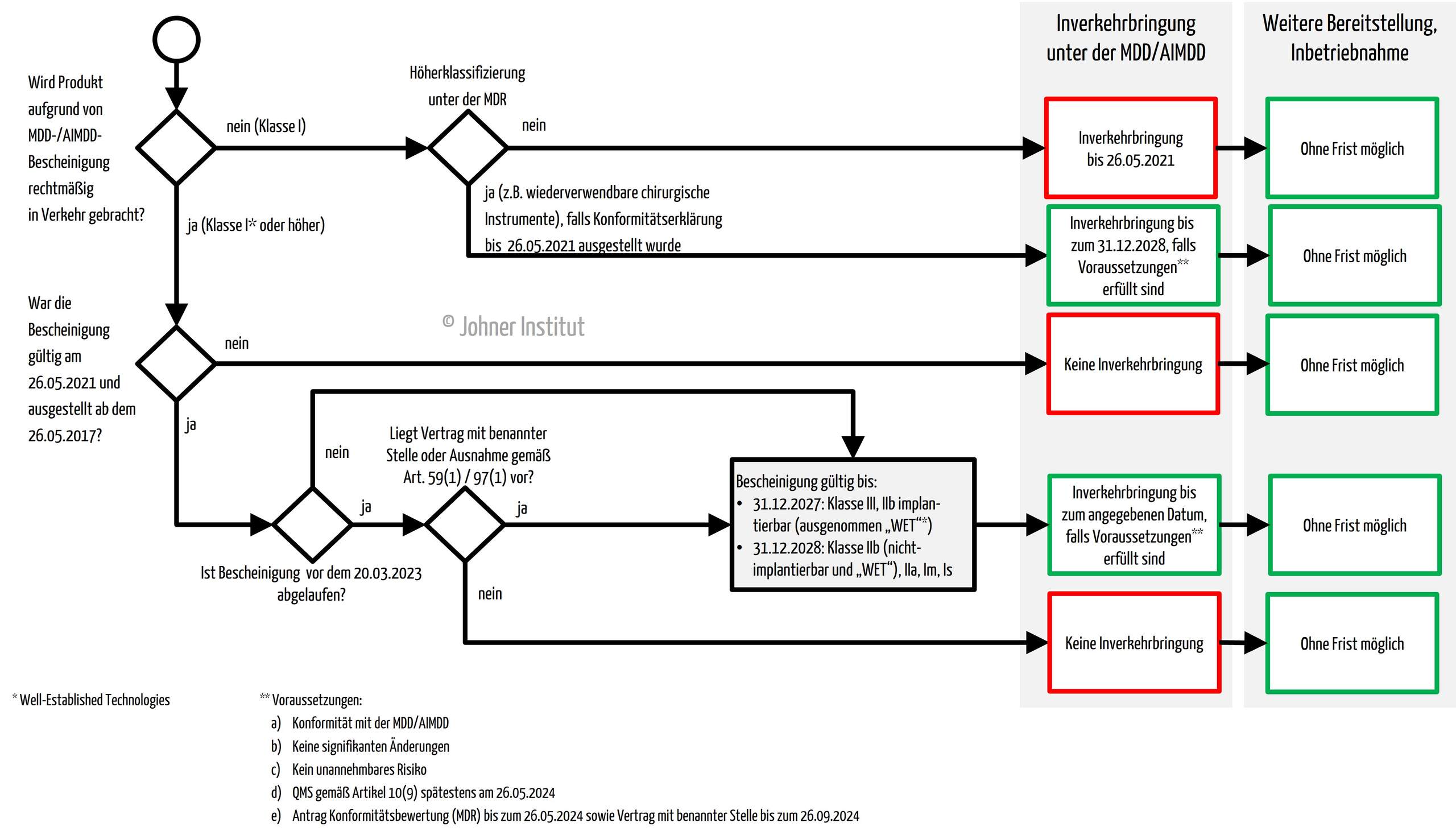
b) Inverkehrbringung, Bereitstellung und Inbetriebnahme
Wie lange Hersteller ihre bestehenden Produkte unter den alten Richtlinien (MDD/AIMDD) noch in den Verkehr bringen, bereitstellen und in Betrieb nehmen (lassen) dürfen, hängt u.a. von der Klasse der Produkte und der Gültigkeit möglicher Bescheinigungen ab.
Falls Sie noch Fragen zum Umstieg auf die MDR haben, nutzen Sie gerne das kostenlose Micro-Consulting des Johner Instituts.
c) EUDAMED-Pflichten, z. B. Registrierung
| Erste 6 Monate nach Veröffentlichung der Funktionsfähigkeit des jeweiligen EUDAMED-Moduls | Ab 6 Monate nach Veröffentlichung | Bis 12 Monaten nach Veröffentlichung | Bis 18 Monate nach Veröffentlichung | ||
| MDR-Produkte und Legacy-Produkte | Keine Anforderungen | Registrierung der Akteure, Anmeldungen zu klinischen Prüfungen, Vigilanz, PMS, Registrierung neuer MDR-Produkte und Bescheinigungen/Zertifikate durch Benannte Stellen sowie Upload der SSCPs (s. Artikel 123(3)) | Abschluss der Registrierung von Legacy-Produkten und MDR-Produkten, welche bereits vor den 6 Monaten nach Veröffentlichung in Verkehr gebracht wurden | Abschluss der Registrierung der Bescheinigungen/Zertifikate durch Benannte Stellen sowie Uploads der SSCPs für Produkte, welche bereits vor den 6 Monaten nach Veröffentlichung in Verkehr gebracht wurden |
Für Wirtschaftsakteure mit Sitz in Deutschland gelten gemäß dieser Veröffentlichung im Bundesanzeiger gesonderte Übergangsbestimmungen. Demnach müssen sich Hersteller und deren Bevollmächtigte bereits seit dem 26.05.2021 direkt in EUDAMED registrieren und nicht in der nationalen Datenbank des BfArMs, dem DMIDS. Gleiches gilt für Importeure von MDR-konformen Medizinprodukten.
d) Übergangsfristen für die UDI
Der Zeitpunkt, zu dem der UDI-Träger aufgebracht werden muss, hängt von der Klasse des Produkts ab:
| Klasse | Zeitpunkt |
| I | 26.05.2025 |
| IIa | 26.05.2023 |
| IIb | 26.05.2023 |
| III und implantierbare Produkte | 26.05.2021 |
Bei wiederverwendbaren Produkten, bei denen der UDI-Träger auf dem Produkt selbst zu platzieren ist, gewährt die MDR zwei zusätzliche Jahre.
Diese Übergangsfristen bezüglich UDI gelten jedoch nicht für Legacy-Produkte, also Produkte „which are compliant with the Medical Device Directives (MDD and AIMDD) and placed on the market after the application date of the Regulations“. Hierfür gibt es zwei weitere Dokumente zu beachten:
Im FAQ schreibt die EU:
In order to facilitate the transition to the new system, the new Regulations give manufacturers the possibility to place products on the market after the general application dates of the new
FAQ der EU
Regulations (and until 26 May 2024 at the latest) by virtue of valid Directive certificates. These legacy devices are not subject to UDI obligations but they should be registered in the Eudamed database. Timelines for registration as described under question 6 also apply to these products. More information on the operational aspects of the registration of legacy devices is available at the MDCG 2019-5 guidance document.
e) Übergangsfrist für die Verantwortliche Person
Wenn Hersteller keine Produkte unter der MDR in den Verkehr bringen, sondern nur „Legacy-Produkte“ nach Art 120 (3), so gilt nach MDCG 2021-25 der Artikel 15 der MDR noch nicht. Dies bedeutet, dass diese Hersteller noch keine Verantwortliche Person benennen müssen.
Bisher hatten wir und auch viele andere dies auf andere Art interpretiert. Der Grund dafür war, dass die Pflichten für die Post-Market Surveillance und die Vigilanz von allen Herstellern ab dem 26.05.2021 erfüllt sein müssen. Die Verantwortliche Person ist für einen wirksamen Post-Market Surveillance- und Vigilanz-Prozess verantwortlich. Die Leitlinie MDCG 2021-25 betrachtet diesen Aspekt auch, kommt aber zu dem Schluss, dass Artikel 15 noch nicht von Herstellern von Legacy-Produkten anwendbar ist.
Alle Hersteller müssen in jedem Fall wirksame Prozesse für die Post-Market Surveillance und Vigilanz implementieren, nur die Überwachung durch die Verantwortliche Person ist noch nicht zwingend vorgeschrieben für Hersteller von Legacy-Produkten.
Details finden Sie auch im Artikel zur PRRC im Abschnitt „Übergangsfristen“.
3. Die MDR-Artikel 120 bis 123
a) MDR Artikel 120 Absatz 2
Originaltext
Der Artikel 120 trägt den Titel „Übergangsbestimmungen“. Er nennt darin auch die Übergangsfristen. Besonders relevant ist der zweite Absatz:
Bescheinigungen, die von Benannten Stellen vor dem 25. Mai 2017 gemäß den Richtlinien 90/385/EWG und 93/42/EWG ausgestellt wurden, bleiben bis zu dem in der Bescheinigung angegebenen Zeitpunkt gültig, außer im Fall von Bescheinigungen gemäß Anhang 4 der Richtlinie 90/385/EWG bzw. gemäß Anhang IV der Richtlinie 93/42/EWG, die spätestens am 27. Mai 2022 ihre Gültigkeit verlieren.
Bescheinigungen, die von Benannten Stellen ab dem 25. Mai 2017 gemäß den Richtlinien 90/385/EWG und 93/42/EWG ausgestellt wurden, die am 26. Mai 2021 noch gültig waren und die anschließend nicht zurückgezogen wurden, behalten ihre Gültigkeit nach dem Ende des auf der Bescheinigung angegebenen Zeitraums bis zu dem Datum, das in Absatz 3a des vorliegenden Artikels für die jeweilige Risikoklasse der Produkte festgelegt ist. Bescheinigungen, die von Benannten Stellen im Einklang mit diesen Richtlinien ab dem 25. Mai 2017 ausgestellt wurden, am 26. Mai 2021 noch gültig waren und vor dem 20. März 2023 abgelaufen sind, gelten nur dann bis zu den in Absatz 3a des vorliegenden Artikels festgelegten Zeitpunkten als gültig, wenn eine der folgenden Bedingungen erfüllt ist:a) Vor Ablauf der Bescheinigung haben der Hersteller und eine Benannte Stelle eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 der vorliegenden Verordnung über die Konformitätsbewertung des Produkts, für das die abgelaufene Bescheinigung gilt, oder eines Produkts, das dazu bestimmt ist, dieses Produkt zu ersetzen, unterzeichnet;
b) eine zuständige Behörde eines Mitgliedstaats hat eine Ausnahme von dem anwendbaren Konformitätsbewertungsverfahren gemäß Artikel 59 Absatz 1 der vorliegenden Verordnung gewährt oder den Hersteller gemäß Artikel 97 Absatz 1 der vorliegenden Verordnung aufgefordert, das anzuwendende Konformitätsbewertungsverfahren durchzuführen
MDR Artikel 120 (2)
Interpretation dieses Textes
Falls der Hersteller ein Zertifikat hat, welches unter den Richtlinien vor dem 26.05.2017 ausgestellt wurde, bleibt dieses bis zum angegebenen Datum gültig. Ausgenommen hiervon sind Zertifikate gemäß Anhang IV der Richtlinien (Batch-Zertifizierung), die bis zum maximal 27.05.2022 gültig waren.
Wenn das Zertifikat der Benannten Stelle hingegen am oder nach dem 26.05.2017 ausgestellt wurde und am 26.05.2021 noch gültig war, behält es seine Gültigkeit bis zu den genannten Fristen (siehe Absatz 3(a)). Wenn dieses Zertifikat allerdings vor Inkrafttreten der Änderungsverordnung, d.h. vor dem 20. März 2023, abgelaufen ist, gilt die verlängerte Gültigkeit nur, falls:
- der Hersteller VOR Ablauf des Zertifikats einen Vertrag mit einer Benannten Stelle zur MDR-Zertifizierung abgeschlossen hat (gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 MDR) und dieser unterzeichnet wurde
- ODER dem Hersteller eine Sondergenehmigung durch die nationale Behörde (in Deutschland das BfArM) gemäß Artikel 59 Absatz 1 MDR erteilt wurde
- ODER eine weitere Inverkehrbringung durch die zuständige Behörde (in Deutschland sind das die Landesbehörden) auch ohne gültiges Zertifikat unter Fristsetzung gestattet wurde (Artikel 97 Absatz 1 MDR).
b) MDR Artikel 120 Absatz (3) – (3b)
Originaltext
(3) Abweichend von Artikel 5 und unter der Voraussetzung, dass die in Absatz 3c genannten Bedingungen erfüllt sind, dürfen die in den Absätzen 3a und 3b genannten Produkte bis zu den in diesen Absätzen genannten Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden.
(3a) Produkte, für die gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG eine aufgrund von Absatz 2 des vorliegenden Artikels gültige Bescheinigung ausgestellt wurde, dürfen bis zu den folgenden Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden:
a) 31. Dezember 2027 für alle Produkte der Klasse III und für implantierbare Produkte der Klasse IIb, ausgenommen Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Zahn- bzw. Knochenplatten, Drähte, Stifte, Klemmen und Verbindungsstücke;
b) 31. Dezember 2028 für andere Produkte der Klasse IIb als die unter Buchstabe a des vorliegenden Absatzes genannten, für Produkte der Klasse IIa und für Produkte der Klasse I, die in sterilem Zustand in Verkehr gebracht werden, oder mit Messfunktion.
(3b) Produkte, für die das Konformitätsbewertungsverfahren gemäß der Richtlinie 93/42/EWG nicht die Mitwirkung einer Benannten Stelle erforderte, für die die Konformitätserklärung vor dem 26. Mai 2021 ausgestellt wurde und für die das Konformitätsbewertungsverfahren gemäß der vorliegenden Verordnung die Mitwirkung einer Benannten Stelle erfordert, dürfen bis zum 31. Dezember 2028 in Verkehr gebracht oder in Betrieb genommen werden.
MDR Artikel 120 (3)
Interpretation dieses Textes
Der dritte Absatz des Artikel 120 (a) und (b) lässt sich wie folgt deuten:
Legacy-Produkte mit gültigem Richtlinien-Zertifikat dürfen abhängig von der Risikoklasse bis zu den genannten Fristen in Verkehr gebracht werden.
- Klasse III, implantierbare Produkte der Klasse IIb (ausgenommen die Produkte mit „Well-Established-Technology“): 31.12.2027
- Sonstige Klasse IIb, IIa, Is und Im Produkte: 31.12.2028
- Produkte der Klasse I unter den Richtlinien, welche unter der MDR höher klassifiziert wurden und deshalb eine Benannte Stelle im Konformitätsbewertungsverfahren einbezogen werden muss (z. B. wiederverwendbare chirurgische Instrumente, viele Software-Produkte): 31.12.2028
Diese erweiterten Übergangsfristen gelten allerdings nur unter bestimmten Bedingungen. Diese sind in Absatz 3 (c) genannt.
c) MDR Artikel 120 Absatz (3c) – (3d)
Originaltext
(3c) Produkte gemäß den Absätzen 3a und 3b des vorliegenden Artikels dürfen nur bis zu den in diesen Absätzen genannten Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden, wenn die folgenden Bedingungen erfüllt sind:
a) Die Produkte entsprechen weiterhin der Richtlinie 90/385/EWG bzw. der Richtlinie 93/42/EWG;
b) es liegen keine wesentlichen Veränderungen der Auslegung und der Zweckbestimmung vor;
c) die Produkte stellen kein unannehmbares Risiko für die Gesundheit oder Sicherheit der Patienten, Anwender oder anderer Personen oder für andere Aspekte des Schutzes der öffentlichen Gesundheit dar;
d) der Hersteller hat spätestens am 26. Mai 2024 ein Qualitätsmanagementsystem gemäß Artikel 10 Absatz 9 eingerichtet;
e) der Hersteller oder der Bevollmächtigte hat spätestens am 26. Mai 2024 bei einer Benannten Stelle einen förmlichen Antrag gemäß Anhang VII Abschnitt 4.3 Unterabsatz 1 auf Konformitätsbewertung für ein in dem Absatz 3a oder 3b genanntes Produkt oder für ein Produkt, das dazu bestimmt ist, dieses Produkt zu ersetzen, gestellt, und die Benannte Stelle und der Hersteller haben spätestens am 26. September 2024 eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 unterzeichnet.
(3d) Abweichend von Absatz 3 des vorliegenden Artikels gelten die Anforderungen dieser Verordnung an die Überwachung nach dem Inverkehrbringen, die Marktüberwachung, die Vigilanz, die Registrierung von Wirtschaftsakteuren und von Produkten für Produkte gemäß den Absätzen 3a und 3b anstelle der entsprechenden Anforderungen der Richtlinien 90/385/EWG und 93/42/EWG.
MDR Artikel 120 (3)
Interpretation dieses Textes
Der dritte Absatz des Artikel 120 (c) beschreibt die Bedingungen, welche für die Anwendung der erweiterten Übergangsfristen erfüllt sein müssen.
Voraussetzung 1
Das Produkt entspricht weiterhin den Anforderungen der MDD bzw. AIMDD. Das klingt logisch und sollte durch die Überwachung der Benannten Stellen und Behörden auch sichergestellt werden.
Voraussetzung 2
Der Hersteller hat keine wesentlichen Änderungen an dem Produkt oder an seiner Zweckbestimmung vorgenommen. Was man genau darunter versteht, erklärt das MDCG im Dokument MDCG 2020-03.
Besonderen Anlass zu Diskussionen bietet die Frage, wie groß Änderungen sein dürfen, damit sie nicht als „wesentlich“ betrachtet werden müssen. Beachten Sie hierzu die Artikel zu Design Changes und Software Changes.
Voraussetzung 3
Dass die Produkte kein unannehmbares Risiko für die Sicherheit und Gesundheit darstellen, sollte durch Einhaltung der Richtlinien sichergestellt sein. Dennoch ist dies eine Überwachungsaufgabe der Benannten Stellen und Überwachungsbehörden. Das stellt auch das MDCG im Guidance-Dokument MDCG 2022-04 klar. Dieses enthält Empfehlungen zu den Überwachungstätigkeiten von Legacy-Produkten während der Übergangsfristen. Es heißt dort:
„In cases where the audit activities reveal a major non-conformity, which may present an
unacceptable risk to the health or safety of patients, users or other persons, the notified body
needs to take action, i.e. suspend, restrict or withdraw the certificate, and inform the relevant
competent authority.“
Voraussetzung 4
Die Forderungen nach einem vollständigen QM-System gemäß Artikel 10 Absatz 9 wurde durch die Änderungsverordnung ergänzt. Somit reicht es nicht mehr aus, nur die Prozesse für die Überwachung nach Inverkehrbringen und Vigilanz gemäß MDR zu implementieren. Letztere müssen bereits seit dem 26.05.2021 MDR-konform implementiert sein (siehe 3(d)).
Voraussetzung 5
Auch diese Bedingung ist nicht zu unterschätzen und darf nicht vernachlässigt werden. Hersteller müssen bis zum 26.05.2024 einen Antrag auf MDR-Zertifizierung bei einer Benannten Stelle gestellt haben. Unklar ist, ob bis dahin der Antrag nur gestellt sein muss oder ob dieser durch die Benannte Stelle auch positiv geprüft worden sein muss. Dies lässt Raum für Interpretation und Meinungsverschiedenheiten. Wir gehen aktuell davon aus, dass dieses Datum allein die Antragstellung betrifft, aber nicht die Prüfung durch die Benannte Stelle. Interessant ist die Möglichkeit, die MDR-Zertifizierung nicht für das eigentliche Legacy-Produkt, sondern für ein „ersetzendes Produkt“ zu beantragen. Unklar ist dabei allerdings, welche Voraussetzungen dafür gelten. Muss das ersetzende Produkt äquivalent sein oder darf unterschiedliche Technologie eingesetzt werden? Auch dies lässt leider Raum für Interpretation. Darüber sollte die MDCG schnellstmöglich Klarheit schaffen.
