
Ein Original Equipment Manufacturer (OEM) ist eine Firma, die Produkte herstellt (entwickelt, produziert), aber nicht notwendigerweise unter eigenem Namen in Verkehr bringt. Man spricht auch vom White-Labeling der Produkte.
Dieser Artikel untersucht die regulatorischen Anforderungen und die Verantwortlichkeiten der OEM sowie der Firmen, die jene Produkte unter eigenem Namen in Verkehr bringen. Letztere nennt man Private Label Manufacturer (PLM).
Hersteller erhalten wertvolle Hinweise, wie sie mit den gesetzlichen Änderungen umgehen und juristische Probleme vermeiden können.
Die MDR erlaubt OEM-PLM-Konstrukte nur noch in engen Grenzen. Erfahren Sie, welche Optionen Sie haben.
1. Begriffe: OEM, PLM, White Label
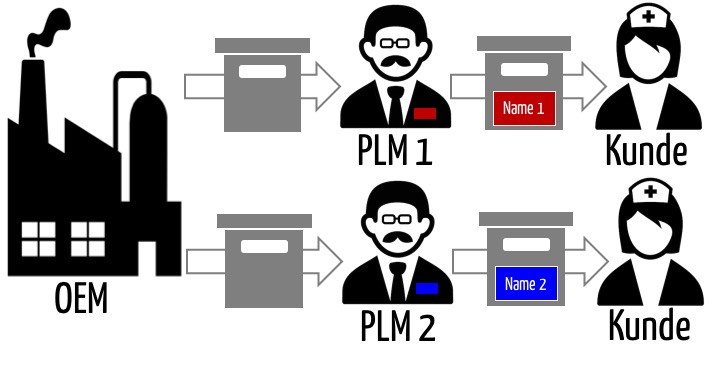
Häufig bringen Firmen (Medizin-)Produkte unter eigenem Namen in den Verkehr, die sie nicht selbst hergestellt haben. Im Sinn des Medizinprodukterechts sind diese Firmen dennoch die Hersteller. Man bezeichnet sie auch als Quasihersteller oder Private Label Manufacturer (PLM). Ein PLM ist eine Firma, die Produkte von einem OEM bezieht und sich bei der Zulassung auf die Zulassung des OEM bezieht.
Beachten Sie, dass in diesem Artikel mit dem Akronym PLM immer der Private Label Manufacturer gemeint ist und nicht das Product Lifecycle Management.
Die Unternehmen, die die Produkte tatsächlich entwickeln, produzieren (und ggf. unter eigenem Namen zulassen), nennt man Original Equipment Manufacturer (OEM), übersetzt: Originalhersteller. So definiert das auch die ZLG:
„Unternehmen, das fertige Produkte für einen Privat Label Manufacturer produziert und in diesem Fall nicht als Hersteller im Sinne des Medizinprodukterechtes auftritt.“
OEM bringen das Produkt nicht unter dem eigenen Label in den Markt. Daher gibt es zwei Möglichkeiten, um das Label anzubringen:
- Ein OEM überlässt das „Labeling“ den PLM. Mit anderen Worten: Ein OEM bringt das Medizinprodukt als „White-Label“ in den Verkehr. Das illustriert die Abb. 1.
- Der OEM bringt für den oder die PLM bereits die jeweiligen Labels auf.
In der Automobil-Industrie wird der Begriff OEM anders verwendet: Hier sind die großen Automarken wie VW, Mercedes und BMW die OEM, also diejenigen, welche die Produkte unter eigenem Namen in den Markt bringen.
1.1. Vorteile einer Zusammenarbeit von PLM und OEM
Quasihersteller (PLM) und Originalhersteller (OEM) können von einer Zusammenarbeit profitieren:
Vorteile für OEM
- Die Originalhersteller können sich auf ihre Kernkompetenzen beschränken: die Entwicklung und Produktion.
- Sie müssen keine eigene Marke etablieren oder eine (weltweite) eigene Vertriebsorganisation aufbauen.
- Dadurch, dass OEM ihre Produkte an viele PLM liefern, ergeben sich Skaleneffekte. Die Stückkosten sinken.
- OEM müssen sich nicht um die Zulassung in den verschiedenen Märkten und die dortige Marktüberwachung kümmern. Somit besteht keine Notwendigkeit, entsprechendes regulatorisches und marktspezifisches Wissen zu etablieren.
Vorteile für PLM
- Viele PLM kaufen OEM-Produkte zu und können so ihr eigenes Portfolio abrunden.
- Es besteht für sie keine Notwendigkeit, eigenes Entwicklungs- und Produktions-Knowhow aufzubauen.
- Falls sie über eine starke Marke verfügen, lassen sich die preisgünstig zugekauften Produkte unter der eigenen, teuren Handelsmarke weiterverkaufen.
Bei den unter der MDD zulässigen OEM-PLM-Konstellationen erscheint nur der PLM und dessen CE-Kennzeichen auf dem Label. Der OEM ist nicht erkennbar.
1.2. Varianten von OEM-PLM-Konstellationen
1.2.1 Quasihersteller verkauft nur
Es gibt Quasihersteller, deren Geschäftmodell ausschließlich auf zugekauften Produkten basiert.
Im Medizinproduktemarkt treten viele Importeure und EU-Repräsentanten als Quasihersteller (PLM) von außereuropäischen, insbesondere asiatischen Herstellern (OEM) auf.
1.2.2 Hersteller ist auch OEM
Der Hersteller tritt gleichzeitig als OEM auf: Er beliefert andere Hersteller mit seinen „umgebrandeten“ Produkten. Das kann in folgenden Situationen sinnvoll sein:
- Billigmarke für andere Märkte etablieren, die die eigene Hauptmarke nicht kannibalisiert
- Überproduktionen, ältere Produktversionen oder Waren minderer Qualität abverkaufen, ohne die Hauptmarke zu schädigen
- Skaleneffekte in der Produktion nutzen
1.2.3 Andere Branchen
Solche Konstruktionen sind in anderen Branchen üblich. Beispielsweise stellt die BSH Hausgeräte GmbH als OEM Küchengeräte für Markenhersteller (PLM) wie Bosch, Siemens und Gaggenau her. Im Discounter-Markt finden sich „White-Label-Versionen“ von Markenprodukten.
1.3. Abgrenzungen: Distributoren und Lieferanten
Eine OEM-PLM-Konstellation unterscheidet sich von einer Hersteller-Distributoren-Konstellation. Bei letzterer verkauft der Distributor die Produkte des Herstellers unverändert unter der Handelsmarke des Herstellers weiter. Der Hersteller bleibt Hersteller im Sinne der Medizinprodukterichtlinie bzw. -Verordnung.
Weiterhin gilt es, OEM und Lieferanten zu unterscheiden: Ein OEM fertigt das komplette Medizinprodukt, hält die vollständige Technische Dokumentation vor und lässt das Produkt auch zu. Hingegen stellt ein Lieferant „nur“ Produkte, Komponenten oder ausgelagerte Prozesse zur Verfügung. Die Technische Dokumentation erstellt der Hersteller. Im Gegensatz dazu macht ein PLM eine „Identitätsaussage“ und verweist auf die Technische Dokumentation, die beim OEM liegt.
2. Regulatorischer Rahmen
2.1. Europa
2.1.1 Überblick / Allgemeines
Weder die Medizinprodukterichtlinie MDD noch die Medizinprodukteverordnung MDR kennt die Begriffe OEM und PLM.
Die EU-Kommission schrieb bereits 2013 in ihrer Empfehlung 2013/473/EU im Abschnitt „Allgemeine Empfehlungen für die Auslagerung der Produktion auf Unterauftragnehmer oder Lieferanten“ (letzte Seite):
„Die benannten Stellen werden darauf hingewiesen, dass die Hersteller: […]
b) ihrer Verpflichtung, die vollständige technische Dokumentation und/oder ein Qualitätssicherungssystem zur Verfügung zu haben, nicht dadurch nachkommen können, indem sie auf die technische Dokumentation eines Unterauftragnehmers oder Lieferanten und/oder deren Qualitätssicherungssystem verweisen;“
2.1.2 MDR bzw. IVDR
Unter MDR und IVDR gibt es keine OEM-PLM- oder White-Labeling-Konstrukte mehr. Die EU-Verordnungen erlauben nur die folgenden Rollen:
Konkret beschreiben MDR und IVDR im jeweiligen Artikel 16 die Pflichten von Importeuren, Händlern und anderen Personen, die Produkte unter eigenem Namen bzw. eigener Marke bereitstellen. In diesem Fall gehen die Herstellerpflichten auf diese Akteure über. Es gibt aber Ausnahmen:
- Der (Original-)Hersteller steht auf der Kennzeichnung des Produkts und hat mit dem Importeur bzw. Händler eine Vereinbarung getroffen, dass er (der Hersteller) für die Einhaltung der Anforderungen verantwortlich bleibt.
- Der Importeur bzw. Händler stellt nur die Begleitinformationen (einschließlich Übersetzung) bereit, die im jeweiligen Mitgliedsland regulatorisch gefordert sind.
- Der Importeur bzw. Händler ändert die äußere Verpackung des Produkts, weil dies im jeweiligen Mitgliedsland regulatorisch gefordert ist.
Die MDR verlangt, dass der Hersteller vollen Zugriff auf die Technische Dokumentation hat. Genau das möchten aber viele OEM verhindern. Damit können die meisten OEM-PLM-Konstrukte in dieser Form nicht weiterbestehen.
Auch ein vollständiges White-Labeling von Medizinprodukten ist nicht mehr möglich.
Interessanterweise schreibt das EU-Parlament als Antwort auf die Petition No 0061/2021:
Also under the new MDR, PLM/OEM relations are indeed possible, without any impact on the placing on the market of devices nor on exports to markets outside the EU, provided that all involved parties comply with their legal obligations and responsibilities, including those on the technical documentation, registration, CE marking and declaration of conformity.
Allerdings sieht man diese Antwort kritisch und entgegnet:
The initial reply of the Commission to the Petitions Committee (August 2021) and its additional reply (April 2022) clarify that the OBL is considered as the (legal) manufacturer of the device in accordance with the MDR. Therefore, the OBL is responsible for compliance with all obligations of manufacturers pursuant to the MDR. This includes the obligation to draw up the technical documentation and to keep it available to the competent authority. The Commission’s replies do not specify how the manufacturer must comply with its obligations, as long as the manufacturer ensures that the obligations are complied with.
Hinweis: Mit OBL meint die Kommission den „Own Brand Labeller“, was dem PLM entspricht.
2.1.3 MHRA
Die britische Gesundheitsbehörde MHRA folgt in einem Guidance-Dokument im Wesentlichen der eben genannten Empfehlung der EU. Sie gestattet aber, dass unter Umständen Informationen vom OEM zurückgehalten werden dürfen, wenn diese für die Bewertung von Sicherheit und Leistungsfähigkeit des Produkts nicht wesentlich sind. Damit erkennt die Behörde prinzipiell das Interesse der OEM an, das geistige Eigentum zu schätzen. Die Grenzen sind aber sehr eng gesteckt.
2.1.4 MDD (mit MDR hinfällig)
Die Medizinprodukterichtlinie MDD verlangte bei Herstellern, die Produkte über ein QM-System gemäß Anhang II in Verkehr bringen, eine angemessene Beschreibung
„falls Auslegung, Herstellung und/oder Endkontrolle und Prüfung des Produkts oder von Produktbestandteilen durch einen Dritten erfolgt: Methoden zur Überwachung der wirksamen Anwendung des Qualitätssicherungssystems und insbesondere Art und Umfang der Kontrollen, denen dieser Dritte unterzogen wird;“
Ansonsten regelte die MDD diese Konstellationen nicht so umfangreich, weshalb die „Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten“ (ZLG) im EK-Med 3.9 B16 die Anforderungen an OEM-PLM-Konstellationen detailliert beschrieb.
2.1.5 ZLG (mit MDR hinfällig)
Dabei unterschied die ZLG folgende Fälle:
| Konstellation | Anforderungen OEM | Anforderungen PLM |
| OEM ist nicht als Hersteller im Sinne der Richtlinie tätig und verfügt nicht über eine Bescheinigung nach Richtlinie 93/42/EWG für das betreffende Produkt |
|
|
| OEM ist für das betreffende Produkt auch selbst Hersteller im Sinne des Medizinprodukterechts und dementsprechend nach Richtlinie 93/42/EWG von einer Benannten Stelle zertifiziert. „Hersteller“ bedeutet in diesem Kontext nicht, dass der OEM das Produkt auch selbst verkauft, sondern „nur“, dass er über eine entsprechende Zertifizierung verfügt. | Die Pflichten des OEM hängen von der Klasse des Produkts ab und von der Existenz und dem Umfang des QM-Systems bzw. Richtlinienzertifikats beim OEM. | Für den Fall, dass der OEM über ein vollständiges QM-System nach Anhang II oder ein anderes Richtlinienzertifikat verfügt, gelten folgende Anforderungen an den PLM:
|
Keine OEM-PLM-Ketten: Die ZLG hatte sich in dem Dokument 3.9 B16 auch zu OEM-PLM-Ketten geäußert. Kurz gesagt: Sie waren verboten. Der OEM musste das Produkt selbst herstellen und durfte es nicht von einem weiteren OEM beziehen bzw. selbst PLM sein.
Die ZLG schrieb dazu:
„Die in diesem Dokument beschriebenen Regelungen sind nicht anwendbar für den Fall, dass ein PLM die von ihm in den Verkehr gebrachten Medizinprodukte nicht von einem OEM, sondern von einem anderen PLM kauft (doppelte OEM-PLM-Konstellation).“
OEM-PLM-Ketten waren vertraglich auszuschließen. Darauf achteten die Benannten Stellen.
2.2 USA
Auch die FDA verfolgt das Prinzip, dass es keine Lücke im Entwicklungs- und Produktionsprozess geben darf, die nicht den regulatorischen Anforderungen z. B. des 21 CFR part 820 genügt.
Zusätzlich fordert die FDA, dass u. a. folgende Beteiligte registriert werden müssen:
- Inverkehrbringer (Hersteller)
- Auftragsfertiger (Contract Manufacturer)
- Hersteller von Zubehör
- Firmen, die Produkte „relabeln“ oder „repackagen“
- Firmen, die die Entwicklung verantworten (Specification Developer)
Dies regelt v. a. 21 CFR part 807. OEM fallen beispielsweise unter „Specification Developer“ und „Contract Manufacturer“. Eine Übersicht über diese Rollen finden Sie auf den Seiten der FDA.
3. OEM-PLM-Konstrukt: Alternativen?
3.1 Variante 1: TÜV als Anbieter von Escrow-Services
Es hat sich nie durchgesetzt, dennoch halten wir es in diesem Kontext für erwähnenswert: Ein TÜV plante angeblich die Gründung einer Firma, die die Technischen Dokumentationen vorhält, damit der TÜV sie einsehen kann. Damit könnten auch unter der MDR die OEM-PLM-Konstellation weiter bestehen.