Zusätzlich zur Antragsstellung muss bis zum 26.09.2024 ein unterschriebener Vertrag mit einer Benannten Stelle zur MDR-Zertifizierung vorliegen.
d) MDR Artikel 120 Absatz (3e)
Originaltext
(3e) Unbeschadet des Kapitels IV und des Absatzes 1 des vorliegenden Artikels bleibt die Benannte Stelle, die die Bescheinigung gemäß Absatz 3a des vorliegenden Artikels ausgestellt hat, für die angemessene Überwachung aller geltenden Anforderungen an die von ihr zertifizierten Produkte verantwortlich, es sei denn, der Hersteller ist mit einer Benannten Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, übereingekommen, dass sie eine derartige Überwachung durchführt.
Spätestens am 26. September 2024 ist die Benannte Stelle, die die schriftliche Vereinbarung gemäß Absatz 3c Buchstabe e des vorliegenden Artikels unterzeichnet hat, für die Überwachung der unter die schriftliche Vereinbarung fallenden Produkte verantwortlich. Betrifft die schriftliche Vereinbarung ein Produkt, das dazu bestimmt ist, ein Produkt zu ersetzen, für das eine Bescheinigung gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG ausgestellt wurde, so wird die Überwachung in Bezug auf das ersetzte Produkt durchgeführt.
Die Vorkehrungen für die Übertragung der Überwachung von der Benannten Stelle, die die Bescheinigung ausgestellt hat, auf die Benannte Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, werden in einer Vereinbarung zwischen dem Hersteller und der Benannten Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, und, soweit durchführbar, der Benannten Stelle, die die Bescheinigung ausgestellt hat, klar geregelt. Die Benannte Stelle, deren Benennung gemäß Artikel 42 erfolgt ist, ist nicht für Konformitätsbewertungstätigkeiten verantwortlich, die von der Benannten Stelle, die die Bescheinigung ausgestellt hat, durchgeführt werden.
MDR Artikel 120 (3)
Interpretation dieses Textes
Im Absatz 3(e) werden die Überwachungstätigkeiten und -zuständigkeiten der Benannten Stellen geregelt. Im Regelfall ist demnach die Benannte Stelle, die das Richtlinien-Zertifikat ausgestellt hat, für die weitere Überwachung innerhalb der Übergangsfristen zuständig. Allerdings dürfen Hersteller ihre Benannte Stelle wechseln, welche dann die Überwachungstätigkeiten übernehmen. Dies kann z. B. notwendig sein, wenn die aktuelle Benannte Stelle (noch) nicht unter der MDR benannt wurde. Spätestens zum 26.09.2024 muss dann eine Benannte Stelle, designiert unter der MDR, die Überwachungsverantwortung innehaben.
e) MDR Artikel 120 Absatz (3f)
Originaltext
(3f) Abweichend von Artikel 5 dürfen implantierbare Sonderanfertigungen der Klasse III bis zum 26. Mai 2026 ohne eine von einer Benannten Stelle nach dem Konformitätsbewertungsverfahren gemäß Artikel 52 Absatz 8 Unterabsatz 2 ausgestellte Bescheinigung in Verkehr gebracht oder in Betrieb genommen werden, sofern der Hersteller oder der Bevollmächtigte spätestens am 26. Mai 2024 bei einer Benannten Stelle einen förmlichen Antrag gemäß Anhang VII Abschnitt 4.3 Unterabsatz 1 auf Konformitätsbewertung gestellt hat und die Benannte Stelle und der Hersteller spätestens am 26. September 2024 eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 unterzeichnet haben.
MDR Artikel 120 (3)
Interpretation dieses Textes
Absatz 3(f) wurde mit der Änderungsverordnung ergänzt. Nun gewährt die MDR auch für implantierbare Sonderanfertigungen der Klasse III eine erweiterte Übergangsfrist bis zum 26.05.2026. Die Bedingungen sind in diesem Fall eingeschränkt. So verlangt die MDR „nur“ einen förmlichen Antrag zur Konformitätsbewertung bis zum 26.05.2024 und den unterzeichneten Vertrag bis zum 26.09.2024.
f) MDR Artikel 120 Absatz (4)
Originaltext
Der vierte Absatz ist vergleichsweise gut verständlich:
(4) Produkte, die vor dem 26. Mai 2021 gemäß den Richtlinien 90/385/EWG und 93/42/EWG rechtmäßig in Verkehr gebracht wurden, und Produkte, die ab dem 26. Mai 2021 gemäß den Absätzen 3, 3a, 3b und 3f in Verkehr gebracht wurden, dürfen weiter auf dem Markt bereitgestellt oder in Betrieb genommen werden.
MDR Artikel 120(4)
Interpretation des Textes
Damit ist der Abverkauf zeitlich nicht mehr limitiert. Diese Regelung soll sicherstellen, dass „sichere und wichtige Medizinprodukte, die bereits in Verkehr gebracht wurden, den Gesundheitssystemen sowie den Patientinnen und Patienten, die darauf angewiesen sind, weiterhin zur Verfügung stehen.“
Zu unterscheiden sind hierbei die Begriffe „Bereitstellung“ und „Inverkehrbringung“. Eine Definition der Begriffe finden Sie in unserem Artikel zur Inverkehrbringung.
g) MDR Artikel 122 – Gedankenstrich 1 und 2
Artikel 122 erzeugt bei vielen Lesern nur Kopfschütteln. Wer soll das beim ersten Lesen verstehen?
„- Artikel 8 und 10, Artikel 10b Absatz 1 Buchstaben b und c, Artikel 10b Absätze 2 und 3 der Richtlinie 90/385/EWG sowie Artikel 10, Artikel 14a Absatz 1 Buchstaben c und d, Artikel 14a Absätze 2 und 3 und Artikel 15 der Richtlinie 93/42/EWG und den in den entsprechenden Anhängen der genannten Richtlinien festgelegten Pflichten zur Vigilanz und zu den klinischen Prüfungen, die gegebenenfalls mit Wirkung von dem in Artikel 123 Absatz 3 Buchstabe d dieser Verordnung genannten Datum in Bezug auf die Anwendung der Pflichten und Anforderungen im Zusammenhang mit den in Artikel 33 Absatz 2 Buchstaben e und f genannten elektronischen Systemen aufgehoben werden,
– Artikel 10a, Artikel 10b Absatz 1 Buchstabe a und Artikel 11 Absatz 5 der Richtlinie 90/385/EWG sowie Artikel 14 Absätze 1 und 2, Artikel 14a Absatz 1 Buchstaben a und b und Artikel 16 Absatz 5 der Richtlinie 93/42/EWG und den in den entsprechenden Anhängen dieser Richtlinien festgelegten Pflichten zur Registrierung von Produkten und Wirtschaftsakteuren und zur Meldung von Bescheinigungen, die gegebenenfalls mit Wirkung von dem in Artikel 123 Absatz 3 Buchstabe d dieser Verordnung genannten Datum in Bezug auf die Anwendung der Pflichten und Anforderungen im Zusammenhang mit den jeweiligen in Artikel 33 Absatz 2 Buchstaben a bis d der vorliegenden Verordnung genannten elektronischen Systemen aufgehoben werden,“
MDR Artikel 122, Gedankenstrich 1 und 2
Interpretation des Textes
Die unter den beiden Gedankenstrichen genannten Anforderungen der alten Richtlinien betreffen folgende Verpflichtungen:
- Meldung von Vorkommnissen
- Registrierung der Wirtschaftsakteure
- Produktregistrierung
- Bescheinigungen der Benannten Stellen
- Meldung/Genehmigung von klinischen Prüfungen
- Melde- und Beobachtungsverfahren der Behörden
Artikel 122 sagt somit aus, dass die genannten Verpflichtungen aus den alten Richtlinien weiterhin gültig sind, bis die entsprechenden Module in EUDAMED funktionsfähig sind und dies von der Kommission verkündet wurde (siehe Artikel 123). Somit sind z.B. Registrierungen von Akteuren und Produkten weiterhin national durchzuführen.
Für alle, die Artikel 122 noch genauer verstehen wollen, hier wichtige Referenzen:
- AIMDD (90/385/EWG)
- Artikel 8: Informationen über Vorkommnisse nach dem Inverkehrbringen
- Artikel 10a: Registrierung des Herstellers
- Artikel 10b Absatz 1 Buchstabe a: Bescheinigungen in Datenbank
- Artikel 10b Absatz 1 Buchstabe b und c: Bescheinigungen in Datenbank (Angaben aus dem Beobachtungs- und Meldeverfahren, Angaben zu klinischen Prüfungen)
- Artikel 11 Absatz 5: von Benannten Stellen ausgestellte und zurückgezogene Bescheinigungen
- MDD (93/42/EWG)
- Artikel 10: Informationen über Vorkommnisse nach dem Inverkehrbringen
- Artikel 14 Absätze 1 und 2: Meldung der für das Inverkehrbringen verantwortlichen Personen
- Artikel 14a Absatz 1 Buchstaben a und b: Europäische Datenbank (Hersteller, Bevollmächtigte, Bescheinigungen)
- Artikel 14a Absatz 1 Buchstaben c und d: Europäische Datenbank (Angaben aus dem Beobachtungs- und Meldeverfahren, Angaben zu klinischen Prüfungen)
- Artikel 15: Klinische Prüfungen
- Artikel 16 Absatz 5: von Benannten Stellen ausgestellte und zurückgezogene Bescheinigungen
h) MDR Artikel 123 („Inkrafttreten und Geltungsbeginn“)
Der Artikel 123 nennt weitere Fristen, die die Wirtschaftsakteure betreffen, z. B.:
- Absatz 3 Buchstabe d: Im Kontext der EUDAMED sind hier alle Pflichten aufgelistet, die spätestens sechs Monate nach dem Zeitpunkt erfüllt werden müssen, an dem das jeweilige EUDAMED-Modul als funktionsfähig erklärt und dies im Amtsblatt der EU verkündet wird.
- Absatz 3 Buchstabe e: Für die Registrierung von Legacy-Produkten und MDR-Produkten, welche bereits vor den unter d genannten 6 Monaten nach Verkündung in Verkehr gebracht wurden, haben Hersteller 12 Monate Zeit.
- Absatz 3 Buchstabe ea und eb: Für die unter e genannten Produkte, haben Benannte Stellen 18 Monate Zeit für die Hinterlegung von Bescheinigungen und Hochladen von SSCPs.
- Absatz 3 Buchstabe ec: Falls Hersteller eine Meldung abgeben müssen (z.B. schwerwiegendes Vorkommnis, Trend, Sicherheitskorrekturmaßnahme im Feld) und die entsprechenden Produkte noch nicht in EUDAMED registriert sind, muss dies unmittelbar erfolgen. Dies gilt nur für MDR-Produkte, nicht für Legacy-Produkte.
- Absatz 3 Buchstabe f: Der UDI-Träger muss bei implantierbaren Produkten sowie Produkten der Klasse III ab dem 26.05.2021 angebracht werden, bei Produkten der Klasse IIa und IIb ab dem 26.05.2023 und für Produkte der Klasse I ab dem 26.05.2025. Für die Vergabe der UDI gilt diese Ausnahmeregelung nicht.
- Absatz 3 Buchstabe g: Für wiederverwendbare Produkte, für die ein „direct marking“ notwendig ist, verlängern sich die unter f genannten Fristen und jeweils 2 Jahre.
4. Sonstige Anforderungen
Folgende Dokumente sind regulatorisch relevant und sollten beachtet werden:
- Application of transitional provisions concerning validity of certificates issued in accordance to Directives 90/385/EEC and 93/42/EEC
- MDCG 2019-15 GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES
- MDCG 2022-4 Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR with regard to devices covered by certificates according to the MDD or the AIMDD
5. Fazit und Zusammenfassung
Der Druck war groß, denn die Beschwerden über die negativen Auswirkungen der MDR konnte die EU-Kommission nicht ignorieren. Daher hat sie mehrfach die Übergangsfristen verschoben. Die Probleme sind dadurch aber nur teilweise behoben. Viele Schäden sind bereits eingetreten:
- Hersteller haben aufgegeben.
- Produkte wurden vom Markt genommen.
- Neue Produkte werden zuerst in den USA zugelassen.
- Die Entwicklung neuer Produkte wurde aufgegeben oder zurückgestellt.
Dennoch war die Verlängerung der Übergangsfristen ein wichtiger Schritt. Die mehrfach überarbeiteten Vorgaben sind allerdings inzwischen schwer zu überblicken und lassen noch Fragen offen.
Änderungshistorie
- 2024-10-11: Überarbeitung aufgrund der Änderungsverordnung zum gradual roll-out von EUDAMED
- 2024-06-07: Kapitel 3 c): Referenz und Zitat aus dem MDCG 2022-4 aktualisiert
- 2023-08-25: In Kapitel 2 a) Flowchart der EU ergänzt
- 2023-03-20: Datum des Inkrafttretens der Änderungsverordnung ergänzt
- 2023-02-24: Artikel komplett überarbeitet nach Verabschiedung durch Parlament
- 2023-02-16: Hinweis, dass Vorschlag dem Parlament vorliegt, ergänzt
- 2023-01-12: Ausblick sowie Link zur Kommentierungsmöglichkeit ergänzt
- 2023-01-10: Hinweis zu geplanten Änderungen der Übergangsfristen ergänzt
- 2022-03-04: Hinweis zu MDCG 2022-4 ergänzt
- 2021-06-14: Link zum überarbeiteten Artikel zur PRRC eingefügt, der die diesbezüglichen Übergangsfristen thematisiert
- 2021-05-17: In Kapitel 4d) die Hinweise zu den UDI-Anforderungen bei Legacy-Devices ergänzt



„Allerdings muss die Bescheinigung der Benannten Stelle am oder nach dem 25.05.2017 ausgestellt sein. Zudem endet ihre Gültigkeit spätestens am 27.05.2022, selbst wenn das Gültigkeitsdatum auf der Bescheinigung nach dem 27.05.2022 liegen sollte.“
Ich glaube hier liegt ein Tippfehler vor – wenn die Bescheinigung nach dem 25.05.2017 ausgestellt wurde, dann ist diese doch höchstens bis am 27. Mai 2024 gültig (und nicht wie geschrieben 2022)?! Steht auch so im 2. Absatz von 120.2.
Liebe Grüsse
Sie haben absolut Recht. Danke, lieber Herr Wettstein!
Ich habe das korrigiert. In der Grafik hatte ich es bereits so dargestellt.
Nochmals vielen Dank!
Herzliche Grüße, Christian Johner
Allerdings muss die Bescheinigung der Benannten Stelle am oder nach dem 25.05.2017 ausgestellt sein. Zudem endet ihre Gültigkeit spätestens am 27.05.2022, selbst wenn das Gültigkeitsdatum auf der Bescheinigung nach dem 27.05.2022 liegen sollte.
Ich nehme an, dass 2022 ein Tippfehler ist – sollte dies nicht 27.05.2024 heissen? Die Bescheinigung wird ja nach dem 25.05.2017 ausgestellt. Also gilt der zweite Absatz von 120 (2), der den 27.05.2024 referenziert.
Hallo Herr Prof. Johner,
ich habe eine Frage bezüglich des Corrigendums und der Anpassung des Artikels 120 (3).
Folgender Fall beschäftigt mich, da ich diesen nicht ganz einordnen kann:
-> Ein MP-Hersteller vertreibt ausschließlich wiederverwendbare Produkte der Klasse I gemäß MDD (nach MDR würden diese in die Risikoklasse Ir neu eingestuft werden)
-> Diese Produkte liefen unter der MDD nicht über eine benannte Stelle
-> Der Hersteller besitzt demnach kein CE-Zertifikat einer benannten Stelle und stellt seine Konformitätserklärungen als Hersteller selbst aus, womit er die Einhaltung der 93/42/EWG bestätigt
-> Dieser Hersteller beruft sich nun auf die Fristverlängerung des Artikels 120 (3).
Jetzt meine Frage:
Im Corrigendum des Artikels 120 (3) heißt es: „…darf ein Produkt, das ein Produkt der Klasse I gemäß der Richtlinie 93/42/EWG ist, für das vor dem 26. Mai 2020 eine EU-Konformitätserklärung erstellt wurde und für das das Konformitätsbewertungsverfahren gemäß der vorliegenden Verordnung die Mitwirkung einer Benannten Stelle erfordert oder für das eine Bescheinigung gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG besteht…“
Ich verstehe den Teil „…und für das das Konformitätsbewertungsverfahren gemäß der vorliegenden Verordnung die Mitwirkung einer Benannten Stelle erfordert…“ nicht ganz, da die Mitwirkung einer benannten Stelle bisher bei Klasse I Produkten noch nicht erforderlich war. Ebenso deute ich den Satz: „…oder für das eine Bescheinigung gemäß der Richtlinie 90/385/EWG oder der Richtlinie 93/42/EWG besteht…“ so, als ob eine Bescheinigung (CE-Zertifikat) vorhanden sein muss, um die Fristverlängerung zu „erhalten“. Hersteller für ausschließliche Klasse I Produkte haben eine solche Bescheinigung gemäß MDD nicht vorliegen…
Wie kann ich den Abschnitt des korrigierten Artikel 120 (3) nun deuten? Gilt das auch, wenn kein CE-Zertifikat einer benannten Stelle vorliegt, da es de facto bisher nie nötig war bzw. wenn das Konformitätsbewertungsverfahren nicht über eine benannte Stelle lief?
Ich werde aus dem Abschnitt ehrlich gesagt nicht ganz schlau. Denn ansonsten könnte von Herstellerseite die ausgestellte EU-Konformitätserklärung mit dem Datum 25.05.2020 datiert werden und somit die Übergangsfrist bis 26.05.2024 genutzt werden? Oder liege ich da falsch? Ich hoffe ich konnte es verständlich erklären und Sie können mir an dieser Stelle weiterhelfen…
Sehr geehrter Herr Bauer,
danke für Ihre spannende Frage!
Das Corrigendum hat man in gewisser Weise für die Klasse-Ir-Hersteller verfasst. D.h. genau diesen Produkten möchte man die Übergangsfrist gewähren.
Die Einbeziehung der benannten Stelle bezieht sich auf die MDR. Daher heißt es auch „der vorliegenden Verordnung“. Gemeint ist die MDR. Dass unter der MDD diese Einbeziehung nicht notwendig war (weil es ein Klasse-I-Produkt war), ist korrekt und sogar die Voraussetzung dafür, von dieser Fristverlängerung zu profitieren.
Falls ich die Frage noch nicht ausreichend beantwortet haben sollte, haken Sie gerne nach.
Viele Grüße, Christian Johner
Im Text wird mehrfach auf die Notwendigkeit der Registrierung von Sonderanfertigungen verwiesen und dabei Artikel 29 Abs. 4 MDR referenziert.
Wenn ich den Artikel 29 lese, steht dort an allen Stellen: „Bevor ein Produkt, bei dem es sich nicht um eine Sonderanfertigung handelt, in Verkehr gebracht wird, gibt der Hersteller…“ – so wie ich es verstehe, sind damit doch explizit Produkte gemeint, die KEINE Sonderanfertigungen sind oder wo habe ich da einen Denk-/Lesefehler?
Sehr geehrter Herr Klahn,
Sie haben absolut Recht! Das war nicht Ihr Denkfehler sondern meine Übermüdung nach einem Tag des MDR-Lesens :-). Der Fehler ist nun behoben.
Danke für Ihren Hinweis!
Beste Grüße, Christian Johner
@Tobias Bauer:
„…und für das das Konformitätsbewertungsverfahren gemäß der vorliegenden Verordnung die Mitwirkung einer Benannten Stelle erfordert…“ Die „vorliegende Verordnung“ ist ja die MDR, wenn also mein MD nach MDD Klasse I war und nach MDR höher klassifiziert wird, dann braucht es neu nach MDR eine benannte Stelle. In diesem Fall darf ich gemäss Corrigendum 2, Aritkel 120 (3) von der Übergangsfrist für mein MD profitieren wenn ich die Konformitätserklärung vor dem 26. Mai 2020 ausgestellt habe.
Klar könnte es nun jemand „zurückdatieren“ (und ich denke/befürchte, das wird wohl der eine oder andere im Juni/Juli auch tun). Da wir die Dokumente Lenken müssen, dürfte es im Falle einer Gerichtsverhandlung schwierig werden das zurückdatierte Datum aufrecht zu erhalten, was wohl dann entsprechende Strafen nach sich ziehen könnte respektive wird.
Lieber Herr Johner,
mir ist die Übergangsfrist bei den Klasse I Produkten irgenwie immer noch nicht so ganz klar.
Wir haben folgenden Fall:
MD der Klasse I nach MDD mit einer Konformitätserklärung aus 2018 von uns als Hersteller ausgestellt, der NB war dabei nicht involviert.
Die Klassifizierung ändert sich unter der MDR nicht.
Darf ich dieses MD nun unter dem Artikel 120 der MDR bis zum Ende der Übergangsfrist weiter in Verkehr bringen / vertreiben?
Vielen Dank für Ihren Kommentar.
Beste Grüße
Thomas Steinberger
Sehr geehrter Herr Steinberger,
wenn Ihr Produkt in der Klasse I war und unter der MDR auch bleibt, profitieren Sie nicht von den Übergangsfristen. Allerdings brauchen Sie dann auch unter der MDR keine benannte Stelle involvieren.
Beste Grüße, Christian Johner
Lieber Herr Johner,
herzlichen Dank für die ausführliche Analyse des Artikel 120.
Es gibt einen kleinen aber feinen Haken in 120 (3). Dort heißt es nämlich auch nach dem zweiten Korrigendum „…..und sofern keine wesentlichen Änderungen der Auslegung und der Zweckbestimmung vorliegen.“
Da der Text unverändert geblieben ist, in englisch, französisch, schwedisch der gleichen Logik folgt und auch im Niederländischen sogar eindeutiger ist, muss besonderer Augenmerk auf das „und“ gelegt werden.
Ich bin mir im Klaren darüber, dass dies zu spannenden Diskussionen und evtl. später gerichtlichen Auseinandersetzungen führen wird – aber wie ich gelernt habe ist es nicht nur im Steuerrecht wichtig, den Gesetzestext so anzuwenden, wie er formuliert ist; auch wenn er der „vernünftig, menschlichen“ Denkweise völlig zuwider läuft.
Dieses „und“ bewirkt, dass nur dann die weitere Vermarktung verboten ist, wenn eine Änderung der Auslegung !UND! der Zweckbestimmung vorliegt. D.h:
– wenn Zweckbestimmung geändert und Auslegung bleibt, darf weiter vermarktet werden
– wenn Auslegung geändert wird und die Zweckbestimmung bleibt, darf weiter vermarktet werden.
Ich habe dazu auch einen Artikel auf LinnkedIn geschrieben: https://www.linkedin.com/pulse/mdr-second-corrigendum-more-confusion-eckhard-jokisch
Viele Grüße
Eckhard Jokisch
Das ist ein sehr spannender Hinweis, lieber Herr Jokisch!
Ihrer Argumentation kann ich folgen. Bei einigen benannten Stellen sieht man das anders. Die Begründung ist, dass bei Änderungen an der Auslegung die Konformität neu bewertet werden muss. Diese sollte dann aber nach den neuen Spielregeln erfolgen, weil man sonst die Produkte ständig weiterentwickeln könnte, solange die Zweckbestimmung gleich bleibt.
Wie Sie sagen: Es wird zu Diskussionen führen. Warten wir gespannt die Gerichtsurteile ab.
Nochmals vielen Dank für Ihren wichtigen Input!
Viele Grüße, Christian Johner
Hallo Herr Johner,
ich bin für Ihre klärenden Ausführungen immer sehr dankbar, aber oben schreiben Sie:
„Die Hersteller müssen Sonderanfertigungen in EUDAMED registrieren (Artikel 29 Absatz 4)“
Ich lese in Artikel 29, Absatz 4 MDR jedoch:
„Bevor ein Produkt, bei dem es sich *nicht* um eine Sonderanfertigung handelt, in Verkehr gebracht wird, gibt der Hersteller die […] genannten Angaben in Eudamed ein […]
Damit sind Sonderanfertigungen doch explizit von der Registrierung in der EUDAMED-Datenbank ausgeschlossen, oder habe ich etwas übersehen?
Sie haben absolut Recht, Herr Ertner! Ich habe das „keine“ vergessen.
Danke Ihrer Hilfe konnte ich den Fehler sofort beheben. Danke für den Hinweis!
Beste Grüße, Christian Johner
Lieber Herr Johner,
ich verstehe immer noch nicht wie das mit den MD der Klasse I ist.
Wir stellen unter MDD MD der Klasse I her, diese werden auch unter der MDR weiterhin Klasse I sein. Wir als Hersteller haben die Konformitätserklärung ausgestellt, somit war der NB nicht beteiligt.
Können wir nun diese Klasse I Produkte weiterhin unter dem Artikel 120 in Verkehr bringen?
Beste Grüße
Thomas Steinberger
Sehr geehrter Herr Steinberger,
nein, Sie dürfen die Produkte mit der gegebenen Konformitätserklärung nicht mehr in den Verkehr bringen, weil es keine Übergangsfrist gibt.
Sie können allerdings ohne Beteiligung einer benannten Stelle eine neue Konformitätserklärung ausstellen, falls Ihre Produkte auch die Anforderungen der MDR erfüllen und falls Sie auch alle Anforderungen der MDR erfüllt sind. Beachten Sie insbesondere die Anforderungen an die technische Dokumenation, an eine verantwortliche Person, an die Post-Market Surveillance und an die Vigilanz.
Beste Grüße, Christian Johner
Lieber Herr Johner,
danke für die Rückmeldung, die Antwort haben sie schon im Kommentar 10 gegeben.
Beste Grüße
Thomas Steinberger
Im Text wird mehrfach auf die Notwendigkeit der Registrierung von Sonderanfertigungen verwiesen und dabei auf Artikel 29 Abs. 4 MDR referenziert.
Wenn ich Artikel 29 lese, steht dort an allen Stellen: „Bevor ein Produkt, bei dem es sich nicht um eine Sonderanfertigung handelt, in Verkehr gebracht wird…“ – so wie ich es verstehe, sind damit doch explizit Produkte gemeint, die KEINE Sonderanfertigungen sind oder wo habe ich da einen Denk-/Lesefehler?
Ich habe jetzt erst gesehen, dass der Irrtum mit den Sonderanfertigungen bereits bereinigt ist… bitte entschuldigen Sie die doppelte Anfrage!
So wie ich den Text verstehe, sind die MDD Produkte damit von der UDI Markierung ausgenommen, sprich ein Klasse III Produkt welches noch ein MDD Zertifikat hat bis z.B Mai 2023 muss erst nach der MDR Zertifizierung mit UDI gelabelt werden (Artikel 10 ausgenommen für MDD Produkte ausser PMS usw, sprich UDI gehört nicht dazu)
Hallo Herr Prof. Johner,
vielen Dank für die dringend notwendige, verständliche Aufbereitung!
Im Abschnitt zu Art. 120 Absatz 3 hat sich noch ein Fehler ein geschlichen, das Enddatum ist 26.05.2024 und nicht 26.04.2024.
Grüße, Raymond Lohrmann
Super, danke, lieber Herr Lohmann!
Das fixe ich sofort! Danke!
Herzliche Grüße, Christian Johner
PS: Ich finde in diesem Abschnitt keinen 26.04. Könnten Sie ein „Hard Reload“ machen? Vielleicht ist es ein Caching Problem. Ich möchte aber sicherstellen, dass ich nichts übersehe.
Kommentare zu Johner – Artikel über die Übergangsfristen in der MDR
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihre Interpretationshilfe. Ich hätte noch einige Korrekturanmerkungen – oder habe an diesen Stellen die MDR nicht richtig verstanden …
Zu „a) MDR Artikel 120 Absatz 2“: Zunächst: Statt „muss die Bescheinigung der Benannten Stelle am oder nach dem 25.05.2017 ausgestellt sein“ muss es doch heißen: „muss die Bescheinigung der Benannten Stelle am oder nach dem 26.05.2017 ausgestellt sein“ (einen Tag später, selbst wenn das nicht bedeutend sein wird).
Ich finde es hier irritierend, dass Sie in der Interpretation nur auf den zweiten Teil des Zitats eingehen (oder sehe ich das falsch?). Ich würde es so interpretieren:
Falls der Hersteller eine aktuelle „Zulassung“ hat, bleibt diese erst einmal gültig.
Wenn die Bescheinigung der Benannten Stelle vor dem 26. Mai 2017 ausgestellt wurde, gilt diese bis zu dem in der Bescheinigung genannten Datum. [Anmerkung: Da Bescheinigungen nicht länger als 5 Jahre gültig sind, verliert die Bescheinigung spätestens am 26. Mai 2022 ihre Gültigkeit.]
Wenn die Bescheinigung der Benannten Stelle am oder nach dem 26.05.2017 ausgestellt wurde, endet ihre Gültigkeit spätestens am 27.05.2024, selbst wenn das Gültigkeitsdatum auf der Bescheinigung nach dem 27.05.2024 [Anmerkung: Dies sollte wegen der maximalen Gültigkeit von 5 Jahren an sich nicht möglich sein.] liegen sollte.
Zur Interpretation von Absatz (3): Statt „Ab dem 25.05.2020 dürfen“ muss es heißen: „Ab dem 26.05.2020 dürfen“.
Weiter schreiben Sie: „2. Der Hersteller hat bereits vor dem 26.05.2020 ein Produkt der Klasse I (MDD) in den Verkehr gebracht, …“ Dieser Fall wird aber doch vollständig von Absatz (4) abgedeckt, auch wenn es Überschneidungen mit Absatz (3) gibt. Ich würde etwa so formulieren: „2. Der Hersteller hat bereits vor dem 26.05.2020 für ein Produkt der Klasse I (MDD), das unter der MDR in eine der Klassen I*, IIa, IIb oder III fallen würde, eine EU-Konformitätserklärung erstellt. Das ist beispielsweise …“ Hier ersetzt also das Ausstellen der EU-Konformitätserklärung die Ausfertigung der Bescheinigung der Benannten Stelle, auf die im hinteren Teil von Absatz (3) eingegangen wird.
Übrigens fehlt in Absatz (3) vor „oder für das“ ein wichtiges Komma; im Englischen und Französischen steht es. In Ihrer Interpretation des Textes haben Sie dieses Komma implizit auch eingefügt – die Version ohne Komma wäre kaum sinnvoll.
Zu Artikel 123 Absatz (d): Statt „25.05.2020 (Geltungsbeginn der MDR)“ bitte zu „26.05.2020 (Geltungsbeginn der MDR)“ korrigieren.
Im Absatz zu den FAQ der NAKI ist bei „Die Möglichkeit, vor dem 26.05.2020 bereits (erstmalig) in Verkehr gebrachten Produkte bereitzustellen/in Betrieb zu nehmen, ist nicht durch Art. 120 Abs. 4 MDR zeitlich befristet.“ das „nicht“ falsch: Absatz 4 setzt genau diese Befristung fest.
Eine Anmerkung auch zur Anfrage von Herrn Steinberger und Ihrer Antwort darauf: Ja, ein (erstmaliges) Inverkehrbringen ist ab dem 26. 5. 2020 nicht mehr möglich. Vertrieb und Inbetriebnahme sind jedoch nach Absatz (4) bis 26. 5. 2025 möglich – oder?
Schließlich sind in der Übersichtsgrafik noch ein paar Daten um einen Tag falsch:
Zeile 2: Inverkehrbringung bis _25._05.2020 gestattet.
Zeile 2: Bereitstellung und Inbetriebnahme bis _26._05.2025.
Zeile 3: Inverkehrbringung bis _26._05.2024.
Zeile 3: Bereitstellung und Inbetriebnahme bis _26._05.2025.
Zeile 4: Wurde die Bescheinigung vor dem _26._05.2017 ausgestellt?
Viele Grüße
K. Roeseler
Sehr geehrter Herr Roeseler,
besten Dank für Ihre umfangreichen Anmerkungen! Ob der Menge brauche ich ein paar Tage, um alle sorgfältig durchzugehen. Das mache ich schnellstmöglich, antworten und aktualisiere den Artikel.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Roeseler,
herzlichen Dank für Ihre großartiges Lektorat. Dank Ihrer Hilfe konnte ich die Fehler (insbesondere die um einen Tag verrutschten Daten) korrigieren. Ich habe mir auch erlaubt, einen Satz von Ihnen zu übernehmen.
Mit nochmaligem Dank und mit vielen Grüßen, Christian Johner
Hallo Herr Johner,
auch mir sind die Übergangsfristen für Klasse I-Produkte noch immer unklar.
MD, die bereits vor dem 26.05.2020 in Verkehr gebracht wurden und nach diesem Datum unverändert verkauft werden (Gültigkeit Konformitätserklärung entsprechend 93/42 bis März 2022) für diese ist noch eine TD nach 93/42 ausreichend ?(Bereitstellung bis 2025).
Die vollen Anforderungen (hinsichtlich TD) entsprechend MDR betreffen m.E. nur neue Produkte der Klasse I. (Inverkehrbringung bis 26.05.2020) Habe ich das richtig verstanden?
Vielen Dank für Ihre Hilfe!
Sehr geehrter Herr Stickel,
für Produkte der Klasse I, die auch unter der MDR in die Klasse I fallen (nicht in Klasse I* oder höher), gibt es keine(!) Übergangsfrist bezüglich der Inverkehrbringung. D.h. die weitere Inverkehrbringung ist nicht gestattet, auch nicht wenn sie unverändert bleiben. Diese Produkte müssen ab dem 26.05.2020 die Anforderungen der MDR erfüllen.
Für Produkte der Klasse I, die unter der MDR in die Klassen I* oder höher fallen, hat das 2. Corrigendum die im Artikel beschriebenen Übergangsfristen ergänzt.
Die Übergangsfristen gibt es auch für Produkte, für die eine Bescheinigung vorliegt.
Vielleicht kann das Ablaufdiagramm etwas zur Klärung beitragen.
Viele Grüße, Christian Johner
Hallo Herr Johner, wieder mal eine erstklassige Zusammenfassung. In den Diskussionen mit benannten Stellen und anderen Herstellern ist mir aufgefallen, dass es unterschiedliche Meinung dazu gibt, welche Berichte zwingend notwendig sind für „legacy devices“ ab dem 26.05.2020. Unstrittig ist z.B. das Vorhandensein eines PMCF-Plans; beim PSUR gibt es „hitzige Diskussionen“ uws.. Es ware toll, wenn Sie ggf. den Artikel mit einem Kapitel oder Tabelle ergänzen könnten, der/die die notwendigen Berichte gemäß MDR für die legacy devices beschreibt. Weiterhin gibt es viel Diskussionen bzgl. der aufzusetzenden Prozesse bzgl. der Registrierung der Wirtschaftsakteure und Produkte für legacy devices. Muss bis 26.05.2020 ein Prozess aufgesetzt sein, der die Registrierung in der EUDAMED beschreibt, obwohl diese noch nicht existent ist? Oder muss wenigstens ein Prozess stehen, der die Registrierung in die nationalen Datenbanken beschreibt (wenn ja, wie präzise muss dies sein, da sich bis zum Mai national noch einiges ändert –> MDG etc.?)?
Sehr geehrter Herr Matz,
danke für Ihre Rückmeldung!
Ich habe in der Tabelle gerne ergänzt, dass die Bericht als Teil des PMS-Systems ohne Übergangsfrist zu erstellen sind.
Es gibt keine Pflicht, einen Prozess aufgesetzt zu haben, der beschreibt, wie man Daten in die noch nicht funktionsfähige und nicht zugängliche EUDAMED einzugeben hat.
Auf eine Vorgabe (dass kann eine Verfahrens- oder Arbeitsanweisung sein), wie man die Produkte und Hersteller registriert (derzeit eben noch national) sollten Sie aber nicht verzichten.
Beste Grüße, Christian Johner
Hallo Herr Johner, vielen Dank für die schnelle Rückmeldung. Sie haben jetzt „nur“ den PSUR beispielhaft ergänzt. Nach meinem/unseren Verständnis ist der PMCF-Plan Pflicht. Zum PSUR bzw. PMS-R gibt es unterschiedliche Meinungen („PSUR muss final vorliegen“ –> bis hin zu „PSUR kann in den ersten 12 Monaten nach Geltungsbeginn der MDR finalisiert warden“). Wie sieht es mit dem SSCP aus? Da in der MDR für die PMS steht „…update the summary of safety and clinical performance…“, könnte man der Meinung sein, dass ja ein SSCP für legacy devices schon vorhanden sein muss, um das Update ab Mai 2020 durchzuführen. Was ist Ihre Erfahrung aus den Kontakten zu den benannten Stellen? Sind PMCF-P, PSUR/PMS-R, SSCP Pflicht? Beste Grüße. Torsten Matz
Sehr geehrter Herr Matz,
Sie haben Recht, ich bin nich auf alle Details eingegangen. Wir schaffen das einfach nicht mehr. In den letzten 20 Monaten haben wir über 2900 Anfragen kostenfrei beantwortet….
Dennoch möchte ich gerne weitere Informationen und zumindest „Antwortfragmente“ geben:
Ich hoffe, ein wenig geholfen zu haben.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
erst einmal vielen Dank für den hilfreichen Artikel. Allerdings habe ich eine Frage:
In der Tabelle in Abschnitt 4c (EUDAMED, Registrierung) wird angegeben, dass Legacy Produkte nach 6 Monaten in Eudamed registriert werden müssen, neue Produkte erst nach 24 Monaten.
Ich hatte bisher gedacht, dass es genau andersherum ist. Im Dokument MDCG 2019-5 (Registration of legacy devices in EUDAMED) steht zum Beispiel:
„Legacy devices […] should be registered in Eudamed without a Basic UDI-DI and UDI-DI. The registration deadlines for those devices is clearly the one referred to in Article 123(3)(e): 18 month after the date of application (provided that Eudamed is fully functional on time).”
Außerdem erscheint es mir logisch, dass Legacy Produkten eine längere Übergangsfrist gewährt wird. Oder habe ich da einen Denkfehler?
Viele Grüße
Carolin Riesenberg
Sehr geehrter Prof. Johner,
bitte überprüfen Sie aus aktuellem Anlass in
„Abb. 3: Entscheidungsdiagramm zur Bestimmung der Übergangsfristen für die Inverkehrbringung, Bereitstellung und Inbetriebnahme“
die Aussage
„Inverkehrbringung bis zum 25.05.2020 gestattet“
mvG
Eckart Klobe
Sie haben absolut Recht, lieber Herr Klobe!
Die Grafik war noch nicht aktualisiert. Dank Ihres Hinweises konnte ich das gleich nachholen.
Vielen Dank!
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für die aufschlussreiche Zusammenstellung.
Ihre Ausführungen zu den Übergangsfristen für die UDI-Kennzeichnung haben bei mir eine Frage hervorgerufen:
Ihre Argumentation, die menschenlesbare Version der UDI sei von der Übergangsfrist ausgeschossen erschließt sich mir nicht ganz.
In der MDR wird ja nicht explizit zwischen UDI-Träger (in ihrer Lesart nur die maschinenlesbare Variante) und der menschenlesbaren Text-Variante unterschieden, wenn es um die Kennzeichnungsfrist geht (Art.123.f)
Ich verstand die Formulierung des UDI-Trägers bislang als Bezeichnung der Kennzeichnung als Ganzes betrachtet (menschen + maschinenlesbare Variante). Wir sind daher davon ausgegangen, die UDI-Kennzeichnung in einem Schritt im Rahmen der Übergangsfrist implementieren zu können.