Die Gültigkeit dieses Konstrukts war sehr fragwürdig, da die Forderung der MDR nicht erfüllt worden wäre, dass der Inverkehrbringer vollen Zugriff auf die Akte hat. Dies hält die Kommission aber für wichtig, um fundierte Entscheidungen treffen zu können.
3.2 Variante 2: Händler statt PLM
Die Motivation vieler Firmen, eine OEM-PLM-Beziehung einzugehen, bestand darin, dass der PLM die Produkte unter eigenem Namen verkaufen kann. Daher bestand eine häufige Änderung (nur) darin, die Farbe, das Logo und die Anschrift zu ändern. Händler hingegen durften am Produkt überhaupt nichts ändern.
Genau an dieser Stelle ändert die MDR die Vorgaben: Die Änderung der Verpackung sowie der Begleitinformationen versteht die MDR nicht mehr als eine Produktänderung, die dazu führt, dass Konformität des Produkts neu nachgewiesen werden muss (Artikel 16(2)).
Doch es gibt Pflichten:
- Der Händler muss identifizierbar sein.
- Er benötigt ein Qualitätsmanagementsystem (Artikel 16(3)), das durch die Benannte Stelle begutachtet sein muss. Derzeit ist noch unklar, ob dies eine Zertifizierung bedingt und ob die Begutachtung sich nur auf die entsprechenden Prozesse des „Re-Labelings“ bezieht oder auf alle Prozesse, die eine ISO 13485 vorschreibt. Ebenfalls bleibt unklar, ob diese Bewertung einmalig oder fortlaufend erfolgen muss.
- Der Händler muss die Behörde über die Inverkehrbringung informieren (Artikel 16(4)).
- Der Händler muss den Hersteller darüber informieren, in welchem Land er das Produkt in Verkehr bringen wird. Der Hersteller muss damit nicht einverstanden sein, muss aber – da in Kenntnis – diese Länder in seine Post-Market Surveillance einschließen (Artikel 16(4)).
Es sind mehrere Varianten dieser neuen Beziehung zwischen Hersteller (ex OEM) und Händler (ex PLM) denkbar:
- Der Hersteller liefert die Produkte als „White Label“ aus, der Händler übernimmt das Labeling.
- Der Hersteller liefert die Produkte bereits in der Verpackung des Händlers aus.
Es ist sogar denkbar, dass der Hersteller „Bulk Ware“ an Händler schickt, der dann erst die Verpackung vornimmt.
Der Originalhersteller muss aber in jedem Fall als solcher erkennbar und auf dem Label (zusätzlich) genannt sein. Das ist ein Unterschied zu den bisherigen OEM-PLM-Konstrukten.
3.3 Variante 3: PLM wird Hersteller
Der (bisherige) OEM übergibt dem (bisherigen) PLM die vollständige Akte. Diese beinhaltet neben der Technischen Dokumentation auch Aufzeichnungen aus dem QM-System sowie Informationen über Zulieferer und ausgelagerte Prozesse des „OEM“.
Genau das versuchen die OEM üblicherweise zu verhindern, da der „PLM“ (der neue Inverkehrbringer) dann über alle Informationen verfügen würde, um das Produkt bei einem anderen Produzenten fertigen zu lassen.
In dieser Variante müsste der bisherige OEM nicht mehr auf dem Label erscheinen. Der „PLM“ hat alle Herstellerpflichten übernommen.
3.4. Variante 4: Gemeinsamer Vertrauter
Es wurde diskutiert, ob ein gemeinsamer Vertrauter des „PLM“ und „OEM“ eine Lösung darstelle. Dieser Vertraute würde als ausgelagerter Prozess des Inverkehrbringers (ex PLM) vollen Zugriff auf die Dokumentation haben und somit dem „PLM“ zugerechnet werden. Der Vertraute wäre zudem über ein NDA mit dem „OEM“ gebunden, welches es verbieten würde, vertrauliche Informationen an andere Personen „seiner Organisation“ weiterzugeben.
Lesenswert ist das Positionspapier von Medical Mountains. Beachten Sie, dass es jedoch keine rechtlich bindenden Aussagen enthält.
4. Verantwortlichkeiten von OEM und PLM
4.1 Beispiel für die Aufteilung
Der Quasihersteller (PLM) und der Originalhersteller (OEM) müssen die Verantwortlichkeiten regeln, beispielsweise in Form einer Verantwortlichkeitsmatrix.
| Tätigkeit | OEM | PLM |
| Entwicklung | X | |
| Produktion | X | |
| Konformitätsbewertung | X | |
| Konformitätserklärung bzw. Zulassung | X | |
| Marktüberwachung | X | |
| Vigilanz (Behördenmeldungen, Rückrufe) | X | |
| Wartung, Schulung, Installation | X | |
| Aufrechterhaltung eines QM-Systems | X | X |
| Gewährleistung, Haftung | X (gegenüber PLM) | X (gegenüber Kunden) |
| … |
Die Aufteilung gemäß Tabelle 2 ist unter der MDR und IVDR hinfällig. Sie kann aber dennoch helfen, Aufgaben zu regeln, beispielsweise wenn der PLM als Hersteller auftritt und der OEM als Entwicklungsdienstleister fungiert, der unter dem QM-System des Herstellers arbeitet. Die Verantwortung verbleibt beim Hersteller.
4.2 Vertrag zwischen OEM und PLM
Ein Vertrag zwischen OEM und PLM musste laut ZLG folgende Punkte regeln (Zitat):
- Geltungsbereich der Vereinbarung (betroffene Produkte/-gruppen)
- Geltungsdauer der Vereinbarung
- Detaillierte Spezifikationen für die jeweiligen Produkte
- Regelungen, wer für welche Dokumentation (Technische Dokumentation, DHR (Device History Record) etc.) verantwortlich ist, inkl. Sprache und Aufbewahrungsfristen, auch nach Ende der Vereinbarung
- Rückverfolgbarkeit von Rohmaterial/Komponenten
- Einfluss des PLM auf das Produktdesign
- Regelungen über das Verfahren, wie Änderungen am Produkt und im Herstellungsprozess veranlasst, freigegeben, durchgeführt, dokumentiert und kommuniziert werden
- Recht zur Einsicht in die bzw. Vorlage der Technischen Dokumentationen für die Benannte Stelle und die zuständige Behörde des PLM
- Regelungen zur Zusammenarbeit bei Vorkommnissen/Meldepflichten/Rückruf, auch nach Ende der Vereinbarung
- Zugangsrecht der Benannten Stellen und Behörden zu den Betriebsstätten des OEM und dessen Zulieferer/Unterauftragnehmer
- Informationspflicht bei Änderungen am Status der Bescheinigungen des OEM und/oder PLM
- Umgang mit Kundenreklamationen sowie Korrektur- und Vorbeugemaßnahmen
- Verantwortlichkeitsmatrix (s. oben).
Lesen Sie hier mehr zum Thema Qualitätssicherungsvereinbarungen (QSV).
5. FAQ zu PLM und OEM
5.1 Welche Änderungen darf der PLM am Produkt vornehmen?
Generell dürfen die PLM, von der Kennzeichnung (inkl. Handelsname und Logo) abgesehen, keine Änderungen am Produkt vornehmen. Dies ist nachvollziehbar, weil jede Änderung eine Auswirkung auf die Sicherheits- und Leistungsfähigkeit des Produkts haben kann. Selbst eine Änderung des farblichen Designs kann Auswirkungen auf die Gebrauchstauglichkeit des Produkts zur Folge haben.