(Die IFA etwa, gibt in ihrer ‚Spezifikation UDI‘ an: „UDI-DI und UDI-PI sind grundsätzlich auf dem so genannten UDI-Träger zur Kennzeichnung maschinenlesbar (AIDC) und in vom Menschen lesbarer Form (HRI)“ aufzubringen)
Können Sie bitte nochmal ausführen, warum der UDI-Träger Ihrem Verständnis nach nur den maschinenlesbaren Code bezeichnet, demzufolge bezüglich der Kennzeichnung auf dem Produkt separat von der HRI anzugehen werden müsste.
Vielen Dank im Voraus und viele Grüße,
Bernhard Scholze
Lieber Herr Scholze,
Sie haben Recht. Ich habe den Beitrag aktualisiert. Woher die Informationen stammten, kann ich leider nicht mehr nachvollziehen.
Nochmals besten Dank für Ihre wichtigen Hinweis!
Viele Grüße, Christian Johner
Sehr geehrte Damen und Herren,
Mit der Verschiebung der MDR um ein Jahr, wurden auch die Übergangfsfristen bei der UDI-Kennzeichnung geändert, s. Abschnitt 8 Europ. Amtsblatt vom 24.04.2020.
Die Übergangsfristen verschieben sich in den 3 Kategorien um jeweils 2 Jahre nach hinten. Als Hinweis zur Anpassung in diesem Blockbeitrag und in dem hervorragenden Merkzettel, der als Download zur Verfügung steht. Sehr hilfreich, vielen Dank dafür.
Mit freundlichen Grüßen
R. Lichtenthal
Sehr geehrter Herr Prof. Johner,
ich habe eine Frage zu Bestandsprodukten, die unter MDR weiterhin zugelassen werden sollen. Dazu folgende fiktive Situation:
Ein Bestandsprodukt hat bis 12/2023 eine MDD-Zulassung und bekommt unter optimalen Bedingungen eine Anschluss-Zertifizierung unter MDR.
Können Artikel dieses Produktes, die beispielsweise in 06/2022 produziert wurden (unter MDD-Zertifikat), nach 12/2023 weiterhin in den Markt gebracht werden (unter MDR-Zertifikat) unter der Voraussetzung, dass es keine wesentlichen Änderungen an diesem Bestandsprodukt gab?
Vielen Dank im Voraus und viele Grüße
Heike Hauptmann
Liebe Frau Hauptmann,
danke für die spannende Frage!
Das Zertifikat bezieht sich auf Produkte einer bestimmten Klasse, nicht auf ein einzelnes Produkt. Daher reicht die alleinige Existenz des „Anschlusszertifikats“ nicht aus. Allerdings können Sie mit diesem MDR-Zertifikat die Konformität einfach neu erklären, insbesondere, wenn sich das Produkt nicht geändert hat.
Eine Prüfung, ob die Anforderungen der MDR auch tatsächlich erfüllt sind (schließlich gab es ja Änderungen zwischen MDD und MDR), muss aber erfolgen.
Ich grüße Sie herzlich, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für diese abermals sehr gute Zusammenfassung.
Ich habe trotzdem eine Frage bezüglich des aktuellen Corrigendums des §120 (3) und (4).
Unsere derzeitigen Medizinprodukte (Klasse I) werden nach den neuen Verordnungen MDR und IVDR in Zukunft ein IVD sein.
Wir stellt sich denn hier die Übergangsfristenausnahmen dar, wenn sich ein Medizinprodukt in ein in-vitro Diagnostikum umwandelt? Wurde bzw. kann man hier analog vorgehen oder gibt es diesbezüglich noch keine Klarheit? Meines Wissens gibt es keine Änderung, die in Kraft getreten ist noch irgendeine Art Diskussion über diesen Aspekt.
Mit freundlichen Grüßen
Dr. Breß
Sehr geehrter Herr Dr. Breß,
danke für die wirklich spannende Frage!
Wenn Ihr Produkt derzeit unter die MDD und künftig unter die IVDR fällt, dann greifen die Übergangsfristen der MDR in der Tat nicht. Mir ist auch keine Diskussion dieses Sonderfalles bekannt.
Mein Tipp wäre, dass Sie klären, ob Sie eine Benannte Stelle unter IVDR benötigen. Bei der dafür notwendigen Klassifizierung helfen wir gerne. Wenn Sie eine BS benötigen, würde ich das mit der abstimmen. Alternativ wäre eine Rückfrage bei der zuständigen Behörde hilfreich. Sie könnten um eine Verlängerung bitten.
Ein zweiter Punkt, der neben der Klassifizierung in die Entscheidung über das weitere Vorgehen einfließen sollte, ist die Frage, ob das Produkt bis 2022 geändert werden soll und falls ja, in welchem Umfang.
Mit diesem beiden Informationen können wir noch genauer recherchieren und antworten.
Beste Grüße, Christian Johner
Sehr geehrter Prof. Johner,
als Hersteller von MD Klasse 1 bereiten wir uns auf die Kennzeichnung unserer Produkte mit dem UDI-Träger ab 2025 vor.
Nun habe ich in einem Ihrer Infopapiere gelesen, dass diese Übergangsregelung nicht für die HRI Verison der UDI zutrifft.
Bedeutet das, dass unsere Produkte bereits ab Mai 2021 die UDI in HRI Form (menschen lesbare Zahlenkombination) tragen müssen? Müssen wir also die UDIs, die wir bereits demnächst für unsere Produkte generieren können und die wir ab Mai 2021 in den Dokumenten aufführen, auch auf die Produkte und alle Verpackungen drucken? Und können nur mit dem maschinenlesbaren Code bis 2025 warten?
Für Ihre Aufklärung wäre ich Ihnen sehr dankbar!
Herzlichen Dank,
Dipl.-Ing. Ina Hein
Sehr geehrte Frau Hein,
die Übergangsfristen betreffen die Anbringung der UDI, sowohl das AIDC als auch HRI-Format. Somit müssen Sie bis 2025 keine UDIs in HRI-Form anbringen. Der entsprechende Beitrag wurde vor einiger Zeit bereits aktualisiert und dabei wurde der fehlerhafte Aussage entfernt. Falls Sie diese Information an einer anderen Stelle gefunden haben, wäre ich über einen Hinweis bzw. Link sehr dankbar.
Herzliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
ich danke Ihnen für ihre „Entwarnung“.
Ich habe mir letzte Woche erst auf dieser Homepage das Merkblatt MDR-Übergangsfristen geöffnet, hier steht es unter Punkt 4, letzter Satz.
Dennoch habe ich die Frage für mich einmal aufgedröselt:
In MDR Artikel 123 (Absatz 3 Buchstabe f) Inkrafttreten wird eine Übergangsfrist für die Umsetzung von Artikel 27 Absatz 4 – Anbringen des UDI-Trägers – bis 2025 (für MD Klasse 1) gewährt.
Wir müssen den UDI-Träger also erst ab Mai 2025 auf unsere Produkte und Verpackungsebenen drucken.
Allerdings wird in Artikel 27 Absatz 1 die Kennzeichnung des Produkts mit der UDI (Nummer, nicht Träger) gefordert.
Dieser Absatz ist nicht in der Übergangsfrist in Artikel 123 eingeschlossen.
Daher vermutlich auch Ihr Hinweis in dem Merkblatt.
Anhang VI, Teil C, Punkte 3. und 4. sprechen ebenfalls getrennt einmal von der Zuteilung (nicht „Kennzeichnung“) der UDI und unter Punkt 4 von der Kennzeichnung mit dem UDI-Träger.
Wiederum ist unter Anhang I, Kapitel III, 23.2 Angaben auf der Kennzeichnung der UDI-Träger als Bestandteil der Kennzeichnung aufgeführt, die UDI-Nummer jedoch nicht explizit.
Vielleicht haben Sie noch eine Idee, wie die Macher der MDR das gemeint haben bzw. einen Hinweis, wie andere Firmen das interpretieren!
Herzlichen Dank
Ina Hein
Sehr geehrter Herr Prof. Johner, im November 2020 streben wir die MDR Zertifizieren an. Das aktuelle MDD Zertifikat ist bis 08.02.2021 gültig, eine Verlängerung bis max. 27.05.2024 ist angestrebt. Wenn nach dem Audit das MDR Zertifikat ausgestellt wurde, ist des dann weiterhin möglich MDD Ware bis zum Ablauf des MDD Zertifikats auf den Markt zu bringen, oder darf ab dem MDR Zertifikate nur nach MDR Konforme Ware in den Verkehr gebracht werden. Danke für eine kurze Stellungnahmen. Mit freundlichem Gruß A. Maack
Sehr geehrter Herr Maack,
es ist möglich, zur gleichen Zeit sowohl unter nach MDD als auch MDR zertifiziert zu sein. Hierzu gibt es ein Statement der EC. Ob man als Hersteller allerdings das gleiche Produkt zur gleichen Zeit unter beiden Zertifikaten parallel in Verkehr bringen darf, ist nicht klar in der MDR geregelt. Nach unserer Erfahrung erlauben dies die Benannten Stellen allerdings nicht, d.h. ist ein Produkt oder Produktgruppe vom MDR-Zertifikat abgedeckt, können die entsprechenden Produkte nicht mehr unter der MDD in Verkehr gebracht werden. Ich würde Ihnen raten, dies mit Ihrer Benannten Stelle abzusprechen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Johner
Wir haben folgende Ausgangslage:
– Produkt nach MDD Klasse I, nach MDR fällt es in eine höhere Klasse
– MDD Konformitätserklärung für das Produkt (Hardware und Software) von Mai 2021
Auf dem Merkblatt MDR-Übergangsfristen steht für diesen Fall:
– Erstmalige Inverkehrbringung bis 26.5.21
– Inverkehrbringung auf den EU Markt bis 26.5.24
Nun meine Frage dazu:
müssen die einzelnen Produkte (Hardware) bis zum Geltungsbeginn (26.5.21) bereits fertig produziert und beim Hersteller an Lager sein oder kann der Hersteller die gleichen Produkte auch noch bis zum 26.5.24 nachproduzieren und unter der gleichen Konformitätserklärung in Verkehr bringen?
Besten Dank für Ihre Antwort.
Sehr geehrter Herr Trepp,
danke für die essenzielle Frage! Die Übungsfrist erlaubt Ihnen, die Produkte weiterhin bis zum 26.05.24 in den Verkehr zu bringen, solange sie keine wesentlichen Änderungen daran machen.
Diese Übergangsfrist ist also nicht die sog. Abverkaufsregelung. Sie dürfen weitere identische Produkte produzieren und verkaufen, nicht nur bereits produzierte Produkte „abverkaufen“.
Viele Grüße, Christian Johner
Hallo Herr Johner,
vielen Dank für die wirklich aufschlussreiche Zusammenfassung zu den Übergangsfristen der MDR.
Was mich umtreibt:
Wir vertreiben zur Zeit Klasse I MP gemäß MDD. Diese werden nach MDR in Klasse IIa höhergestuft, daher wollen wir die Produkte bis Ende Mai „einfrieren“. Schaut man sich die Übergangsfristen zur UDI-Kennzeichnung an, so müssten wir bei Klasse IIa Produkten bis zum 26.05.2023 den UDI-Träger aufbringen. Handelt es sich bei der Anbringung des UDI-Trägers dann um eine „wesentliche Änderung“? Dann würde man ja ein Jahr der Übergangsfrist verlieren.
Oder wie kann man das verstehen?
Besten Dank für eine kurze Rückmeldung!
Sehr geehrte Laura,
danke für Ihre wichtige Frage!
Die Pflicht, eine UDI aufzubringen, führt zum Glück nicht dazu, dass man mit dem Aufbringen eine wesentliche Änderungen durchführt, was wiederum die Übergangsfristen aushebeln würde. Also gute Nachricht!
Beste Grüße, Christian Johner
Sehr geehrter Prof. Dr. Johner,
in Art. 120 Abs. 11 MDR gibt es eine Übergangsregelung für klinische Prüfungen. Wann gilt denn eine klinische Prüfung als „eingeleitet“? Ist es der Zeitpunkt der Antragstellung beim BfArM, das positive Votum der Ethik oder der tatsächliche Studienbeginn (erste Patientenaufklärung)?
Danke für Ihre Rückmelung.
Beste Grüße
Sehr geehrter Thomas,
danke für Ihre Frage. Unser Clinical Affairs Team hat mich gerade diese Antwort wissen lassen:
siehe MPDG § 99, Absatz 4:
Unbeschadet des Artikels 120 Absatz 11 Satz 2 der Verordnung (EU) 2017/745 sind für klinische Prüfungen, die vor dem 20. März 2010 begonnen wurden, die §§ 19 bis 24 des Medizinproduktegesetzes in der Fassung der Bekanntmachung vom 7. August 2002 (BGBl. I S. 3146), das zuletzt durch Artikel 1 des Gesetzes vom 14. Juni 2007 (BGBl. I S. 1066) geändert worden ist, weiter anzuwenden.
Begonnen im Sinne von Satz 1 ist eine klinische Prüfung, wenn nach Vorliegen aller Voraussetzungen für den Beginn der klinischen Prüfung der erste Prüfungsteilnehmer in die Teilnahme an der klinischen Prüfung eingewilligt hat. Sie gilt damit als eingeleitet im Sinne von Artikel 120 Absatz 11 der Verordnung (EU) 2017/745.
Hilft das? Falls nicht, haken Sie einfach nach.
Viele Grüße, Christian Johner
Guten Tag,
vielen Dank für den mal wieder sehr hilfreichen Artikel!
Weiter oben wurde die bereits die Aussage getroffen „So wie ich den Text verstehe, sind die MDD Produkte damit von der UDI Markierung ausgenommen…“.
Ich habe auch das Verständnis, dass Legacy-Produkte von der UDI Kennzeichnung ausgenommen sind. Können Sie das bestätigen?
Beste Grüße
Nadine Langguth
Liebe Frau Langguth,
ganz genau. Für Legacy-Produkte müssen Sie keine UDIs vergeben. Erlaubt ist es aber, d.h. man kann zukünftig auch für Legacy-Produkte UDIs in EUDAMED eintragen, wenn man dies als Hersteller möchte.
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore:
Folgende Konstelation:
wir möchten gerne ein Medizinprodukt der Klasse I (Nitril-Untersuchungshandschuhe) mit unserem Markennamen in Europa vertreiben. Der Hersteller ( aus Indien) soll nicht mit seinem Namen auf der Verpackung ersichtlich sein (demnach altes PLM/OEM-Konstrukt). Demnach müssten wir ja als Hersteller alle Pflichten laut Artikel 10 erfüllen. Dieses besagt ja auch u.a. dass die TD des Herstellers komplett bei uns vorliegen muss. Der Hersteller möchte dies nicht.
Nun meine Frage:
1. Wenn ich das MDCG 2020-2 Dokument richtig interpretiere, dann gilt für die MP der Klasse I (keine Messfunktion…)(Konformitätsbewertungsverfahren nach Anhang I und Anhang II MDD laut Konformitätserklärung des OEM vom 11.01.2020) keine verlängerte Übergangsfrist, oder sehe ich das falsch?
2. Was heißt das für uns wenn wir als Hersteller auftretten möchten bzgl. technische Dokumentation und Inverkehr bringen?
Vielen Dank im voraus für ihre Bemühungen
Sehr geehrter Herr Quadt,
zu 1. Es gibt in diesem Fall in der Tat keine erweiterte Übergangsfrist. Sie müssten also für eine Inverkehrbringung ab dem 26.05.2021 MDR-Konformität erklären.
zu 2. Sie müssten sämtliche Verpflichtungen aus Artikel 10 erfüllen (u.a. QM-System etablieren, eine technische Dokumentation erstellen und pflegen gemäß Anhang II und III, klinische Bewertung durchführen, etc.) Sie benötigen demnach die TD des Herstellers/OEMs.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Prof. Dr. Johner,
ich habe eine Frage zum Inhalt der Konformitätserklärung von Lagecy-Produkten. Es ist soweit klar, dass wir Produkte (Software stand alone) Klasse 1 nach MDD über den 26.5.2021 hinaus unverändert verkaufen können. Die Konformitätserklärung bezieht sich weiterhin auf die MDD mit Datum vor dem Stichtag.
Meine Frage ist, ob es besondere Anforderungen an den Inhalt der Konformitätserklärung gibt, die das Produkt z.B. als Lagecy-Produkt identifiziert? Für den Betrachter der Konformitätserklärung ist ja nicht ohne weiters ersichtlich, warum nach MDD erklärt wird, eine Prüdung damit sehr schwierig.
Vielen Dank für Ihre Mühe vorab.
Andreas Schrade
Sehr geehrter Herr Schrade,
für die Konformitätserklärung für Legacy-Produkte gibt es keine zusätzlichen Anforderungen. Die Konformität würden Sie weiterhin gemäß MDD erklären. Eine Begründung, warum Sie noch unter der MDD Konformität erklären dürfen, ist nicht Bestandteil der Konformitätserklärung. Dies sollten Sie im Rahmen Ihres QM-Systems bzw. als Aufzeichnung für eine Behörde vorhalten können.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Prof. Dr. Johner,
ich habe eine Frage zu den Übergangsfristen von Produkten die vorher kein Medizinprodukte waren und durch die MDR zu einem Medizinprodukte werden. In diesem Fall Kontaktlinsen ohne Sehstärke. Diese wurden zwar auf die gleiche Weise wie die Kontaktlinsen mit Sehstärke (waren in der MDD schon Medizinprodukt) produziert wurden, jedoch nach MDD kein Medizinproduktwaren.
Welche Übergansfristen gelten in diesem Fall ?
MFG
Frank Hanninger
Sehr geehrter Herr Hanninger,
die Übergangsfristen für die in Anhang XVI genannten Produkte finden Sie in Artikel 1:
„(2) Diese Verordnung gilt des Weiteren ab dem Geltungsbeginn der GS gemäß Artikel 9 für die in Anhang XVI aufgeführten Produktgruppen ohne medizinische Zweckbestimmung, wobei der Stand der Technik und insbesondere bereits geltende harmonisierte Normen für analoge Produkte mit medizinischer Zweckbestimmung, die auf ähnlicher Technologie beruhen, zu berücksichtigen sind. Gegenstand der GS für jede in Anhang XVI aufgeführte Produktgruppe sind mindestens die Anwendung des Risikomanagements gemäß Anhang I für die betreffende Produktgruppe und erforderlichenfalls die klinische Bewertung der Sicherheit.
Die erforderlichen GS werden bis zum 26. Mai 2021 erlassen. Sie gelten nach Ablauf von sechs Monaten ab dem Zeitpunkt ihres Inkrafttretens oder ab dem 26. Mai 2021, wobei das spätere Datum maßgebend ist.“
D.h. sobald die Gemeinsamen Spezifikationen erlassen wurden, gibt es eine Übergangsfrist von 6 Monaten.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Dr. Johner,
Sehe ich das richtig, dass ein MP der Klasse IIb nach MDD welches nach dem 26.05.2017 in Verkehr gebracht wurde weiterhin bis 27.05.2024 auch ohne weiter Bescheinigungen oder Prüfungen auf dem Markt bleiben darf und erst ab 27.05.2024 eine klinische Prüfung nachgewiesen werden muss? Und müssen dies eigene Studien sein?
Vielen Dank für Ihre Mühe!
V. Stockmar
Sehr geehrte Frau Stockmar,
ein Medizinprodukt der Klasse IIb nach MDD darf noch unter der MDD in Verkehr gebracht werden, solange des entsprechende MDD-Zertfikat der benannten Stelle gültig ist. Dies ist maximal der 26.05.2024. Auch unter der MDD werden klinische Daten gefordert. Ob diese von klinischen Prüfungen mit dem eingenen Produkt stammen müssen, ist abhängig von den verfügbaren klinischen Daten. Die Forderung nach einer klinischen Prüfung gemäß MDR besteht generell nur für Produkte der Klasse III und implantierbare Produkte. Dennoch können abhängig von der Datenlage klinische Prüfungen mit dem eigenen Produkt auch für Produkte mit niedrigerem Risiko notwendig sein.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Dr. Johner,
ich habe eine Frage, die sich mir aus der Lektüre der Leserzuschriften ergeben hat:
Es geht mir um MD’s, die unter MDD Klasse 1 eingestuft waren und auch unter MDR Klasse 1 – Produkte bleiben. Diese profitieren nicht von Artikel 120 (3), für diese gibt es lt. Absatz (3) keine Übergangsfrist.