Die MDR weicht diese strenge Vorgabe auf.
5.2 Was sind die Nachteile und Gefahren für OEM und PLM?
Die Vorteile einer OEM-PLM-Konstellation wurden weiter oben aufgezählt. Diese Form der Zusammenarbeit birgt aber auch Nachteile und Gefahren für beide Parteien:
- Da ein reiner OEM über keinen direkten Marktzugang und keine direkten Kundenbeziehungen verfügt, ist er abhängig vom PLM – und dessen Vertriebserfolg.
- Umgekehrt ist der PLM abhängig von der Belieferung durch den OEM. Das betrifft auch explizit die Versorgung mit Ersatzteilen.
- PLM verfügen häufig nicht über das ausreichende technologische Verständnis, um die Qualität der Produkte und der Dokumentation zu prüfen und damit Risiken für Patienten, aber auch das eigene Unternehmen zu bewerten.
- Dennoch müssen die PLM letztlich für die Produkte geradestehen.
5.3 Was sind die typischen Schwierigkeiten bei einer OEM-PLM-Konstellation?
Einige Tätigkeiten und Aktivitäten lassen sich nicht strikt zwischen OEM und PLM aufteilen. Dazu zählen insbesondere das Risikomanagement und die Bewertung der Gebrauchstauglichkeit. Dies wird zum Problem, da regelmäßig nur die PLM über ein ausreichendes Verständnis des Nutzungskontexts verfügen.
Ein häufiger Streitpunkt ist die Technische Dokumentation: Die OEM möchten das technische Wissen nicht offenbaren. Wenn sie nicht selbst als Hersteller auftreten (s. o.), muss der Hersteller (PLM) aber vollständigen Zugang dazu haben. Escrow Agreements können einen Ausweg aus diesem Dilemma bieten. Allerdings wird dieser Ausweg mit der MDR verbaut. Denn der Hersteller muss über vollen Zugriff auf die Akte verfügen.
5.4 Werden OEM auch auditiert?
Wie oben dargelegt, hängt die Antwort auf diese Frage davon ab, ob
- der OEM auch Hersteller im Sinne der Richtlinie ist und
- der OEM über ein vollständiges und zertifiziertes QM-System gemäß Anhang II oder anderes Richtlinienzertifikat verfügt.
Details regelte das oben referenzierte Dokument EK-Med 3.9 B 16. Allerdings ist dieses Dokument nur für die MDD gültig, nicht mehr für die MDR/IVDR. Bei der MDR/IVDR unterliegt der Hersteller der Überwachung durch die Behörden und ggf. durch die Benannte Stelle.
5.5 Wer kann verklagt werden, wenn einem Patienten etwas wegen eines Produktfehlers zustößt?
Generell kann jeder (z. B. Patienten, Krankenhäuser, Wettbewerber) jeden (OEM, PLM) verklagen. Die Wahrscheinlichkeit einer Klage hängt ab von der „Greifbarkeit“ und der Finanzstärke des potenziellen Beklagten.
6. Fazit, Zusammenfassung
Spätestens mit der MDR und IVDR sind die Möglichkeiten genommen, dass sich Hersteller ihrer Verantwortung entziehen, indem sie sich hinter einem OEM-PLM-Konstrukt verstecken. Derjenige, der das Produkt erstmalig in den Verkehr bringt, ist in der Verantwortung. Und das ist auch gut so.
Änderungshistorie
- 2025-04-22: Zweite Hälfte von Kapitel 2.1.2 (Bewertung der EU-Kommission und des EU-Parlaments) ergänzt. Zusammenfassung zu Beginn des Artikels geändert. Die Aussage abgeschwächt, dass das OEM-PLM-Konstrukt unter der MDR generell nicht mehr möglich sei
- 2024-12-11: Artikel aktualisiert (einige Absätze gelöscht, andere hinzugefügt, andere überarbeitet)
- 2019-10-17: Erste Version des Beitrags veröffentlicht



Hallo Christian
Auch unter der MDD wird es immer schwieriger, mit OEM/PLM Konstrukten zu arbeiten. So hat z.B. die Benannte Stelle SQS letzte Woche ihren Kunden mitgeteilt, dass das ZLG-Dokument nicht mehr weiter berücksichtig wird und sie ab sofort erwarten, dass der PLM vollständigen Zugriff auf die TechDoc hat. Hier ergibt sich also schon vor der Einführung der MDR grossen Handlungsbedarf für viele PLM. Wir empfehleun unseren Kunden schon jetzt, nicht mehr auf „klassische“ OEM/PLM Konstrukte zu setzen. Lösungen wie http://www.decomplix.ch können da eine valable Alternative sein.
Danke, Hansjörg! Ich habe den Artikel entsprechend ergänzt.
Hallo Hansjörg,
Hallo Christian,
das stimmt so nicht.
Der Originaltext der SQS ist folgender:
[ZITAT – Start]
Der OBL muss mit dem Medizinprodukte Hersteller (Inverkehrbringer oder OEM) vertraglich sicherstellen, dass die SQS als Notified Body des OBL Zugriff zur Technischen Dokumentation des Medizinprodukte Herstellers bekommt und diese im Rahmen des EG-Konformitätsbewertungsverfahrens des OBL vor einer Zertifikatsfreigabe prüfen kann.
[ZITAT – Ende]
Der PLM muss demnach nicht zwangsläufig Zugriff die komplette techn. Dok. bekommen, sondern diese nur der benannten Stelle (in diesem Fall SQS) zur Verfügung stellen. Das muss dann allerdings zwischen OEM und PLM vertraglich geregelt werden. Somit sehe ich die ganze Sache weniger dramatisch als sie dargestellt wird. Eine vertragliche Regelung ist durchaus machbar und bedeutet viel weniger Aufwand als angedroht.
LG Tobias
Herzlichen Dank für den wichtigen Beitrag! Ich habe den OEM-Artikel darauf nochmals überarbeitet.
Fair enough, da hast du Recht Tobias, die TD muss nicht dem PLM zur Verfügung gestellt werden. Danke für diese wichtige Präzisierung. Es ist aber so oder so eine deutliche Verschärfung der Praxis und wird zu einigen bösen Überraschungen führen. Kommt dazu, dass doppelte Konstellationen (Kaskaden) ganz ausgeschlossen werden.
Interessante Diskussion!
Mich interessiert in welchem Artikel der MDR explizit (bzw. wahrscheinlich verklausuliert) die verschärften Durchgriffsrechte auf die TechDoc des OEM gefordert werden und darüber hinaus, ob der äquivalente Passus auch in der IVDR auftaucht. Artikel 16 der MDR (und der IVDR) hat mir diesbezüglich keine Aufklärung gebracht.
Hallo Patrick,
ich könnte mir vorstellen das die MDR unter Artikel 14 Abschnitt 6 darauf verweist. Aber wie du schon sagst, es wird da unheimlich viel verklausuliert und schwammig beschrieben. Einen OEM oder PLM / OBL kennt die MDR nicht. Es wird darin nur von Herstellern, Händlern, Importeuren oder ganz allg. von Wirtschaftsakteuren geschrieben.
Die MDR ist und bleibt immer noch ein Monster, aber wir versuchen es zu bändigen. 😉
LG Tobias
Hallo Forenleser,
ich weiß nicht, ob es noch interessant ist, aber bereits die Empfehlung der Kommission 2013/473/EU für Benannte Stellen stellt klar:
Abschnitt:
Allgemeine Empfehlungen für die Auslagerung der Produktion auf Unterauftragnehmer oder Lieferanten
Die benannten Stellen werden darauf hingewiesen, dass die Hersteller:
b) ihrer Verpflichtung, die vollständige technische Dokumentation und/oder ein Qualitätssicherungssystem zur Verfügung zu haben, nicht dadurch nachkommen können, indem sie auf die technische Dokumentation eines Unterauftragnehmers oder Lieferanten und/oder deren Qualitätssicherungssystem verweisen;
Der SNCH (Luxemburg) hat sich bereits 2013 direkt und strikt dran gehalten und akzeptierte eine Vertragliche Regelung mit Zugriffsrecht auf die Tech. Doku. vom OEM nicht. Mit einer vorliegenden Rumpfdokumentation konnte man ebenfalls nicht argumentieren.
Ich kann mir nicht vorstellen, dass mit der MDR in diesem Bereich mit einer Lockerung zu rechnen ist. Leider hat die ZLG Ihre entsprechenden Papers nie angepasst.
VG,
Joachim
Besten Dank, lieber Herr Schneider, für diese Leseempfehlung! Ich werde den Blog entsprechend ergänzen.
Viele Grüße, Christian Johner
Ich hätte eine Frage zu folgendem Fall:
Ich vertreibe ein Produkt auf dem ich als Vertrieb gekennzeichnet bin und der Hersteller Firma XYZ ist. Der Produktname enthält u.a. allerdings meinen Firmennamen, dies hat mir der Hersteller ermöglicht.
Aus meiner Sicht ist dies kein OEM-PLM Verhältnis sondern ein Hersteller-Distributor Verhältnis.
Allerdings irritiert mich die Ausdrucksweise:
„bring ein Händler/Distributor ein Produkt unter seinem Namen in Verkehr […], so geht die Herstellerverantwortung auf ihn über“
Ich gehe aber davon aus, dass die Verantwortung an die Herstellerangabe geknüpft ist und nicht im wörtlichen Sinn der Produktname gemeint ist.
Sehe ich das richtig?
Über einen Kommentar wäre ich dankbar!
LG
Christine
Das würde ich genauso sehen, wie Sie schreiben. Mit „unter seinem Namen“ ist nicht der Produktname gemeint, sondern der Name des Inverkehrbringers.
Hallo Herr Dr. Johner,
zum Thema MDR Hack:
Artikel 16(2) bezieht sich auf „Änderung eines bereits im Verkehr befindlichen oder in Betrieb genommenen Produkts“. Läßt sich der Artikel auf Produkte anwenden, die neu produziert werden?
Erlaubte Änderungen wären demzufolge die Gebrauchsanweisung und die äußere Verpackung. Was ist mit Logos auf dem Produkt und Änderungen am Typschild mit dem PLM als Hersteller?
Der Gedanke ist hier, dass der Händler ein Produkt (nicht notwendigerweise die Instanz eines Produkts), das der Hersteller sonst unter eigenem Namen verkauft, umlabelt. Wichtig ist, dass der Hersteller es bereits unter eigenem Namen in Verkehr gebracht hat. Wie gesagt, es geht nicht um ein einzelnes Produkt.
Die Änderung des Logos ist okay, das Typenschild muss aber weiterhin den ursprünglichen Hersteller nennen und erkenntlich machen.
Hallo Herr Johner,
Den oben genannten 2. Hack als Alternative zum OEM-PLM-Konstrukt kann ich leider nicht nachvollziehen.
Grundsätzlich gilt, dass der Hersteller, der das Produkt unter eigenem Namen in Verkehr bringt genannt werden muss.
Wenn es einem PLM darum geht, das Produkt unter eigenem Namen in Verkehr zu bringen, verbleibt bei Ihm die Herstellerverantwortung und die Verpflichtung die Dokumentation vorhalten zu können. „Unter eigenem Namen“ heißt für die meisten PLM auch, dass auf dem Produkt nicht der Name des eigentlichen Herstellers auftaucht und so das Produkt aus dem „Stall“ des PLM scheint.
Artikel 16 bedeutet meiner Meinung nach lediglich, dass nun nach der MDR der Händler Tätigkeiten ausführen kann, die ihn nach der MDD zum Hersteller gemacht haben. Meines Erachtens der Händlername muss Aufgrund der Tätigkeiten zusätzlich zum Herstellername genannt werden, ersetzt aber nicht den Herstellername. Das eigentliche PLM Problem scheint aber so nicht gelöst zu werden.
Viele Grüße
Hallo Herr Mohr,
genau diese Ansicht vertreten wir auch.
Das labeln des PLM ohne Angabe des tatsächlichen Herstellers wird auch mit diesem 2. „Hack“ unserer Meinung nach unter der MDR nicht möglich sein. Aus diesem Grund wird es wahrscheinlich der mittelfristige Tod des OEM-PLM Geschäfts bedeuten. Wir sehen zumindest noch keinen nachvollziehbaren Weg aus diesem Dilemma.
Liebe Grüße
Sehr geehrter Herr Mohr, sehr geehrter Herr Bauer,
ich bin ganz Ihrer Meinung, dass der „echte Hersteller“ als solcher genannt bleiben muss. Offensichtlich war der Text diesbezüglich nicht klar genug formuliert. Dies ist nun geändert und in fett formatiert.
Herzlichen Dank für Ihre sehr wichtigen Hinweise!
Viele Grüße, Christian Johner
unter b) steht im ersten Satz des Kastens
„Die MDR verlangt, dass der Hersteller vollen Zugriff auf die technische Dokumentation hat.“
Wird diese Aussage aus der MDR Artikel 16 (1) „Ein Händler, Importeur (…) hat die Pflichten des Herstellers bei Ausführung folgender Tätigkeiten: (…)“ entnommen? Oder steht die Formulierung mit dem Zugriff auf die tech. Dok. explizit an einer anderen Stelle in der MDR?
beste Grüße
B. Bressler
Diese Aussage gilt unabhängig vom Kontext. Ohne vollen Zugriff auf die technische Dokumentation sind die Anforderungen des Anhangs I nicht nachweisbar erfüllt und es wird gegen die Anforderungen des Anhangs II verstoßen.
Es kann gut sein, dass ich mich irre, aber soweit ich das versteh, macht die TÜV-Firma keinen juristischen Unterschied bei der Geschichte:
Wenn doch die 2013/473/EU fordert, dass der Hersteller die vollständige technische Dokumentation und/oder ein Qualitätssicherungssystem zur Verfügung haben muss und auch nicht dadurch nachkommen kann, indem auf die technische Dokumentation eines Unterauftragnehmers oder Lieferanten und/oder deren Qualitätssicherungssystem verweisen wird, wie soll denn dann der TÜV-Hack Abhilfe bei der Geschichte schaffen? Genau diesen Verweis stellt der Hack doch dar ?!
Durch die TÜV-Firma ist nämlich nicht sichergestellt, dass der Hersteller (im Sinne der MDR) voll-umfänglichen Zugriff auf die tech. Dok. hat. Letztendlich erfolgt eben doch nur ein Verweis auf die tech. Dok. und die TÜV-Firma stellt dabei einen Unterauftragnehmer mit der technischen Dokumentation dar.