In Art. 120 (4) aber wird direkt nur noch über „Produkte“ allgemein geschrieben, und es wird nicht unterschieden, ob eine Benannte Stelle notwendig ist, und am Ende des Absatzes wird auf Art. 120 Absatz 2 verwiesen, quasi als Gegenstück zu Absatz 4 (Absatz 4: (auch) Artikel, für die keine Benannte Stelle erforderlich ist, also Klasse 1). Diese „Produkte“ dürfen bis 27. Mai 2025 bereitgestellt und in Betrieb genommen werden.
Produkte der Klasse 1, die unter 93/42/EWG vor dem 26. Mai 2021 (Legacy-Produkte) rechtmässig in Verkehr gebracht wurden, entsprechen diese nicht genau der Definition in Art. 120 Absatz (4)?
Auch wurde in diesem Absatz der Begriff „Bescheinigung“ weggelassen, möglicherweise um klarzustellen, dass es sich in diesem Absatz nicht um Produkte handelt, für die eine benannte Stelle erforderlich ist (also alle, außer Klasse 1).
Dieser Absatz sagt auch nichts darüber aus, ob diese Produkte ab dem 26. Mai 2021 eine neue Konformitätserklärung nach (EU) 2017/745 benötigen.
In einer Diskussion vertrat man die Meinung, dass Klasse 1 – Produkte, die auch unter MDR Klasse 1 – Produkte bleiben, ab dem 26. Mai 2021 eine Konformitätserklärung nach (EU) 2017/745 benötigen, andernfalls nicht mehr in
Verkehr (einschließlich bereitgestellt und/oder in Betrieb) genommen werden dürfen. Dies wurde mit dem Text aus dem Artikel 120 (4) begründet. Ich kann diese Interpretation dieses Artikels nicht nachvollziehen, da in diesem Artikel nicht einmal überhaupt verschiedene Klassen genannt werden. Ist denn die o.g. Auffassung mit Artikel 120 (4) vereinbar?
Es scheint so, dass dieser Absatz für die Behandlung von Klasse 1 – Produkten gemacht worden zu sein, die unter der MDR in Klasse 1 bleiben. Etwas plakativ formuliert: War das Klasse 1 – Produkt schon rechtmässig vor 26. Mai 2021 im Markt, und ändert sich bei diesem weder die Zweckbestimmung, noch die Auslegung, kann dieses bis 27. Mai 2025 bereitgestellt, in Betrieb genommen (und auch bis dahin nachproduziert) werden. Bis dahin muß die 2017/745 Konformität vollumfänglich hergestellt und erklärt worden sein (ab 26. Mai 2021 bereits die Anforderungen an Vigilanz, TD, PMS, Verantwortliche Person, jöhrliche Reports, usw., jedoch ohne dies extra zu erklären). Die Konformitätserklärung nach 93/42/EWG bliebe bis zum Ende Ihrer Laufzeit (spätestens bis zum 26. Mai 2024) gültig.
Gäbe es nicht diesen Artikel 120 (4), dann würde ich den Beschreibungen hinsichtlich „Produkt profitiert nicht von Artikel 120 (3)“ zu 100 Prozent folgen können. Aber es gibt ihn nun mal.
Vielen Dank vorab für Ihre Mühe und
mit freundlichen Grüßen
M. Schmidt
Sehr geehrter Herr Schmidt,
vielen Dank für Ihre ausführliche Darlegung. Bezüglich Ihrer ersten Frage umfasst nach unserer Auffassung Art. 120 Absatz (4) auch Produkte der Klasse I jedoch nicht ausschließlich. Dies ist darin begründet, dass der referenzierte Absatz 3 auch Produkte höher Klasse I umfasst.
Hinsichtlich Ihrer zweiten Fragestellung kann ich leider auch nicht nachvollziehen, wie diese Schlussfolgerung(en) getätigt wurden. Gemäß unserer Interpretation dürfen Produkte der Klasse I gemäß 93/42/EWG, welche auch unter der 2017/745 Produkte der Klasse I sind, nur bis zum 26.05.2021 auf Basis der „alten“ Richtlinie (93/42/EWG) Inverkehr gebracht werden. Die Bereitstellung und Inbetriebnahme ist jedoch bis zum 27.05.2025 gestattet (Siehe Abbildung 3).
Viele Grüße
Sehr geehrte Damen und Herren,
wie verhält es sich mit Medizinprodukten (Röntgentherapiegerät), die ohne Konformität und CE erstmals in Verkehr gebracht wurden und nun wieder in Betrieb genommen werden sollen (Standortwechsel)?
Grüße D. Hammer
Sehr geehrte Frau Hammer,
Produkte die nicht rechtmäßig in Verkehr gebracht wurden, dürfen nicht am Menschen eingesetzt werden. Dies ändert sich auch nicht durch einen Standortwechsel und einer neuen Inbetriebnahme. Wenn das Produkt durch ein gültiges MDD-Anhangszertifikat abgedeckt ist und eine entsprechende Konformitätserklärung existiert, könnte ein Rückruf und eine Inverkehrbringung unter der MDD erfolgen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Dr. Johner,
im Juli wurde die 4. Berichtigung der MDR veröffentlicht, für einige Sprachen (unter anderem englisch) gibt es jedoch keine.
Gibt es keine, weil die Änderungen die englische Version nicht betreffen?
Gibt es wie bei Normen Übergangsfristen bei Berichtigungen der MDR?
Viele Grüße
L. Kuipers
Mein Kollege Christopher Seib war so nett, diese Frage zu beantworten:
Genau, das 4. Corrigendum betrifft nur die Korrektur der Übersetzungen (Rechtschreibfehler, Wording). Sofern in der Berichtigung nicht anders festgehalten, gilt sie mit Veröffentlichung im Amtsblatt der Europäischen Union.
Herzliche Grüße
Anja Segschneider | Redaktion
Sehr geehrte Damen und Herren,
vielen Dank für Ihren informativen und hilfreichen Beitrag. Eine Unklarheit hat sich mir noch ergeben, bezüglich Legacy Devices und deren Verpflichtungen und Fristen.
In einem EU Artikel mit dem Titel „Management of Legacy Devices – MDR EUDAMED“ vom 15.2.21, werden, wie bei vielen MDCG Artikeln, die Legacy Devices erwähnt, allerdings mit einem expliziten Verweis:
„Legacy Devices with the risk class I that are not sterile and/or with a measuring function under the Directives cannot be considered as Legacy Devices because they do not require a certificate issued by a Notified Body. These must be registered only as Regulation Devices in EUDAMED within the 18 months after the date of application (or 24 months after the date of publication of the notice referred to in Article 34(3) if EUDAMED is not fully functional before the date of application of the MDR).“
Was heißt das für Klasse I („MDD-Produkte“, die vor dem 26.5.21 „deklariert“ wurden), z.B. „Standalone“-Software? Sind diese dann nur „Legacy Produkte“ im Sinne der MDR und dem Artikel 120 und nicht gemäß der Definition der MDCG Legacy-Device-Dokumente und dessen Fristen/Verpflichtungen? Oder bezieht es sich in diesem spezifischen Artikel ausdrücklich nur auf die Verpflichtung für die Eudamed Registrierung, und ich habe etwas missverstanden? Was hieße das jetzt für die Verpflichtungen von solchen Produkten bezüglich UDI und Eudamed?
Vielen Dank und liebe Grüße
D. Pagel
Sehr geehrter Herr Pagel,
im MDCG-Dokument ist die Rede von Klasse I-Produkten. Für diese gibt es ja keine Übergangsfrist gemäß MDR. Somit fallen diese eben nicht unter die Definition „legacy device“, da sie seit dem 26.05.2021 nur noch unter MDR-konform in Verkehr gebracht werden dürfen. Hier wurden aber die Klasse I-Produkte übersehen, die gemäß MDR höher gestuft werden. Diese dürfen sehr wohl noch bis zum 26.05.2024 in Verkehr gebracht werden unter der MDD, wenn die Konformität vor dem 26.05.2021 erklärt wurde. Diese zählen eben auch zu den Legacy-Devices. Somit gelten hier dieselben Anforderungen an die Registrierung, d.h. eine UDI-Vergabe ist nicht notwendig, eine Registrierung ist notwendig nach Ablauf der Übergangsfristen.
Freundliche Grüße
Luca Salvatore
Lieber Herr Johner,
eine Frage: betrifft die Deadline 2024 nur das in Betrieb nehmen oder betrifft das auch die Nutzung? Sprich, darf der Kunde unsere MDD Produkte auch nach Mai 2025 noch für die Diagnose verwendet oder müssen unsere System dann stillgelegt werden?
Herzlichen Dank!
Frederik Bender
Lieber Herr Bender, vielen Dank für diese spannende Frage.
Die Nutzung des Produktes durch Anwender ist davon unabhängig. Sie als Hersteller legen selbst fest, wie lange die Lebensdauer des Produktes ist und wie lange Sie support bieten möchten. Mehr dazu finden Sie in diesem Artikel:
https://www.johner-institut.de/blog/regulatory-affairs/inverkehrbringung/
Beste Grüße, Sebastian Grömminger
Sehr geehrter Herr Prof. Dr. Johner,
ich habe eine Frage zu den Übergangsfristen, speziell in Verbindung mit den 3 Klassen.
So wie ich die ganze Thematik verstanden habe, reden wir von einer MDR Konformitätsvorgabe bei Klasse I Produkten ab dem 26.05.2021. Würde für mich als Wirtschaftsakteur „Händler“ bedeuten, dass ich bei zum Bespiel einer Lieferung (Deutscher Hersteller beliefert unseren Großhandel) von OP-Masken der Klasse I einer komplette Konformität laut MDR verlangen kann. Jetzt ist bei der 50er OP Masken Verpackung keine UDI drauf. Ich habe dann wiederrum in mehreren weiteren Artikeln gelesen, dass die UDI Vorgabe seit dem 21.05.2021 erst für Klasse III Produkte gilt. Ab 2023 dann für IIa und b und für Klasse I dann ab 2025…
Nach meinem Verständnis gehört die UDI jedoch in die Bewertung der MDR Konformität dazu oder? Meine Frage zielt also in diesem speziellen Fall darauf ab, welche Übergangsfrist für Klasse I dann gilt.
Gilt jetzt generell (ink. UDI): (Hersteller an Großhändler- Herstellungsstufe)
Klasse I: 2021
Klasse Ix -III: 2024
Und ab Handelsstufe: Abverkauf (Bereitstellung) bis 2025
ODER:
die oben beschriebenen sich auf die UDI beziehenden Daten (21/23/25)
FAZIT: Wenn die UDI nicht in die MDR-Konformitätsbewertung (was totaler Quatsch wäre) mit einbezogen werden soll, dann kann ich mit allen Daten mein QMS anpassen. Wenn es jedoch, was nahe liegt, mit einbezogen werden soll, kann nur eine von beiden Varianten betrachtet werden.
Vielen Dank im Voraus für Ihre Mühe und viele Grüße
Sebastian Föllmann
Lieber Herr Föllmann,
bitte haben Sie noch etwas Geduld. Wir werden Ihnen zeitnah antworten.
Herzliche Grüße
Anja Segschneider | Redaktion
Guten Tag Herr Johner,
bezgl. meiner Frage oben welche noch nicht beatwortet wurde, wollte ich nochmals kurz und knackig nachfragen.
Also, wenn wir über Klasse I nach MDR sprechen, dann müssen wir als Wirtschaftsakteur „Händler“ prüfen, ob vollständige MDR-Konformität besteht (CE, UDI, Herstellerangaben, EC-REP usw.) da es keine Übergangsfristen gibt oder?
Ich habe es so verstanden, dass die UDI ganz klarer Bestandteil der MDR-Konformität ist. Ist es dann nicht wiedersprüchlich, das die Produkte der Klasse I ab dem 26.05.2021 mdr konform in den Verkehr gebracht werden müssen (Lieferung Hersteller an Händler), aber eine UDI erst 2025 haben müssen? Ist die UDI nicht das Aushängeschild in Bezug auf MDR-Konformität?
Vielen Dank im Voraus und viele Grüße
Sebastian Föllmann
Sehr geehrter Herr Föllmann,
danke für Ihre Frage zur MDR und den Übergangsfristen.
Es ist in der Tat etwas verwirrend, was „MDR Konformität“ bedeutet. Letztlich meint man damit die Konformität mit den Anforderungen der MDR, die zum jeweiligen Zeitpunkt gelten. Es ist also nicht die Konformität gemeint mit allen Anforderungen, die zu einem späteren Zeitpunkt gültig sind.
Ganz konkret: Wenn der Hersteller noch keine UDI aufbringen muss, dann müssen Sie als Händler auch nicht prüfen, ob diese aufgebracht wurde. D.h. dass Sie als Händler die Übergangsfristen zumindest indirekt auch betreffen.
Mit herzlichen Grüßen, Christian Johner
Guten Tag,
ich wollte fragen, ob irgendwo steht, die Übergangsfristen von IVDR-Legacy devices?
Vielen Dank im Voraus!
MfG
rONY
Sorry! Ich meine Übergangsfristen zur Registrierung von Legacy devices in EUDAMED.
MFG
Rony
Hallo Rony,
danke für Ihre Frage zur Registrierung von IVD-Legacy-Devices.
Art. 110 der IVDR besagt, dass mit Geltungsbeginn der IVDR am 26. Mai 2022 die Registrierung von Wirtschaftsakteuren und Produkten auch für Legacy Devices gemäß IVDR zu erfolgen hat.
Die Übergangsfristen der IVDR wurden erst kürzlich angepasst, diese Forderung bleibt jedoch erhalten. Mehr dazu finden Sie auch in unserem Artikel: https://www.johner-institut.de/blog/johner-institut/uebergangsfristen-der-ivdr/
Das MDCG-Dokument MDCG 2019-5 unterstreicht dies und gibt zusätzliche Hinweise, was bei der Registrierung von Legacy Devices (sowohl Medizinprodukte als auch IVD) in der EUDAMED zu beachten ist.
Beste Grüße
Sophie Bartsch
Sehr geehrter Herr Prof. Dr. Johner,
vielen Dank für Ihre informativen und den hilfreichen Beitrag.
Können Sie mir sagen, woran ein Importeur eines MDD Klasse I Produktes (ohne r,s,m Funktion) entscheiden kann, ob er es nach dem 26.05.2021 in Verkehr bringen darf. Für ein „reguläres“ MDD Klasse I Produktes wäre dies nicht erlaubt. Falls eine Höherstufung nach MDR vorliegen würde, wäre das Inverkehrbringen wiederum erlaubt. Meine Frage ist, woran kann ein Importeur die Höherstufung nach MDR erkennen? In meinem Verständnis befindet sich diese Information nicht auf der Konformitätserklärung des Herstellers.
Viele Grüße
Reinhard Köhler
Sehr geehrter Herr Köhler,
Artikel 13 der Medizinprodukteverordnung (MDR) definiert, welchen Pflichten Importeure nachkommen müssen. Unter Punkt (2) ist hierbei folgendes definiert:
[…] Ist ein Importeur der Auffassung oder hat er Grund zu der Annahme, dass ein Produkt nicht den Anforderungen dieser Verordnung entspricht, darf er dieses Produkt nicht in Verkehr bringen, bevor die Konformität des Produkts hergestellt ist; in diesem Fall informiert er den Hersteller und den Bevollmächtigten des Herstellers […]
Somit ist der Importeur verpflichtet, die entsprechende Information (Medizinprodukte Klassifizierung) von dem Hersteller einzuholen, um seinen Pflichten nachzukommen.
Viele Grüße
Sehr geehrter Hr. Johner,
ich hätte ein Frage zur Kennzeichnung des Importeurs.
Wir erfüllen für einen Hersteller aus einem Drittland sowohl die Rolle des EU-REP als auch die Rolle des Importeurs.
Zusätzlich haben wir exklusive Vertriebsrechte für ganz Europa.
Besteht in diesem Fall die Möglichkeit, auf die Angabe des Importeurs zu verzichten?
Grüße
H. Simon
Lieber Herr Simon,
bitte haben Sie noch etwas Geduld. Wir werden Ihnen zeitnah antworten.
Herzliche Grüße
Anja Segschneider | Redaktion
Sehr geehrter Herr Simon,
aus meiner Sicht können Sie auf die Angabe des Importeurs nicht verzichten. Artikel 13 Punkt (3) der MDR definiert, welche Kennzeichnungspflichten Importeure erfüllen müssen. Eine Ausnahme von diesen Pflichten kann ich Artikel 13 nicht entnehmen.
Herzliche Grüße
Sehr geehrter Herr Prof. Johner,
es geht um ein Produkt – in diesem Fall eine medizinisch-pharmaklogische Software – die bisher kein MP ist aber nach der neuen MDR zertifiziert werden muss.
Hat dieses Produkt noch eine Übergangsfrist oder ist diese bereits abgelaufen?
Vielen Dank & freundliche Grüße,
Lars Frohn
Sehr geehrter Herr Frohn,
für Produkte, die keine MPs waren, gab und gibt es keine Übergangsfrist. Daher müssen Sie den „normalen“ Weg gehen.
Geben Sie einfach Bescheid, wenn wir dabei helfen können.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Prof Johner,
Ich hätte eine Frage zu einem Medizinprodukt der Klasse I, das vor dem Beginn der MDR unter MDD konform erklärt wurde (2020) und nun weder für MDR noch als Legacy device unter MDD weiter angemeldet wurde. Es soll(te) sozusagen „auslaufen“. Die Konformitätserklärung wurde mit einem Gültigkeit auf 3 Jahre versehen, d.h. bis 2023.
Wie lange dürfen die verkauften MP, die sich bereits im Mark befinden noch von den Benutzer angewendet werden? Bis zum Ablauf der Konformitätserklärung?
Vielen Dank und viele Grüße
Sehr geehrte Frau Schulz,
die MDR macht keine Vorgaben für die Verwendung der Produkte, sondern nur für
D.h. wenn es eine Begrenzung gäbe, dann würde sie von den Herstellern kommen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ich habe die vielen Anfragen dieses Blogs durchstöbert, aber keine Frage+Antwort zur MDD-Konformitätserklärung wie folgt gefunden (ich hoffe, mir ist nichts entgangen):
Beispiel Klasse IIa MDD-Produkt:
Die Abverkaufsregelung erlaubt es den Händlern, MDD-Produkte bis 26.05.2025 abzuverkaufen.
Nun kann eine MDD-Konformitätserklärung allerdings nur bis maximal 26.05.2024 ausgestellt werden (abhängig von der zugrunde liegenden Frist der relevanten Bescheinigung).
Wenn nun der Händler uns als Hersteller ab 27.05.2024 nach einer Konformitätserklärung eines noch-nicht-MDR-Produkts fragt, können wir ihm nichts anderes als die abgelaufene Konformitätserklärung vorweisen.
-> Ist dies eine Art Definitionslücke?
Laut Richtlinie 93/42/EWG muss ein auf dem Markt befindliches Medizinprodukt ja eine gültige Konformitätserklärung besitzen. Aber die setzt bei Klasse IIa eine Bescheinigung voraus, die ja aber nur bis 26.05.2024 gültig ist.
Mit freundlichen Grüßen
Uwe Boetcher
Sehr geehrter Herr Boetcher,
es muss hierbei zwischen „Inverkehrbringung“ und „Bereitstellung, Inbetriebnahme“ unterschieden werden. Die Inverkehrbringung ist bis zum Zeitpunkt des Datums der Bescheinigung jedoch maximal bis zum 27.05.2024 möglich. Die „Bereitstellung, Inbetriebnahme“ ist darüber hinaus bis zum 27.05.2025 möglich. Somit würden die Händler im Rahmen des Abverkaufs (Bereitstellung, Inbetriebnahme) die zum damaligen Zeitpunkt der Inverkehrbringung gültige Konformitätserklärung mit dem spätesten Datum 27.05.2024 erhalten.