Bitte klären Sie mich auf, wenn ich das völlig falsch verstehe
Grüße
Sehr geehrter Herr Bressler,
Sie haben eine guten Punkt. Die Auslegung der MDR-Forderung ist auch etwas widersprüchlich. Die benannten Stellen (zumindest einige) interpretieren das so, dass der Hersteller Zugang zur Dokumentation haben muss, damit die benannten Stellen diese prüfen können. Es würde — so die Sichtweise — also gar nicht primär darum gehen, dass die Dokumente ihm alle selbst vorliegen, sondern dass er der benannten Stelle Zugang zu allen Dokumenten gewähren muss. Das wäre über diesen „Escrow-Ansatz“ möglich. Ob die benannten Stellen, die dies anbieten wollen, eher von ökonomischen Gedanken oder eher vom Streben nach Rechtskonformität für die „Hersteller“ getrieben sind, lässt sich schwer beurteilen.
Wie sich dieser Ansatz in der Praxis und v.a. Rechtssprechung künftig darstellen wird, ist derzeit unklar.
Beste Grüße, Christian Johner
Vielen Dank für ihreAntwort, Herr Johner!
Sehr geehrter Herr Johner,
Ich habe zwei Fragen zu der OEM-PLM-Konstellation.
1) Wie weit geht die Forderung nach Zugriff auf die TD des OEM-Herstellers? Schließt diese tatsächlich die kompletten DHF, DMR bis hin zu Produktionsaufzeichnungen mit ein?
2) Ich setze einmal voraus, dass ich als PLM vollumfänglichen Zugriff auf die TD des OEM erhalte.
Kann ich nach der MDR in meiner Funktion als PLM, d.h. nach Übernahme der Herstellerverantwortung, dem OEM immer noch komplett die Konformitätsbewertung überlassen? Oder anders gefragt wie und in welchem Umfang müssen wir als PLM die TD noch einmal prüfen und bewerten?
VG
Susanne Wolpert
Sehr geehrte Frau Wolpert,
danke für die wichtigen Fragen:
ad 1) Ja
ad 2) Die Konformitätsbewertung des OEMs erspart Ihnen nicht die eigene Konformitätsbewertung. Wenn Sie allerdings den Vollzugriff haben, sollte das kein Problem sein, es ist ja alles gemacht. Es wird nicht verlangt, dass Sie z.B. Tests wiederholen. Um die Prüfung der Unterlagen kommen Sie aber nicht umhin.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
bei mir hat sich nach einiger Recherche über das von Ihnen beschriebene Thema unter anderem die folgende Frage ergeben, auf die ich bisher keine Antwort finden konnte.
Die Medizinprodukte müssen unter der neuen MDR mit einer UDI gekennzeichnet und ausführlich gelabelt werden, dabei muss der wirkliche Hersteller (OEM) auf dem Label erkennbar sein. Wenn ich jetzt der PLM in diesem Falle bin und das Produkt weiter verkaufen möchte, darf ich dann meine eigenen Angaben zusätzlich auf das Label bringen, also meine eigenen Seriennummern etc oder ist dies nicht möglich.?
Über eine Antwort wäre ich sehr dankbar!
Viele Grüße
Ihre S.K.
Sehr geehrter S.K.,
Sie dürfen das! Sie wären nur nicht mehr PLM (das Konzept möchte die EU nicht mehr), sondern Distributor. Der hat aber mehr Freiheiten, was die Gestaltung (optisch) des Produkts z.B. Logos, Farben betrifft.
Beste Grüße, Christian Johner
Hallo!
Mein Kommentar kommt etwas spät, aber vielleicht liest es noch jemand:
MDR sagt (Annex II, 1.1):
„…a general description of the key functional elements, e.g. its parts/components (including software if
appropriate), its formulation, its composition, its functionality and, where relevant, its qualitative and
quantitative composition…“
Die viel diskutierte Rezeptur/Herstellungszusammensetzung ist meiner Meinung nach damit nicht unbedingt weiterzureichen. Nur, wenn dies für die Konformitätsbewertung relevant ist.
ebenso Annex II 1.2:
„… an overview of the previous generation or generations of the device produced by the manufacturer, where such
devices exist; “
Ich sehe hier kein Bedürfnis meine ganze Entwicklungsakte freizugeben an einen legalen Hersteller (der mein PLM Kunde ist). Ich sehe die Pflicht ihn über vorige Informationen zu informieren und einen Überblick zu schaffen.
Selbes für Annex II 3:
„…identification of all sites, including suppliers and sub-contractors, where design and manufacturing activities are
performed. “
Keiner meiner Zulieferer erfüllt Design- oder Herstellungsaufgaben (mit Ausnahme von Sterilisatoren und die muss man ja ohnehin bekannt geben). Für Produkte, die rein mit Chemikalien im Eigenbau produziert werden, sehe ich keinen Grund warum der legale (PLM-) Hersteller die Identität meiner Lieferanten kennen muss.
Ich habe mich auf jeden Fall selbst überzeugt. Jetzt fehlt nur noch mein Notified Body 😉
LG
DS
Guten Morgen,
wir hatten für Kl.1 Produkte bisher tlw. eine OEM/PLM-Konstruktion, tlw. auch Eigenproduktion und somit schon TDs. Diese erweitern wir nun und erstellen für alle Produktgruppen TDs nach MDR – nun meine Frage:
Klinische Bewertung, Risikoanalyse, Biologische Beurteilung etc. sind ja einheitlich – aber was ist mit den Dokumenten zur Herstellung (Herstellabläufe, Materialzeugnisse etc), wenn wir das gleiche Produkt in Zukunft bei unterschiedlichen Lieferanten einkaufen wollen? Kann ich „unterschiedliche“ Unterlagen von mehreren Herstellern in meiner Akte hinterlegen? (die Produktionsschritte, Hilfsstoffe etc. sind ja ggf. unterschiedlich) Wie muss ich vorgehen? Wie sind die vertraglichen Beziehungen (wir wollen als Hersteller auftreten)? Im Voraus vielen Dank!
Gruß Rainer
Sehr geehrter Rainer,
ich verstehe Ihre Frage so, dass Sie wissen wollen, ob eine technische Dokumentation sowohl Dokumente enthalten kann, die für mehrere Produkte einer Produktklasse (gleiche Basic UDI-DI) gelten, und Dokumente, die für jedes Produkt (gleiche Basic UDI-DI und eigene UDI-DI) spezifische sind.
Die kurze Antwort lautet „ja“. Der Gedanke mit der Basis UDI-DI war genau so etwas zu ermöglichen.
Beste Grüße Christian Johner
Sehr geehrter Herr Johner,
ich habe eine Frage in Bezug auf Änderung / Anpassung unserer PLM/OEM Konstellationen. Wir möchten bei bestimmten Produkten nur noch als Händler auftreten, allerdings mit unseren eigenen Etiketten. Der Hersteller wird selbstverständlich mit seinem CE auf diesem Etikett genannt und wir sind als Händler ebenso eindeutig erkennbar. Alle weiteren Daten sind die des vom Hersteller freigegeben Etiketts, aber… Wir möchten natürlich unsere eigenen Artikelnummern verwenden, was vielerlei Hintergründe hat (Katalog, Werbematerial, Kundenhistorie, etc.).
Wir selbst praktizieren dies so bereits mit eigenen Kunden, die auch schon vom wackligen „OEM-Zug“ abgesprungen sind und jetzt ebenso nur noch als Händler auftreten. Auch diese Kunden verwenden eigene Artikelnummern, welche wir vertraglich mit einer entsprechenden Vergleichsliste geregelt haben.