Herzliche Grüße
Sehr geehrter Hr. Johner,
die Fragen hatte ich schon bereits gestellt gehabt, aber leider noch keine Antwort. Ich würde mich über eine kurze Stellungnahme freuen.
Fallbeispiel: Hersteller ABC aus USA hat 1000 Stück von Produkt A und 800 Stück von Produkt B produzier. Der Hersteller hat bereits von Produkt A (100 Stück) vor 25.05.2021 nach Frankreich versendet gehabt und von Produkt noch gar nichts. Nun möchte der Hersteller seine Produkte A und B (Klasse I nach MDD und MDR; CE und ISO 13485 läuft 01.06.2024 aus) nach DEU verkaufen. Konformitätserklärung ist noch nach MDD wurde 01.06.2020 erstellt.
1 Frage: Da der Hersteller Produkt A nach Frankreich vor 25.05.2021 verkauft hat, ist es damit in Verkehr gebracht?
Somit..
2. Frage: Darf der Hersteller die Reste (900 Stück) von Produkt A an uns liefern mit einer MDD Konformitätserklärung (Artikel 120 (3) MDR) oder muss der Hersteller eine Konformitätserklärung nach MDR ausstellen, da es für Produklasse I keine Bennante Stelle braucht. Könnte der Hersteller es bis 2025 abverkaufen?
3. Frage: Nun wir benötigen mehr als 900 Stück von Produkt A, darf der Hersteller ohne irgw. Veränderungen an Produkt o. Zweckbestimmung weiter produzieren?
4. Verstehe ich Bue Guide 2022 richtig: ich darf nur die Menge an Charge verkaufen, die vor 25.05.2021 produziert und in EU inverkehr gebracht wurde? Wie wurde man Inverkehr nachweisen (Lieferschein?)
Ausschnitt aus der Bue Guide 2022: Hinsichtlich der „Bereitstellung“ bezieht sich der Begriff „Inverkehrbringen“ nicht auf eine Produktart, sondern auf jedes einzelne Produkt, unabhängig davon, ob es als Einzelstück oder in Serie hergestellt wurde. Folglich kann das Inverkehrbringen in der Union für jedes einzelne Produkt nur einmal in der gesamten EU und nicht in jedem einzelnen Mitgliedstaat erfolgen. Daher muss jedes einzelne Produkt eines Produktmodells oder einer Produktart, das nach dem Inkrafttreten neuer Anforderungen in Verkehr gebracht wird, diese erfüllen, auch wenn die Bereitstellung des Produktmodells oder der Produktart vor dem Inkrafttreten neuer Harmonisierungsrechtsvorschriften der Union mit neuen obligatorischen Anforderungen erfolgte.
Ich bedanke mich herzlich und freue mich auf Ihre Antwort
Viele Grüße
J. Müller
Sehr geehrte Frau Müller,
vielen Dank, dass Sie sich so intensiv mit unseren Fachartikeln auseinandersetzen und sich tief in die Thematik eindenken! Es ist uns eine Herzensangelegenheit, auf Verständnisfragen zu unseren Beiträgen möglichst schnell zu reagieren. Bitte haben sie jedoch Verständnis, dass die Vielzahl und detailtiefe Ihrer Fragestellungen die kostenfreien Möglichkeiten übersteigen, weshalb ich Ihnen hierbei ein Beratungsgespräch empfehlen würden. So können wir dann viel gezielter auf Ihre Situation eingehen und Ihnen bestmöglich weiterhelfen.
Herzliche Grüße
Liebe Frau Müller,
mögen Sie diesbezüglich einmal Kontakt mit unseren Berater:innen aufnehmen? In einem Gespräch können diese Ihnen sicherlich besser weiterhelfen. Telefon: +49 (7531) 94500 20 oder per E-Mail an [email protected] .
Herzliche Grüße
Anja Segschneider | Redaktion
Hallo liebes Johner Team,
ich stelle mir die Frage, ob es einen Tippfehler im Schaubild zu den Übergangsfristen gibt. Für ein MDD Klasse I Produkt, dass gemäß MDR in eine höhere Klasse kommt wird dort als Datum für die Bereitstellung der 26.05.2024 angegeben, also das gleiche Datum wie für die Inverkehrbringung.
Der Fristenrechner liefert aber den 26.05.2025 :
Bescheinigung
Wird das Produkt auf Basis einer Bescheinigung einer benannten Stelle rechtmäßig in Verkehr gebracht?
Ihre Auswahl: Nein (Klasse I)
Änderung unter MDR
Änderung der Klasse unter der MDR?
Ihre Auswahl: fällt unter MDR in höhere Klasse z.B I* oder IIa
Inverkehrbringung
Inverkehrbringung bis 26.05.2024 gestattet.
Bereitstellung, Inbetriebnahme
Inbetriebnahme bis 26.05.2025 gestattet.
Was stimmt nun?
Viele Grüße
Claus Leiendecker
Sehr geehrter Herr Leiendecker,
vielen Dank für Ihren wichtigen Hinweis. In der Tat liefert der Fristenrechner hierbei ein falsches Datum. Diesen Fehler werden wir umgehend korrigieren.
Für den von Ihnen geschilderten Fall gilt, dass die Inverkehrbringung und die Bereitstellung, Inbetriebnahme bis zum 26.05.2024 gestattet ist.
Herzliche Grüße
Guten Morgen Herr Gerhart,
herzlichen Dank für die Stellungnahme. Dann wäre das PDF Merkblatt welches oben verlinkt ist vermutlich auch nicht ganz korrekt, oder. Darin findet sich eine Korrektur des Klasse I Zweiges im Schaubild. Bereitstellung & Inbetriebnahme wurde vom 26.05.2024 auf den 26.05.2025 geändert. Scheinbar auf Basis Anpassungen zur MDR vom April 2020. Alles ziemlich verwirrend. 🙂
Viele Grüße
Claus Leiendecker
Sehr geehrter Herr Leiendecker,
nochmals herzlichen Dank für Ihr wachsames Auge. Auch dies werden wir direkt mit prüfen.
Herzliche Grüße
Hallo liebes Johner-Team,
zunächst vielen Dank für Ihren Artikel zu den MDR-Übergangsfristen! Wir diskutieren gerade folgendes Problem aus Betreibersicht: ein nach MDD Klasse II a Medizinprodukt wurde in Übereinstimmung mit den Übergangsfristen in 2022 gekauft und in Betrieb genommen. Das Produkt hat ein nach dem 25.05.2017 ausgestelltes Richtlinienzertifikat (93/42/EWG) , welches eine Laufzeit bis zum 26. Mai 2024 hat. Eine „Rezertifizierung“ nach MDR 2017/745 wird herstellerseitig nicht erfolgen – das MP ist nach MDR in die Klasse II b einzuordnen und muss lt. Herstellerangaben „an die Anforderungen der MDR 2017/745 angepasst“ werden.
Welche Risiken ergeben sich für den Betreiber, wenn er das MP NACH dem 26.05.2024 weiter betreibt? Nach MPDG und MPBetreibV dürfen MP nur betrieben und angewendet werden, wenn sie den gesetzlichen Vorgaben entsprechen … das hier gegenständliche MP entspricht den Vorgaben der MDR 2017/745 jedoch nicht und die Übergangsfrist ist abgelaufen. In § 11 MPDG heißt es: „Medizinprodukte dürfen nicht betrieben oder angewendet werden, wenn sie Mängel aufweisen, durch die Patienten, Beschäftigte oder Dritte gefährdet werden können.“ Ist der Fakt, dass ein MP den Vorgaben der MDR nicht entspricht, ein „Mangel“ i.S.d. § 12 MPDG? Und was ist nun mit § 77 MPDG (Überprüfung der „Beachtung medizinprodukterechtlichen Vorschriften)?
Kommt es bei Anwendung des MP zu einem Patientenschaden, ist der Betreiber u.a. dafür beweispflichtig, dass das MP den geltenden Sicherheitsanforderungen entspricht – wie soll er das tun, ohne auf eine gültige Konformitätsbescheinigung verweisen zu können?
Hören wir hier „die Flöhe husten“ oder besteht hier tatsächlich ein erhöhtes Haftungsrisiko für den Betreiber?
Herzlichen Dank und beste Grüße
Gerhild Klinkow
Sehr geehrte Frau Klinkow,
danke für die spannenden, umfangreichen und komplexen Fragen, welche ich in diesem Rahmen leider nicht vollumfänglich würdigen kann. Meine Gedanken hierzu sind, dass Sie das richtige Gespür haben. Es können Haftungsrisiken auf Sie als Betreiber übergehen, wenn Sie beispielsweise die Rahmenbedingungen für den ordnungsgemäßen Betrieb, welcher seitens des Herstellers vorgeben wurde, aufgrund von bspw. technischen Neuerungen anpassen müssen. Des Weiteren dürfen Sie die seitens des Herstellers definierte Produktlebensdauer nicht überschreiten.
Ein Mangel liegt aus meiner Sicht nicht vor, da das Medizinprodukt zum Zeitpunkt der Inbetriebnahme den damalig gültigen Rechtsvorschriften entsprochen hat. Dies bedingt natürlich, dass das Produkt unverändert betrieben wird. Bezüglich der Beweispflicht können Sie auf die damalig gültige Konformitätserklärung verweisen sowie auf die Einhaltung der aktuellen Anforderungen der Medizinproduktebetreiberverordung. Auch dies bedingt, dass keine Veränderungen an dem Produkt vorgenommen oder durchgeführt worden sind.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
vielen Dank für Ihre Antwort, aus der sich wertvolle Anregungen für das QM- u. Risikomanagement ergeben!
Herzliche Grüße
Gerhild Klinkow
Sehr geehrter Herr Gerhart,
der Vorschlag der Kommission vom 6.1.2023 verlängert die Übergangsfristen nur für Produkte mit EU-Bescheinigungen und für solche Produkte, die die Einbindung einer Benannten Stelle erfordern, wie Kl. Ir. Für die restlichen Klasse-I-Produkte, die nicht Is, Im oder Ir sind, lief die Frist am 26.05.2021 ab. Könnten Sie dies bitte präzisieren?
Vielen Dank.
Mit freundlichen Grüßen
Michael Lang
Sehr geehrter Herr Lang,
ich bin unsicher, an welcher Stelle Sie eine Präzision wünschen. Können Sie mir hierbei helfen?
Herzliche Grüße
Sehr geehrter Herr Gerhart,
das Instituts-Journal 01/23 nennt folgende Übergangsfristen:
Restliche Klasse-I-Produkte, für die unter der MDR eine benannte Stelle einbezogen werden muss (z.B. Produkte der Klasse Ir): 31.12.2028
Restliche Klasse-I-Produkte: 26.05.2021 (wie bisher)
Dies steht im Widerspruch zur Aussage im Artikel:
Restliche Klasse-I-Produkte: 31.12.2028
Mit besten Grüßen
Michael Lang
Sehr geehrter Herr Lang,
herzlichen Dank für Ihr wachsames Auge. Ich habe die entsprechende Stelle soeben korrigiert.
Herzlichen Dank und Grüße
Liebes Johner-Team,
ich bin mir nicht ganz sicher, ob ich die UDI-Kennzeichnungspflicht richtig verstanden habe. Wir stellen Medizinprodukte der Klasse IIa und IIb her und haben jeweils noch ein gültiges EG-Zertifikat nach MDD bis Mai 2024.
Gehe ich richtig in der Annahme, dass für diese Legacy Devices die UDI Kennzeichnungspflicht nicht ab Mai 2023, sondern ab Zulassung nach MDR also Mai 2024 besteht?
Wird die UDI-Kennzeichnungspflicht von dem Vorschlag für eine weitere Verlängerung der Übergangsfristen vom Jänner 2023 betroffen sein?
Meine Annahme ist bisher, dass ab dem Zeitpunkt der MDR Zulassung die Produkte mit einem UDI gekennzeichnet sein müssen.
Vielen Dank für Ihre Unterstützung.
Schöne Grüße, Maxi
Sehr geehrter Herr Fürhauser,
der „Unique Device Identification (UDI) System – FAQs“ beantwortet unter Punkt 7, wie mit „legacy devices“ (Produkte, welche ein gültiges Zertifikat besitzen) bzgl. UDI umzugehen ist. Hierbei ist definiert, dass „legacy devices“ nicht den UDI-Verpflichtungen unterliegen.
Bzgl. ihrer weiteren Frage sieht der aktuelle Vorschlag bzgl. der Anpassung der Medizinprodukteverordnung nach meinem Kenntnisstand keine Änderung oder Ergänzung der UDI-Kennzeichnungspflicht vor.
Herzliche Grüße
Hallo liebes Johner Team,
ich hätte eine Frage zum Thema – Kennzeichnung des Importeurs auf beim Produkt.
Lt. Art 120 (3) muss die Registrierung von Wirtschaftsakteuren (dh auch des Importeurs) bereits bei MDD zugelassenen Produkten erfolgen.
Muss der Importeur dann auch schon verpflichtend auf der Kennzeichnung von MDD konformen Produkten (dh „Legacy“-Produkten) angeführt sein?
Vielen Dank für Ihre Rückmeldung & Unterstützung.
Schöne Grüße,
Sonja
Sehr geehrte Frau Holub,
gemäß der Medizinprodukterichtlinie (MDD) muss auf dem Label – bei importierten Produkten – nur der Authorized Representative genannt werden und nicht zusätzlich der Importeur.
Herzliche Grüße
Sehr geehrtes Johner Team,
ich habe eine Frage zu den Zertifikaten:
Wird das MDD Zertifikat ungültig sobald man ein MDR Zertifikat hat (für die gleichen Produkte) oder kann man das MDD- und das MDR Zertifikat parallel nutzen?
Als Beispiel sei die OEM-PLM Konstellation genannt. Kann ich eine bestehende OEM-PLM Konstellation auf Basis eines MDD Zertifikates bis zum Ende der neuen Übergangsfrist betreiben und parallel die Produkte unter eigenem Namen unter der MDR vermarkten?
Herzliche Grüße
Sehr geehrter Herr Steger,
das MDD-Zertifikat ist lediglich solange gültig, wie Sie gebrauch von Artikel 120 der MDR machen. Sobald ein MDR-Zertifikat, für das/die gleiche(n) Produkt(e) vorliegt, verliert das MDD-Zertifikat die Gültigkeit, da Sie von Artikel 120 keinen Gebrauch mehr machen können.
Herzliche Grüße
Hallo Herr Gerhart.
Sie schreiben:
„Sobald ein MDR-Zertifikat, für das/die gleiche(n) Produkt(e) vorliegt, verliert das MDD-Zertifikat die Gültigkeit, da Sie von Artikel 120 keinen Gebrauch mehr machen können.“
Gilt das auch vor dem 26.05.2024, oder nur unter der neuen verlängerten Übergansfrist?
Woraus ist konkret das zu entnehmen?
Ich denke das ist ein sehr wichtiger Frage für viele Hersteller ist, die zum Beispiel nach Erteilung des MDR-Zertifikates nicht ad hoc auf MDR umstellen können oder wollen (z. B. um Restbestände des legacy device abverkaufen zu können, weil die UDI noch nicht implementiert ist etc.)
Recht herzlichen Dank schon im Voraus für Ihre Antwort.
Guten Morgen Herr Flötotto,
ich antworte gerne stellvertretend für Herrn Gerhart.
Die Aussage ist unabhängig von den neuen Übergangsbestimmungen. Wir wissen allerdings, dass benannte Stellen unter Umständen eine Karenzzeit geben, in der das alte MDD-Zertifikat noch gültig bleiben darf, eben genau um die Probleme mit dem Umstellen der Produktion bzw. Umlabeln zu minimieren. Ich empfehle, dies mit Ihrer benannten Stelle zu besprechen.
Über die Konformitätserklärung müssen Sie dann eindeutig auf die entsprechenden Chargen referenzieren und angeben welche Vorschrift (MDD oder MDR) greift.
Herzliche Grüße
Luca Salvatore
Hallo zusammen,
ich möchte zu der Frage von Hr. Flötotto noch hinzufügen, dass wir an der Stelle auch die Non-EU Länder mit unterschiedlichen Registrierungszeiten betrachten müssen, die u. U. auch noch mit MDD Produkten beliefert werden müssen, auch wenn bereits ein MDR EU Zertifikat mit dem entsprechenden Produktscope vorliegt.
Insofern ist Ihre Aussage, Hr. Gerhart regulatorisch korrekt, praktisch aber so nicht umsetzbar.
Und wie ist es, wenn nicht einzelnen Produkte, sondern Produktgruppen mit gemeinsamer EMDN und identischem Intended Purpose (IIb) auf dem EU Zertifikat stehen und die Produkte innerhalb der Produktgruppen erst zu unterschiedlichen Zeitpunkten tatsächlich MDR-ready werden (z. B. bedingt durch Produktionsabläufe)?
Viele Grüße
Wulfgäng
P.S.: Muss leider ein Pseudonym verwenden, da ich von unserer Rechtsabteilung „gebeten“ worden bin, keine firmeninternen Problemstellungen in öffentlich zugänglichen Foren zu diskutieren.
Hallo Wulfgäng,
korrekterweise dürfte die Benannte Stelle das MDR-Zertifikat in diesem Fall nur ausstellen, falls die vom Zertifikat erfasste Produktgruppe insgesamt MDR-konform ist. Sonst wäre es ja theoretisch möglich, dass Sie gewisse Produkte aus dieser Gruppe, welche noch nicht auf MDR-Konformität gebracht wurden, unter dem neuen Zertifikat in Verkehr bringen.
Andererseits müssen Sie der Benannten Stelle im Rahmen der Zertifizierung die Produktgruppen und zugehörigen Produkte nennen, welche Sie umstellen möchten. Diese „Liste“ dürfen Sie dann später (nach Zertifikatserteilung) ergänzen um weitere Produkte, nach Mitteilung an die Benannte Stelle. Somit weiß die Benannte Stelle jederzeit, welche Produkte pro Produktgruppe von welchem Zertifikat erfasst sind und welche (noch) nicht. Somit sollte das genannte Vorgehen in Absprache mit der Benannten Stelle möglich sein.
Freundliche Grüße
Luca Salvatore
Liebes Johner Team,
vielen Dank für die Zusammenfassung des neuen Proposals!
Eine Frage jedoch hierzu:
Woher kommt ihr zu dem Schluss, dass Klasse I Produkte, die Voraussetzung des Antrags bei der benannten Stelle nicht betrifft?
„Antrag bei Benannter Stelle spätestens am 26.05.2024 (betrifft nicht Klasse-I-Produkte)“.
Das Proposal sagt doch:
(3d) Produkte dürfen nur bis zu den in den Absätzen 3b und 3c genannten Zeitpunkten in Verkehr gebracht oder in Betrieb genommen werden, wenn die folgenden Bedingungen erfüllt sind:
[ …]
e) der Hersteller oder ein Bevollmächtigter hat spätestens am 26. Mai 2024 einen förmlichen Antrag gemäß Anhang VII Abschnitt 4.3 Unterabsatz 1 auf Konformitätsbewertung für ein in den Absätzen 3b und 3c genanntes Produkt oder für ein Produkt, das dazu bestimmt ist, dieses Produkt zu ersetzen, gestellt, und die Benannte Stelle und der Hersteller haben spätestens am 26. September 2024 eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 unterzeichnet.
Die Produkte unter 3 c) wären doch eben die Klasse I Produkte, die unter MDR eine benannte Stelle brauchen. Somit müssten auch diese einen Antrag bei der BS einreichen.
Beste Grüße
Philipp Malsch
Sehr geehrter Herr Malsch,
die Aussage „betrifft nicht Klasse-I-Produkte“ innerhalb des Satzes „Antrag bei Benannter Stelle spätestens am 26.05.2024 (betrifft nicht Klasse-I-Produkte)“ bezieht sich nur auf Klasse-I-Produkte und nicht auf Produkte der Klasse Is, Im. Für die zuletzt genannten muss gemäß Abschnitt 3d bzw. indirekt 3b (b) eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 zwischen Hersteller und Benannter Stelle unterzeichnet werden.
Herzliche Grüße
Hallo liebes Johner Team,
bei den geplanten Änderungen zur verlängerung der Übergangsbestimmungen ist mir bei Ihren Aussagen zu den Voraussetzungen eine Frage aufgekommen:
Sie schreiben:
„Antrag bei Benannter Stelle spätestens am 26.05.2024“
Im Vorschlag der Kommission (Proposal for a REGULATION OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL – https://ec.europa.eu/info/law/better-regulation/) steht:
„have signed a written agreement“ was ich mit „haben eine schriftliche Vereinbarung oder Vertrag“ übersetzen würde. Also etwas schärfer formuliert, als Sie schreiben.
Wie würden Sie das interpretieren?
Auch insgesamt ist dieser gesamte Absatz, der unten auf Seite 7 des genannten Dokuments beginnt, wichtig als jetzt neu hinzugekommenen Voraussetzung zur Nutzung der Übergangsfristen und ich denke eine nähere Betrachtung dieses Absatzes würde gut in Ihren Fachartikel passen.
Viele Grüße
Frank Dutschke
Sehr geehrter Herr Dutschke,
wir haben den Artikel inzwischen grundlegend überarbeitet und die neuen Übergangsfristen betrachtet.
In der deutschen Fassung heißt es „förmlicher Antrag“ bis zum 26.05.2024. Der Vetrag hingegen muss bis zum 26.09.2024 unterzeichnet sein.
Unklar ist, ob der Antrag bis zum genannten Datum nur gestellt oder auch bereits durch die Benannte Stelle geprüft und akzeptiert sein muss. Wir gehen aktuell von ersterem aus.
Herzliche Grüße
Luca Salvatore
Sehr geehrtes Johner-Team,
Sie schreiben, dass die UDI Kennzeichnungspflicht für Klasse IIa und Klasse IIb Produkte ab dem 26.05.2023 verpflichtend ist, jedoch bei wiederverwendbaren Produkten 2 zusätzliche Jahre gewährt werden.
Gilt dies auch für die Produktverpackung?
In unserem speziellen Fall handelt es sich um einzeln verpackte und wiederverwendbare Klasse IIa und IIb Produkte. Die Basis-UDI-DI ist bereits generiert. Die GTIN (UDI-DI) sind für die meisten Produkte bereits vergeben.
Sind nun alle wiederverwendbaren Produkte einschließlich deren einzelnen Produktverpackungen der Klasse IIa und Klasse IIb, für die UDI-Kennzeichnung, von der weiteren Übergangsregelung bis zum 26.05.2025 betroffen?
Besten Dank im voraus und viele Grüße nach Konstanz.
Felix Wagner
Guten Tag Herr Wagner,
die zusätzlichen 2 Jahre gelten nur für die direkte Markierung auf dem Produkt. Somit wäre in Ihrem Fall der 26.05.2023 für die Kennzeichnung der Produktverpackung relevant.
Herzliche Grüße
Luca Salvatore
Hallo liebes Johner Team,
ich hätte ein Frage bezüglich MDR Artikel 120 Absatz 3 (e). Der Hersteller muss bis zum 26. Mai 2024 ein Antrag bei einer benannten Stelle einreichen. Was ist wenn das MDD Zertifikat, vor dem 26. Mai 2024 abläuft? Muss vor Ablauf des MDD Zertifikats, ein Vertrag mit deiner benannten Stelle vorliegen oder ist die Frist trotzdem der 26. Mai 2024 und bis dahin ist das Zertifikat weiterhin gültig?
Guten Tag,
das Datum auf dem MDD-Zertifikat ist in diesem Fall nicht mehr relevant. Es behält weiterhin Gültigkeit, wenn die genannten Voraussetzungen erfüllt sind.
Somit ist die Bedingung zum förmlichen Antrag zum 26.05.2024 unabhängig von dem auf dem Zertifikat angegebenem Datum.
Freundliche Grüße
Luca Salvatore
Vielen Dank für die schnelle Antwort. Was ist jedoch, wenn das MDD Zertifikat abläuft, bevor die Entscheidung der Änderung der MDR bzgl Artikel 120 im europäischen Amtsblatt veröffentlicht wird.
In diesem Fall muss entweder vor Ablauf des Zertifikats ein Vertrag zur Konformitätsbewertung nach MDR mit einer benannten Stelle unterzeichnet vorliegen oder eine Sonderzulassung (Artikel 59) durch das BfArM oder Fristsetzung durch die Landesbehörde (Artikel 97).
Freundliche Grüße
Luca Salvatore
Guten Tag liebes Johner-Team,
danke für Ihre großartige Arbeit bei der Aufklärung der MDR-Übergangsbestimmungen.
Mir stellt sich eine Frage als Händler von Medizinprodukten. Wenn ich Ihre Grafik richtig lese, dann können Händler Ware (Klasse I* oder höher), für die keine MDR-Bescheinigung angestrebt wird, bis Mai 2024 auf Lager geben sofern das MDD-Zertifikat noch gültig ist und diese Ware anschließend ohne Frist auf dem Markt bereitstellen. Ist das so korrekt?
Freundliche Grüße
Karin Wiesinger
Guten Tag Frau Wiesinger,
ich bin Ihrer Meinung. Solange die Produkte rechtmäßig in den Verkehr gebracht wurden, können diese im Anschluss ohne Abverkaufsfrist weiter bereitgestellt werden gemäß den neuen Übergangsbestimmungen.
Freundliche Grüße
Luca Salvatore
Hallo liebes Johner Team,
habe ich es richtig verstanden? Der Ansatz in MDCG 2022-18 in Bezug auf Art. 97 MDR ist nur eine Übergangsmaßnahme bis zum Inkrafttreten der Änderungsverordnung? Bedeutet dies, dass, wenn die MDD-Bescheinigung eines Herstellers vor Inkrafttreten des Vorschlags abläuft, der Hersteller nicht mehr Art. 97/MDCG 2022-18 beanspruchen kann und daher nicht von der Verlängerung der Gültigkeit der MDD-Bescheinigung profitieren kann, wenn er nicht zuvor einen Vertrag mit einer benannten Stelle abgeschlossen hat? In der Änderungsverordnung heißt es:
„Am 9. Dezember 2022 veröffentlichte die Koordinierungsgruppe Medizinprodukte ihr Positionspapier MDCG 2022-18, in dem ein einheitliches Vorgehen der zuständigen Behörden bei der Anwendung von Marktüberwachungsmaßnahmen dargelegt wird, um die Lücke zwischen dem Ablaufen der Bescheinigungen gemäß der Richtlinie über Medizinprodukte oder der Richtlinie über aktive implantierbare medizinische Geräte und der Ausstellung von Bescheinigungen gemäß der Verordnung über Medizinprodukte zu überbrücken. Dieser Ansatz soll eine befristete Maßnahme sein, bis die in diesem Vorschlag enthaltenen Änderungen der Rechtsvorschriften wirksam werden. Der Ansatz trägt dazu bei, eine Unterbrechung der Versorgung mit Medizinprodukten auf dem EU-Markt zu vermeiden…“
Viele Grüße
Liebe Marie M.,
falls die MDD-Bescheinigung vor Inkrafttreten der neuen Übergangsbestimmungen abläuft, dann darf der Hersteller entweder Artikel 97 MDR anwenden oder er müsste einen Vertrag mit einer benannten Stelle geschlossen haben vor Ablauf. Dies sind die in der neuen Verordnung genannten Optionen (siehe Artikel 120(2)). Alternativ wäre auch eine Sonderzulassung nach Artikel 59 möglich, welche aber nur in Sonderfällen ausgestellt wird.
Freundliche Grüße
Luca Salvatore
Lieber Herr Salvatore,
am 20.03.2023 wurden neue Übergangsfristen veröffentlicht. In welchen Fällen würde es jetzt noch Sinn machen, das Verfahren für Legacy Devices gemäß Artikel 97 (MDCG 2022-18) zu beantragen?
Freundliche Grüße
S. Aslani
Guten Tag Frau Aslani,
durch die Verlängerung der Übergangsfristen ist eine Anwendung des Artikel 97 im Zusammenhang mit der Umstellung von MDD auf MDR nicht mehr relevant, zumindest solange die MDD-Zertifikate noch Gültigkeit besitzen. Ansonsten kann Artikel 97 zum Einsatz kommen bei bestehenden MDR-Zertifikaten oder neuen Produkten im Rahmen der MDR-Zertfizierung, in Fällen in denen Nichtkonformitäten festgestellt wurden (ohne unvertretbares Risiko) z.B. bei der Begutachtung der technischen Dokumentation.
Freundliche Grüße
Luca Salvatore
Lieber Herr Salvatore,
vielen Dank für die Antwort. Angenommen das bestehende MDD Zertifikat eines Herstellers (Klasse IIb) läuft im Jahr 2024 ab und der Hersteller ist noch nicht bereit für die Zertifizierung nach MDR. Wird in diesem Fall das Zertifikat noch einmal unter der MDD ausgestellt oder sollte der Hersteller den Antrag nach Artikel 97 stellen?
Freundliche Grüße
S.Aslani
Liebe Frau Aslani,
das Zertifikat behält in diesem Fall Gültigkeit bis Ende 2027/2028 (je nach Risikoklasse). Eine erneute Ausstellung des MDD-Zertifikats erfolgt nicht, dennoch bleibt es gültig. Um allerdings nach Mai 2024 weiter in den Verkehr bringen zu dürfen, müssen Sie die in der Verordnung genannten Voraussetzungen erfüllen (u.a. QM-System nach MDR, Antrag auf MDR-Zertifizierung).
Freundliche Grüße
Luca Salvatore
Liebes Johner-Team,
Abbildung 1 legt den Gedanken nahe, dass die Inbetriebnahme bereits inverkehrgebrachter Medizinprodukte ohne Berücksichtigung der Voraussetzungen unter den drei Sternen (*** bei Inverkehrbringen) möglich sein könnte. Ist diese Annahme korrekt?
Muss das QMS lt. Art. 10 (9) (Punkt d) der drei Sterne) zertifiziert sein? Wenn ja, muss es nach MDR zertifiziert sein oder genügt ein ISO 13485:2016-Zertifikat?
Wie immer herzlichen Dank vorab!
Freundliche Grüße
Sonja Oechsler
Liebe Frau Oechsler,
die Bedingungen sind vor der Inverkehrbringung sicherzustellen. Diese sollten demnach vor der Inbetriebnahme erfüllt worden sein durch den Hersteller. Der Anwender kann bei Anbringung des CE-Kennzeichens von einer rechtmäßigen Inverkehrbringung ausgehen. Somit sind diese Bedinungen nicht durch die Person, welche die Inbetriebnahme vornimmt, zu prüfen.
Das QM-System muss die Anforderungen des Artikel 10 (9) erfüllen. Eine Zertifizierung ist nicht notwendig. Dies ist genau der Grund für die erweiterten Übergangsfristen, da eben noch keine MDR-Zertifizierung vorliegt bzw. vorliegen muss.
Freundliche Grüße
Luca Salvatore
Liebes Johner-Team,
vielen Dank für Ihren Fachartikel, der viele offene Fragen beantwortet.
Ist bereits bekannt, welche Behörde am Ende zu bewerten hat, ob ein Unternehmen die Voraussetzung für eine Verlängerung des MDD-Zertifikates erfüllt?
Müssten wir als Hersteller hierfür womöglich einen Antrag stellen, und Nachweise liefern, dass wir die Anforderungen erfüllen?
Die Befürchtung besteht, dass dadurch ein erneuter bürokratischer Bottleneck entstehen könnte.
Vielen Dank im Voraus und viele Grüße
Moritz Freimut
Guten Morgen Herr Freimut,
mit Ihrem MDD-Zertifikat bleiben Sie weiterhin unter der Überwachung durch Ihre benannte Stelle (und auch Ihrer zuständigen Landesbehörde). Die benannte Stelle wird in der Überwachung sicherlich prüfen, ob Sie die Bedingungen erfüllen und im negativen Fall das Zertifikat für ungültig erklären bzw. zurückziehen vor Ende der neuen Übergangsfristen. Wir gehen nicht davon aus, dass ein Antrag bei der Landesbehörde für die Verlängerung des Zertifikats notwendig ist. Dies wäre in der Tat ein bürokratischer Bottleneck.
Herzliche Grüße
Luca Salvatore
Hallo,
kurzer Hinweis: Ihr Flow-Chart berücksichtigt der neuen Art. 120 2a nicht, sondern nur 2b.
Viele Grüße
Alexander Walter
Guten Morgen Herr Walter,
Artikel 120 (2)(a) bezieht sich auf die Bedingung der Unterzeichnung eines Vertrags mit der benannten Stelle zur Konformitätsbewertung unter der MDR bei Zertifikatsablauf vor Inkrafttreten der neuen Übergangsbestimmungen. Dies haben wir im Schaubild beachtet in der Raute unten, mittig („Liegt Vertrag mit benannter Stelle…vor“).
Freundliche Grüße
Luca Salvatore
Liebes Johner Team,
Sie schreiben zu Artikel 120 Absatz 2:
„Wenn das Zertifikat der Benannten Stelle hingegen am oder nach dem 26.05.2017 ausgestellt wurde und am 26.05.2021 noch gültig war, behält es seine Gültigkeit bis zu den genannten Fristen (siehe Absatz 3(a))“
Das eine Zertifizierungsantrag gestellt wurde, wäre demnach (anders als bei Produkten deren Zertifikat bereits abgelaufen ist) keine Bedingung für die Nutzung der verlängerte Übergansfrist (so ist es auch Ihrem Fließdiagramm zu entnehmen).
Das scheint im Wiederspruch zu Artikel 120 Absatz 3(c) e zu stehen, nach dem die Fristen nach Absatz 3a ausdrücklich nur dann gelten, wenn eine Zertifizierungsantrag gestellt wurde.
(Die anderen Ausnahmetatbestände nach Artikel 59 und 97 habe ich der Einfachheit halber hier ausgelassen.)
Ich bitte um Erläuterung.
Herzlichen Dank!
Lieber Herr Flötotto,
das ist in der Tat etwas verwirrend. Artikel 120 Absatz 2 betrifft die Gültigkeit des MDD-Zertifikats. Absatz 3 hingegen regelt die Übergangsfrist der Inverkehrbringung unter der MDD. D.h. das Zertifikat würde demnach die Gültigkeit bis zu den genannten Fristen behalten, unabhängig davon, ob ein Zertifizierungsantrag gestellt wurde. Allerdings wäre ohne das Stellen des Zertifizierungsantrags bis zum 26.05.2024 keine weitere Inverkehrbringung möglich. Somit hätte man zwar ein gültiges Zertifikat, allerdings ohne die Möglichkeit einer Inverkerhbringung. Was der Sinn davon ist, erschließt sich mir leider nicht. Eventuell könnte ein Hersteller das (weiterhin gültigte) Zertifikat für die Vermarktung außerhalb der EU nutzen.
Freundliche Grüße
Luca Salvatore
Die Angabe zu den Registrierungspflichten ist zwar auf europäischer Ebene korrekt. Allerdings hat meines Wissens eine Actor-Registrierung nach deutschen Recht jetzt schon in in EUDAMED zu erfolgen. Siehe z.B.: https://www.bfarm.de/DE/Medizinprodukte/Antraege-und-Meldungen/Uebersicht-Meldewege/_node.html
Sehr geehrter Herr Müller,
ganz genau. Für Wirtschaftsakteure mit Sitz in Deutschland gelten die auf der BfArM-Webseite bzw. im Bundesanzeiger veröffentlichten Registrierungspflichten.
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
danke für die rasche Antwort.
Dann schlage ich vor, das oben kurz zu erwähnen; nicht, dass sich Leser sonst ungerechtfertigt in Sicherheit wiegen.
Viele Grüße
Frank Müller
Guten Tag Herr Müller,
den Hinweis haben wir inzwischen im Artikel ergänzt.
Vielen Dank und freundliche Grüße
Luca Salvatore
Guten Tag,
ist es richtig verstanden, dass für die verlängerten Übergangsfristen die Klassifizierung unter MDD anzuwenden ist? Produkte, die durch die MDR höhergestuft werden, werden dann erst nach 2027 bzw. 2028 höher gestuft, oder?
Viele Grüße
Nicole Schuster
Guten Tag Frau Schuster,
die Klassifizierung bezieht sich auf Anhang VIII der MDR, d.h. im Rahmen der Übergangsbestimmungen sind die MDR-Klassifizierungsregeln anzuwenden.
Wenn Sie noch unter einem MDD-Zertifikat in den Verkehr bringen, dann gilt so lange die Klassifizierung der MDD. Sobald Sie MDR-Konformität erklären, dann sind die MDR-Klassifizierungsregeln anzuwenden.
Freundliche Grüße
Luca Salvatore
Ich habe eine Frage bzgl. Produkten, die weiterhin keine NB einbinden, wie Bsp. Sterilisationscontainer.
Seit dem Geltungsbeginn muss doch bereits eine TD und DoC gemäß MDR vorliegen oder erst dann, wenn tatsächlich das QMS auf MDR-Konformität geprüft wurde? Die Fristverlängerungen betrifft dann auch indirekt Produkte die keine NB involvieren?
Vielen Dank
Guten Tag,
Produkte die gemäß MDR weiterhin in die Klasse I fallen, dürfen seit dem 26.05.2021 nur MDR-konform in den Verkehr gebracht werden. Für diese wird also eine TD, DoC und QMS gemäß MDR benötigt. Diese sind nicht von den neuen Übergangsbestimmungen betroffen.
Freundliche Grüße
Luca Salvatore
Liebes Johner-Team,
vielen Dank für Ihre tolle Arbeit und unglaublich informativen Berichte/ Hilfestellungen.
Da gesetztestexte jedoch nicht immer leicht zu lesen sind und manchmal zu viel Inpretationsspielraum zulassen hätte ich noch eine Frage bzgl. Artikel 1; Absatz 3a a) und b).
Sind Schrauben von den verlängerten Übergangsfristen ausgenommen?
Unsere Leseart war bisher immer das (u.a.) Schrauben (well established-technology) von der Frist in Artikel 3a a) ausgenommen sind und aufgrund deren dortigen Nennung ebenfalls von 3a b) ausgeschlossen wären.
Diese Meinung teilen jedoch nicht alle, weshalb wir nun auf Ihre Expertise angewiesen sind.
Vielen Dank und mit freundlichen Grüßen.
Liebe Frau Schwarz,
Schrauben, im Sinne von „well established technology“, sind gemäß der Änderungsverordnung ausgenommen in Absatz 3a a). Da sie nicht von 3a a) abgedeckt sind, gilt für diese IIb-Produkte entsprechend 3a b) („…31 December 2028, for class IIb devices other than those covered by point (a),… „). Sie sind somit nicht von den Übergangsfristen ausgenommen.
Viele Grüße
Luca Salvatore
Sehr geehrte Damen und Herren,
die Anbringung der UDI am Produkt muss bei Klasse I spätestens ab dem 26.05.2025 erfolgen. Gilt diese Übergangsfrist auch für die Nennung der Basis-UDI in der Konformitätserklärung, oder muss diese bereits jetzt erfolgen?
Sehr geehrter Herr Eder,
für die Vergabe der Basis-UDI gibt es keine Übergangsfrist. Diese muss erfolgen und angegeben werden, sobald MDR-Konformität erklärt wird.
Freundliche Grüße
Luca Salvatore
Vielen Dank für die hilfreichen Erläuterungen. Anlässlich einer konkreten Situation habe ich folgende Frage:
Dürfen „Legacy-Produkte“ mit der Zweckbestimmung „The Medical Diode Laser System is intended to clinically remove excessive body hair for hirsutism patients. Operated by doctor“ an nichtmedizinische, gewerbliche Anwender abgegeben werden?