Aus unserer Sicht wäre dies regulatorisch durchaus durchführbar und machbar, da ansonsten die ganze Co-Labelling / Re-Packer-Konstellation ad absurdum geführt wird, wenn dann auch nur noch die Artikelnummern des Herstellers verwendet werden dürfen. Manche ERP-Systeme kommen nämlich mit bestimmten Nummernkreise nicht zurecht. Das wäre – um es sanft auszudrücken – hanebüchen…
Aber wir haben aktuell exakt diese hanebüchene Situation, da einer unserer Lieferanten bzw. besser gesagt dessen notified body es nicht gestattet, dass wir unsere eigenen Artikelnummern verwenden. Er begründet das mit der fehlenden Verknüpfung zu den ihm vorliegenden Artikelnummern und das im Falle von Vorkommnissen ihm diese (unsere) Nummern nicht bekannt sind…
Wie gesagt, ich finde das sehr unverständlich, denn der Hersteller sowie wir als Händler haben eine vertragliche Regelung in der eindeutig ein Bezug zu den Artikelnummern des Herstellers hergestellt ist.
Mich würde Ihre Meinung an dieser Stelle brennend interessieren bzw. ggf. auch regulatorische Passagen die diesen Bereich in irgendeiner Form genauer beschreiben. Der Artikel 16a der MDR schreibt nur vom eigenen Namen, dem eigenen eingetragenen Handelsnamen oder der eigenen eingetragenen Handelsmarke, jedoch aber nicht direkt von einem Produktcode oder einer Artikelnummer.
Ich hoffe ich konnte alles möglichst verständlich erläutern und hoffe, dass Sie an dieser Stelle einen Input für uns haben?
LG Tobias Bauer
Lieber Herr Bauer,
das ist wirklich eine verzwickte Situation.
Mir ist keine Regularie bekannt, die Ihnen Ihre eigene Artikelnummer verbietet. ABER:
Wichtig ist, das Produkt des Herstellers(!) eindeutig zu charakterisieren. Man will vermeiden, dass ein Händler den Weg zum Herstellen und dessen eindeutigen Produkt „vernebelt“. Die Zuordnung zu den vom Hersteller gemeldeten Produkten (EUDAMED) muss einem Dritten möglich sein, sonst ist der ganze Witz dahin: Besonders bei kritischen Produkten möchte man, dass sogar Endanwender z.B. die Post-Market Surveillance Reports in der EUDAMED einsehen können. Ohne eine klare Zuordnung wird das nicht gelingen.
Fazit: Sie können Ihre Artikelnummer nutzen. Aber die (zusätzliche) eindeutige Bezugnahme (UDI-DI) zum Herstellerprodukt muss gegeben sein.
Ich grüße Sie herzlich, Christian Johner
Sehr geehrter Herr Johner,
wenn ich Waren aus Fernost in die EU importiere, dann gelte ich laut Gesetz offiziell als Hersteller und muss auch als dieser auf dem Produkt genannt werden. Der OEM sitzt in China.
Wie sieht das die neue MDR Regelung? Müssen die Kontaktdaten des OEM auf dem Produkt erscheinen?
Die Definition von Hersteller, Händler usw. ist in der MDR etwas ungenau definiert…
Ich danke im Voraus!
Mit freundlichen Grüßen,
Matthias
Die MDR kennt keine OEM-PLM-Konstrukte mehr.
Die MDR definiert die von Ihnen genannten Begriffe. Wenn Sie dazu Fragen haben, geben Sie gerne Bescheid.
Ein Importeur ist kein Hersteller, aber er zählt zusammen mit den Herstellern zu den Wirtschaftsakteuren.
Der Name des Hersteller, das ist in Ihrem Fall die Firma in Fernost muss auf dem Produkt aufgebracht sein. Es ist aber auch möglich zusätzlich die Angaben des Händlers mit aufzubringen.
Sehr geehrter Herr Johner,
kann ich weiterhin die OEM-PLM Konstellation mit einem Kunden außerhalb der EU fortführen? Wenn ja, muss ich vertraglich festlegen, dass die Produkte unter dem PLM Namen nicht in die EU zurückgeführt werden?
Herzlichen Dank vorab!
Mit freundlichen Grüßen
Petra
Sehr geehrte Frau Petra,
das Ende, das dieser Artikel diskutiert, betrifft nur Produkte, die im EU-Markt in den Verkehr gebracht werden sollen. Wenn EU-Hersteller in Märkten außerhalb der EU ihre Produkte in den Verkehr bringen möchten, dann gelten die Gesetze des entsprechenden Marktes. Hier sind PLM-OEM-Konstruktionen oft noch möglich.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
wie verhält es sich, wenn ein und das selbe Produkt nach MDR der Frima X als OEM für den PLM/Hersteller gefertigt, dieses aber gleichzeitig von der Firma X als Hersteller (Manufacturer nach FDA) an einen Relabeler in die USA verkauft wird?
Vielen Dank vorab
Viele Gürße
Simon
Sehr geehrter Simon,
ich bin nicht sicher, ob ich Ihre Frage verstehe. Das sind zwei Rechtsbereiche. Die Firma X kann ihr Produkt gleichzeitig an zwei Firmen, eine in EU (PLM) und eine in den USA (Relabeler) verkaufen. Da spricht nichts dagegen, außer, dass es in der EU keinen PLM mehr gibt. Es bleibt daher der Firma X (OEM) nichts anderes übrig, als entweder die komplette technische Dokumentation zu öffnen oder den PLM auf einen Händler zu beschränken. Hierbei sollte dieser Artikel 16 der MDR lesen, um seine Freiheitsgrade zu kennen.
Vielleicht ist es mir gelungen, die Frage zu beantworten, falls nicht, einfach nachhaken.
Viele Grüße, Christian Johner
Guten Tag Herr Johner,
wir möchten gerne ein Medizinprodukt der Klasse I (Nitril-Untersuchungshandschuhe) mit unserem Markennamen in Europa vertreiben. Der Hersteller (Indien) soll nicht mit seinem Namen auf der Verpackung ersichtlich sein (demnach altes PLM/OEM-Konstrukt). Demnach müssten wir ja als Hersteller alle Pflichten laut Artikel 10 erfüllen. Dieses besagt ja auch u.a. dass die TD des Herstellers komplett bei uns vorliegen muss. Der Hersteller möchte dies nicht, und wenn ich das MDCG 2020-2 Dokument richtig interpretiere, dann gilt für die MP der Klasse I eine verlängerte Übergangsfrist, oder sehe ich das falsch? Wenn die Ausnahme für Klasse I Produkte gilt, dann könnten wir doch in diesem Fall die in ihrem Beitrag gezeigte e)ZLG Konstelation anwenden?
Mit freundlichen Grüssen
Sehr geehrter Herr Quadt,
wir haben Ihnen hier einen Beitrag zu den Übergangsfristen erstellt. Wie Sie der Grafik ganz unten entnehmen können, gilt die Übergangsfrist bei Klasse-I-Produkten bezüglich der Inverkehrbringung nur für Produkte, wenn diese unter der MDR höher gestuft werden. Solange diese in der Klasse I bleiben, endet die Übergangsfrist. Für die Bereitstellung und Inbetriebnahme bleibt noch etwas mehr Zeit.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
mit großem Interesse habe ich Ihren Blogbeitrag gelesen.