Hintergrund der Frage ist, dass mit der Veröffentlichung der Gemeinsamen Gerätespezifikation MDR Annex XVI und der Durchführungsverordnung 2022/2346 vom 1. Dezember 2022 die 6-monatige Übergangsfrist ausläuft, in der Haarentfernungslaser ohne medizinische Zulassung an gewerbliche Anwender abgegeben werden durften. Der Hersteller hat nun nicht mehr die Wahl, die Haarentfernungsgeräte mit einer nichtmedizinischen Zulassung in der EU in Verkehr zu bringen. Bedeutet dies, dass gewerbliche Anwender, die derzeit eine Ersatz- oder Neuanschaffung planen, mit den Behandlungen warten müssen, bis die ersten Laser der Klasse IIb mit nichtmedizinischer Zweckbestimmung nach Anhang XVI MDR zur Verfügung stehen? Nach meinen Informationen wird dies nicht innerhalb der nächsten 2 Jahre der Fall sein.
Sehr geehrter Herr Freier,
vielen Dank für Ihre Frage!
Die Durchführungsverordnung 2022/2346 legt im Artikel 2 die Übergangsfristen und die daran geknüpften Bedingungen fest. Demnach richtet sich die weitere Inverkehrbringung und Inbetriebnahme von Produkten ohne medizinische Zweckbestimmung nach
– dem Zeitpunkt der ersten Inverkehrbringung
– der Klasse des Produkts
– der Notwendigkeit einer klinischen Prüfung.
Wir haben die möglichen Konstellationen in einem separaten Fachartikel für Sie zusammengefasst und Sie können leicht prüfen, welche Übergangsfrist für Ihr Produkt zutreffend ist.
https://www.johner-institut.de/blog/regulatory-affairs/produkte-ohne-medizinische-zweckbestimmung/
Herzliche Grüße
Manuela Reinhold
Sehr geehrter Herr Johner
Danke Ihnen für die tolle Übersicht.
Kennen Sie sich auch in der Schweiz aus bezüglich Ausnahmeregelung (Art. 59 und Art. 90). Wer sind solche nationale Behörden? Ich nehme mal an das ist das BAG und die Swissmedic. Liege ich damit richtig?
Besten Dank und freundliche Grüsse
Matthias Sägesser
Sehr geehrter Herr Sägesser,
danke für Ihre Frage!
Sie haben die Frage im Kontext des Artikels „Übergangsfristen der MDR“ gestellt. Bezüglich der MDR sind die Behörden in der EU zuständig und zwar bei den Ausnahmen jede für ihr Land. In Deutschland wäre das das BfArM.
Wenn Sie die Übergangsfristen gemäß der MepV meinen, dann ist die entsprechenden Behörde die SwissMedic, nicht das BAG.
Jede Länderbehörde kann nur für ihr Land entscheiden.
In der Hoffnung, Ihre Frage richtig verstanden zu haben und mit vielen Grüßen,
Christian Johner
Sehr geehrter Herr Gerhardt
heisst das, dass alle Produkte, die bereits vor dem 26.05.2021 auf dem Markt, also in Verkehr gebraucht wurden, gelten keine Übergangsfristen (Abverkaufsfristen).
Die neuen Übergangsfristen betreffen aber Produkte, die noch nie auf dem Markt, sprich in Verkehr gebracht worden sind.
Habe ich das korrekt verstanden?
Merci Ihnen und beste Grüsse
Matthias Sägesser
Sehr geehrter Herr Sägesser,
vielen Dank für Ihre Frage!
Die beiden Begriffe „Übergangsfrist“ und „Abverkaufsfrist“ haben eine unterschiedliche Bedeutung. Die verlängerten Übergangsfristen beziehen sich auf das Inverkehrbringen, also die erstmalige Bereitstellung eines Medizinprodukts auf dem Unionsmarkt und betrifft immer jedes einzelne Produkt. Die Abverkaufsfrist limitierte bisher die weitere Bereitstellung bzw. Inbetriebnahme von (bereits in Verkehr gebrachten) Medizinprodukten auf dem Unionsmarkt. Mit der Änderungsverordnung 2023/607 wurden die Fristen für den Abverkauf jedoch vollständig aufgehoben. Demnach dürfen alle Produkte, die vor dem 26. Mai 2021 gemäß den Richtlinien 90/385/EWG und 93/42/EWG rechtmäßig in Verkehr gebracht wurden ohne Frist weiter auf dem Markt bereitgestellt oder in Betrieb genommen werden.
Herzliche Grüße
Manuela Reinhold
Sehr geehrter Herr Johner,
Die OEM-Lieferanten eines Private Label Herstellers planen, ihre MDR-Zertifizierung im Jahr 2024 zu erhalten. Jedoch ist dieser Zeitplan für den PLM realistisch betrachtet nicht umsetzbar. Ist es möglich, dass der PLM während des Übergangszeitraums von den OEM-Lieferanten beliefert wird, wenn die ehemaligen OEM-Lieferanten bereits MDR-zertifiziert sind, während sich der PLM noch im Zertifizierungsprozess befindet? Falls ja, welche Bedingungen müssen erfüllt sein, damit dies möglich ist?
Herzliche Grüsse
Amir Biljali
Sehr geehrter Herr Biljali,
unter der MDR benötigen Sie als PLM eine unabhängige Zertifizierung (inkl. vollständiger Zugriff auf die Technische Dokumentation). Dabei ist es unerheblich, ob der OEM eine Zertifizierung für das Medizinprodukt besitzt.
Wenn Ihre Zertifizierung unter der MDD aktuell auf dem MDD-Zertifikat des OEMs aufbaut (klassische OEM/PLM-Zertifizierung), dann müsste das MDD-Zertifikat des OEMs weiterhin Gültigkeit besitzen, damit Sie rechtmäßig in den Verkehr bringen dürfen. Ich würde empfehlen, dies mit der benannten Stelle zu diskutieren. Eventuell kann das MDD-Zertifikat des OEMS parallel zum MDR-Zertifikat für einen gewissen Zeitraum weiterhin aktiv bleiben.
Freundliche Grüße
Luca Salvatore
Liebes Johner-Team,
Täusche ich mich, oder hat sich der Text der MDR-Artikel 120 – 123 seit Februar / März komplett geändert? Ich finde darin auch keine Angabe zur Übergangsfrist bis 31.12.2028 (siehe Ablaufdiagramm, Abb.1) für Klasse I (Legacy) Produkte mehr. Wurde diese Frist geändert?
Liebe Frau Laube,
die Übergangsbestimmungen wurden Anfang des Jahres angepasst durch die Änderungsverordnung EU 2023/607 gültig seit dem 20.03.2023. In der aktuellen konsolidierten Fassung (https://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=CELEX:02017R0745-20230320#tocId130) finden Sie die neuen Übergangsfristen, u.a. den 31.12.2028.
Herzliche Grüße
Luca Salvatore
Hallo,
von einem Hersteller die Produkte entsprechen weiterhin der Richtlinie 93/42/EWG (MDD) und erfüllen die Anforderungen für die Verlängerung der Gültigkeit der CE Bescheinigung gemäss Artikel 120 der Verordnung (EU) 2017/745 (MDR). Dies Bedeutet der Hersteller kann gebrauch der Erweiterung des EG-Zertifikats gemäß Richtlinie 93/42/EWG (MDD) machen.
nun, dem Hersteller wird jedoch , das Zertifikat am 11/2023 ablaufen. Kann der Hersteller eine Self-Declaration zum Anhang erstellen oder sollte der Notified Body eine Declaration letter erstellen?
Es wird ständig auf die Regulation (EU) 2023/607 referenziert, jedoch sehe ich hierzu keinen Artikel, welches sagt, dass ein Hersteller eine Selbsterklärung zum Anhang erstellen kann?
Freundliche Grüsse
Hallo Kübra,
das Richtlinien-Zertifikat behält in diesem Fall Gültigkeit, auch wenn das auf dem Zertifikat angegebene Datum abgelaufen ist. Dies kann sicherlich zu Verunsicherung bei Kunden und ausländischen Behörden führen, falls dort das Zertifikat genutzt wird. Aus diesem Grund gibt es die Möglichkeit, als Hersteller selbst eine Erklärung auszustellen oder noch besser durch die Benannte Stelle. Dazu gibt es vom Team NB eine entsprechende Vorlage (https://www.team-nb.org/wp-content/uploads/2023/07/Team-NB-PositionPaper-NB-ConfirmationLetterEU2023-607-20230503-V2.pdf).
Viele Grüße
Luca Salvatore
Hallo Luca,
Vielen lieben Dank für die super Rückmeldung.
EIne Frage habe ich jedoch noch. Auch in der Vorlage steht dass die BS diese Vorlage unterzeichnen muss, auch hier habe ich keine Referenz dass ein Hersteller selber dies unterzeichnen darf.
Wo kann ich sehen, dass der Hersteller selbst unterzeichnen darf?
Liebste Grüsse
Hallo Kübra,
diese Vorlage ist speziell für Benannte Stellen gedacht und nicht für die Ausgabe durch Hersteller. Es ist aber möglich, eine ähnliche Erklärung in abgewandelter Form als Hersteller auszugeben. Allerdings werden Kunden und Behörden eher eine Bestätigung einer Benannten Stelle bevorzugen.
Viele Grüße
Luca
Hallo,
ersetzt denn die Selbsterklärung des Herstellers und das Bestätigungsschreiben der BS die bisher vom Hersteller ausgestellte Konformitätserklärung?
Hallo,
nein, die Konformitätserklärung gemäß MDD ist weiterhin gefordert und wird nicht ersetzt.
Freundliche Grüße
Luca Salvatore
Liebes Johner-Team
kann man als Hersteller von Medizinprodukten einem Händler nur den Confirmation Letter der Benannten Stelle für die von der Übergangsfrist betroffenen Produkte vorzeigen, oder muss die Self-Declaration vom Hersteller ebenfalls immer mit vorgelegt werden?
Vielen Dank!!
Liebe Valerie,
es gibt keine Pflicht, Händlern die Self-Declaration vorzulegen. Somit sollte auch ein Confirmation Letter der Benannten Stelle als Nachweis ausreichen, zusammen mit der MDD-Konformitätserklärung.
Viele Grüße
Luca Salvatore
Liebes Team,
wenn die Überwachung der MDD-Produkte während der Übergansfrist von einer neuen Benannte Stelle durchgeführt werden soll (da diese Bennate stelle das neue Zertifizierungsverfahren im Rahmen der MDR durchführt), welches CE-Logo sollte dann auf den Produkten angebracht werden bzw. welche Zertifizierungsstelle sollte genannt werden, diejenige, die die Produkte im Rahmen der MDD zertifiziert hat, oder diejenige, die die Zertifizierungsverfahren bzw. Überwachung vornimmt?
Viele Dank für die Rückmeldung!
Viele Grüße,
Julia Ditker
Liebe Julia,
generell ist es möglich, die CE-Kennzeichnung mit Kennnummer der alten Benannten Stelle weiterhin anzugeben (siehe auch Frage 16 https://health.ec.europa.eu/system/files/2023-07/mdr_proposal_extension-q-n-a.pdf). In der 3-Parteienvereinbarung mit alter und neuer Benannter Stelle kann aber auch geregelt werden, dass ab einem bestimmten Zeitpunkt die Kennnummer der neuen Benannten Stelle angegeben wird.
Herzliche Grüße
Luca Salvatore
Liebes Johner Team,
wir – als Legalhersteller wiederverwendbarer chirurgischer Instrumente (unter MDD Konformität erklärt) stehen vor dem Problem, dass die Konformitätserklärung nun 2024 ausläuft. Wir möchten die Übergangsfrist ausnutzen, stehen aber bereits mit einer BS in Kontakt. Erklären wir nun erneut unter MDD von 2024-2028 Konformität?
Liebe Frau Sachner,
in diesem Fall empfehle ich, ein Addendum zur ursprünglichen Konformitätserklärung zu erstellen und dieser beizufügen. Dort können Sie das neue Gültigkeitsdatum nennen mit Begründung und idealerweise auch einer Erklärung, dass ansonsten keine signifikanten Änderungen an den entsprechenden Produkten vorgenommen wurden.
Herzliche Grüße
Luca Salvatore
Der Link zum Guidance-Dokument MDCG 2022-04 ist verunglückt. Es müsste auf die Revision 2 vom Mai 2024 referenziert werden. Das nachfolgende Zitat ist dann sinngemäß korrekt, tatsächlich ist der Text inzwischen erweitert. Die Vertraulichkeit des Audits wird in diesem Fall durchbrochen.
Lieber Herr Wegener,
vielen Dank für Ihren wichtigen Hinweis!
Der Link führte gar zu einem anderen MDCG-Dokument. Den Fehler haben wir direkt behoben und das Zitat aus der aktuellen Revision 2 des MDCG 2022-04 übernommen.
Freundliche Grüße
Luca Salvatore
Liebes Johner Team,
uns als Händler wurde vom Hersteller mitgeteilt, dass er noch keine schriftliche Vereinbarung mit der Benannten Stelle geschlossen hat, die Unterzeichnung aber zeitnah stattfände. Wir haben im August 2024 vom Hersteller die Produkte nach Art. 120 Abs. 3b MDR erworben und die Abgabe nach dieser Mitteilung des Herstellers gestoppt. Nun unsere Frage: Können wir diese Produkte weiterverkaufen, wenn der Hersteller uns zeitnah eine mit der Benannten Stelle unterzeichnete Vereinbarung vorlegt oder sind die Produkte per se nicht mehr verkehrsfähig, weil die Frist 26.09.2024 abgelaufen ist?
Vielen Dank und beste Grüße
Heleri Binder
Liebe Frau Binder,
wenn bis zum 26.09.2024 noch keine schriftliche Vereinbarung mit der Benannten Stelle vorlag, sind die Übergangsfristen ab diesem Zeitpunkt nicht mehr anwendbar. Somit dürften dann auch die entsprechenden Legacy-Produkte nicht mehr in Verkehr gebracht werden ab diesem Datum. Sie schreiben, dass Sie die Produkte als Händler bereits im August 2024 erworben haben. In der Annahme, dass zu diesem Zeitpunkt eine Inverkehrbringung stattgefunden hat, und die sonstigen Voraussetzungen erfüllt waren (u.a. Antrag bei Benannter Stelle und MDR-konformes QMS bis zum 26.05.2024), wäre eine weitere Bereitstellung („sell-off“-Regelung) dieser Legacy-Produkte durch Sie als Händler unbefristet möglich. Allerdings ist dies ohne eine Erklärung durch die Benannte Stelle oder Hersteller schwer nachzuvollziehen bzw. von Ihnen zu prüfen.
Herzliche Grüße
Luca Salvatore
Liebes Johner Team,
meine Frage bezieht sich auf die „Kenntlichmachung“ des UDI – muss dieser als UDI erkennbar gemacht werden (z.B. durch das UDI-Symbol entspr. ISO 15223-1) oder nur wenn es mehrere als UDI „vermutbare“ Kennzeichnungen gibt?
Vielen Dank im Voraus und Gruß
Kai Grüning
Lieber Herr Grüning,
falls nur ein Datenträger auf dem Produkt bzw. der Verpackung vorhanden ist, dann ist die Anbringung des UDI-Symbols entspr. ISO 15223-1 nicht notwendig. Falls mehrere Datenträger vorhanden sind, dann empfiehlt es sich, das UDI-Symbol in unmittelbarer Nähe des UDI-Trägers anzubringen.
Freundliche Grüße
Luca Salvatore
Liebes Johner Institut-Team, lieber Herr Salvatore,
da die Übergangsfrist für Klasse I Produkte demnächst ausläuft, stellt sich uns folgende Frage. Wie lange dürfen wir unsere Produkte nach alter Ordnung (ohne UDI-Träger) noch verkaufen.
Konkreter Fall:
Wir sind Händler in Deutschland und die Artikel tragen unsere Marke. Der Hersteller sitzt in Malaysia.
Die letzte Sendung befindet sich gerade auf dem Schiff in Richtung Deutschland und trifft vor dem 26.5.25 in DE, bei uns ein. Dürfen wir die erhaltene Ware dann unbegrenzt lange verkaufen, da sie unser Lager in Deutschland pünktlich erreicht hat, oder müssen wir bis zum 26.5 an unsere Handelskunden (z.B. LEH, Drogerien) ausgeliefert haben?
Vielen Dank für Ihre Hilfe.
Freundliche Grüße
Diether Gärstner
Lieber Herr Gärstner,
ich antworte Ihnen gerne stellvertretend für meinen lieben Kollegen Luca Salvatore, der sich derzeit in seinem wohlverdienten Urlaub befindet.
Es kommt in diesem Fall ganz darauf an, welche Rolle Sie in Abhängigkeit Ihrer Tätigkeiten einnehmen. Grundsätzlich bezieht sich die Kennzeichnungspflicht mit der UDI auf die Inverkehrbringung MDR-konformer Produkte. Produkte der Klasse I dürfen demzufolge durch den Hersteller noch bis 26.05.2025 ohne UDI-Kennzeichnung in Verkehr gebracht werden. Sie schreiben jedoch, dass der Hersteller in Malaysia sitzt. In diesem Fall wird die Rolle des Importeurs notwendig, der die Produkte auf dem Unionsmarkt in Verkehr bringt. Derjenige, der die Produkte aus dem Ausland erstmalig auf dem Unionsmarkt bereit stellt, wird damit automatisch zum Importeur.
Sind Sie tatsächlich nur Händler, dann dürfen Sie die Produkte auch weiterhin (ohne UDI-Kennzeichnung) auf dem Unionsmarkt bereit stellen, insofern diese durch den Importeur rechtmäßig bis 26.05.2025 in verkehr gebracht worden sind. Nehmen Sie allerdings die Rolle des Importeurs ein, ist es erforderlich, dass die Produkte ohne UDI-Kennzeichnung noch vor dem 26.05.2025 rechtmäßig in Verkehr gebracht, d.h. an Ihre Handelskunden verkauft werden. Laut Blue Guide der Europäischen Union ist für die Inverkehrbringung nicht zwingend die physische Übergabe eines Produktes erforderlich. Eine Inverkehrbringung setzt aber ein Angebot oder eine (schriftliche oder mündliche) Vereinbarung zwischen zwei oder mehr juristischen oder natürlichen Personen in Bezug auf die Übereignung, die Übertragung des Besitzes oder sonstiger Rechte hinsichtlich des betreffenden Produkte voraus.
Ich hoffe, ich konnte Ihnen damit helfen.
Herzliche Grüße,
Manuela Reinhold
Liebes Team,
vielen Dank für diesen informativen Beitrag!
Ein Aspekt ist jedoch weiterhin unklar: Wie ist die Situation zu bewerten, wenn ein Hersteller die Zulassung eines Produkts gemäß den Anforderungen der MDR geplant und alle erforderlichen Anträge konform bei der benannten Stelle eingereicht hat, sich jedoch im Verlauf der genehmigten Übergangsfrist entscheidet, das Produkt vom Markt zu nehmen, weil der Zulassungsprozess wirtschaftlich nicht mehr tragbar ist? Gelten in diesem Fall weiterhin die Übergangsregelungen, oder verlieren diese ihre Gültigkeit mit der Entscheidung zur Marktrücknahme? Und welche Konsequenzen würden damit einhergehen?
Vielen Dank für Ihre Hilfe und viele Grüße
Guten Tag,
hier nochmal meine Antwort aus unserem Englischen Blog:
Entscheidend in diesem Fall ist, ob die Voraussetzungen für die Übergangsfristen nach MDR weiterhin erfüllt sind. Falls der Antrag bei der benannten Stelle zurückgezogen oder der Vertrag beendet wird, ist die Voraussetzung nach 120(3c) (e) MDR nicht mehr erfüllt, d.h. Sie könnten nicht mehr von den Übergangsfristen profitieren.
Freundliche Grüße
Luca Salvatore