Wir sind eine deutsche Firma und versuchen derzeit, die Produktion des Antigen-Schnell-Tests mit unserem eigenen Patent bei einer Fabrik in China zu veranlassen, die als Hersteller das Produkt in Deutschland zulassen lässt, z.B. durch BfArm. Dürfen wir dann unsere Markennamen auf der Verpackung drucken lassen und als „Patentinhaber“ sowie „Händler/Importeur“ mit unserem Firmennamen und Anschrift auf der Verpackung stehen, auch wenn wir keine EU-Bevollmächtigte von diesem Produkt sind?
Welche Qualifikationsanforderungen (wie QM, Hygiene, Mitarbeiter, CE-Zertifikat) und Verpflichtungen bzw. technische Dokumentation müssen wir hier dann überhaupt noch erfüllen/vorlegen?
Laut meinem Verständnis ist das ja eigentlich keine „OEM/PLM“ Struktur mehr, da der eigentliche Hersteller mit seiner Anschrift dann auf der Verpackung steht und er mit dem Importeur bzw. Händler eine Vereinbarung getroffen hat, dass er (der Hersteller) für die Einhaltung der Anforderungen verantwortlich bleibt. Wir ändern auch nicht mal die Außerverpackung, weil der Hersteller die Produkte bereits in der Verpackung des Händlers ausliefert. Deswegen sollte doch die Verantwortung nur an die Herstellerangabe geknüpft sein.
Allerdings bin ich etwas verunsichert von dem Punkt „2. Variante: Händler statt PLM„ in Ihrem Text. Demnach gibt es Pflichten für den Händler wie z. B. „ein Qualitätsmanagementsystem“, die Behörde über die Inverkehrbringung zu informieren usw. Wenn wir aber als Händler die komplette Produktionskette inkl. Verpackung in China haben, müssen wir trotzdem diesen Pflichten nachkommen?
Vielen Dank vorab
Viele Grüße
(Frau) Qimei Li
Sehr geehrte Frau Li,
danke für Ihre Frage!
Sie dürfen zusätzliche Informationen wie Patente oder Händler mit aufbringen. Sie als Hersteller in der EU müssen aber auch genannt sein.
Sie dürfen auch Prozesse auslagern. Sie müssen diese aber lenken.
Händler haben in der Tag Informationspflichten. In Ihrem Fall scheinen Sie aber nicht der Händler, sondern der Inverkehrbringer selbst zu sein.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich habe eine Frage bez. der Registrierung als „contract manufacturer“ bei der FDA.
Unsere Firma (Lohnfertiger) fertigt Medizinprodukte für eine weitere Firma, die das Produkt (ihre eigene Entwicklung) überprüft und wiederum an einem Inverkehrbringer verkauft.
Haben wir die Pflicht uns bei der FDA zu registrieren?
Würde eine entsprechende QSV ausreichen?
Vielen Dank für Ihre Antwort.
P. Beisiegel
Sehr geehrte Frau Beisiegel,
sobald Sie als Lohnfertiger ein „finished device“ gemäß FDA-Definition herstellen, gelten Sie als „contract manufacturer“ und müssen sich bei der FDA registrieren. Abhängig von den gefertigten Produkten müssen Sie die Quality System Regulation der FDA einhalten (21 CFR 820). Eine QSV ohne Registrierung bei der FDA würde in diesem Fall nicht ausreichen.
Als finished device definiert die FDA: „a finished device means any device or accessory to any device that is suitable for use or capable of functioning, whether or not it is packaged, labeled, or sterilized.“
Herzliche Grüße
Luca Salvatore
Wir sind Hersteller von Medizinische Gesichtsmaske EN14683:2019 Typ IIR, und haben die Produktion am 25.05.2021 eingestellt. Alle Masken wurden bis zum 26.05.2021 abverkauft. Was bedeutet dies für die Technische Dokumentation, muss diese auf die neue MDR angepasst werden, oder reicht eine Aktuallisierung mit den PMS Daten aus?
Muss eine PSUR wie sie die MDR fordert trotzdem erstellt werden?
Ich bedanke mich schon mal vorab für ihre Unterstützung, und hoffe sie können mir weiterhelfen.
Viele Grüße L.M.
Sehr geehrter L.M.
Wenn die Produkte abverkauft wurden, müssen Sie die TD nicht mehr aktualisieren. Die Anforderungen an die PMS gelten jedoch. Dafür gibt es keine Übergangsfristen. Lesen Sie dazu auch den 4. Abschnitt der Antwort auf die Frage 17 im FAQ des NAKI.
Viele Grüße, Christian Johner
Hallo,
gibt es Neuerungen bzw. weiterführende Informationen im Bereich OEM/PLM Konstrukten laut IVDR oder MDR?
Danke
Sehr geehrte Frau Plietz,
Es gibt keine Neuerungen und ich erwarte auch keine. Die PLM-OEM-Konstrukte sind Vergangenheit.
Der Inverkehrbringer braucht vollen Zugriff auf die technische Dokumentation.
Die angedachten Konstrukte mit einem Mittelsmann werden von einigen Behörden nicht akzeptiert.
Beste Grüße, Christian Johner
Dear Prof. Dr. Johner,
Dear Dr. Christian
May I ask you kindly a couple of questions?
If a company A is the OEM but the products of this company are on the market from company B (PLM). In the legal contract however, company A is distributor and company B is the manufacturer. My couple of questions are:
1- Which duties does every one have according to IVDR?
2- Is it allowed for company A to assign the UDIs and resgiter them in EUDAMED?
3- Is it allowed to have on the products the UDIs assigned from company A, or must company B assign the UDIs and put them on the labels on the products?
4- Does this apply for legacy devices as well?
Thanking you!
Best regards,
Rania
Dear Rania,
thank you very much for your interest in that topic. Due to the complexity of your question, I suggest answering that in an individual consultation. Would you mind contacting us via our contact form?
Best regards
Christian
Sehr geehrter Herr Johner,
ab wann kann ein PLM ein Produkt nicht mehr in Verkehr bringen? Wenn für ein Produkt noch ein gültiges PLM EG-Zertifikat (MDD bzw. Legacy Device) vorliegt, aber das Zertifikat des OEMs abläuft, bis wann darf das Produkt in Verkehr gebracht werden? Ablauf EG-Zertifikat OEM oder Ablauf EG-Zertifikat PLM?
Vielen Dank für Ihre Antwort.
Viele Grüße
Nick
Lieber Nick,
das Thema OEM und PLM haben wir ausführlich in zwei Blockbeiträgen aufgegriffen:
https://www.johner-institut.de/blog/regulatory-affairs/oem-original-equipment-manufacturer/
https://www.johner-institut.de/blog/johner-institut/uebergangsfristen-mdr/
Ich würde davon ausgehen, dass der Ablauf eines der beiden Zertifikate sofort die Vermarktungsmöglichkeit beendet!
Da die MDR zumindest in Teilen (PMS, Vigilanz) bereits für Produkte gilt, die noch unter der MDD zertifiziert sind, sollte schon ein Vertrag zwischen beiden existieren, die die neue Situation unter der MDR berücksichtigt und in die Zukunft gerichtet ist.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Johner,
müssen sich contract manufacturer (also früher OEM) in EUDAMED registrieren? Das geht, meiner Meinung nach aus dem IVDR Artikel 10(14) hervor. Wir dachten aber immer, dass contract manufacturer keine Wirtschaftsakteure sind und damit sich nicht in EUDAME registrieren müssen (können).
Vielen Dank für Ihre Antwort.
Viele Grüße
Kristina
Liebe Frau Schenkova,
die Registrierungspflicht beschränkt sich gemäß IVDR zunächst tatsächlich auf (legale) Hersteller, Importeure und Bevollmächtigte.
Wenn man als (legaler) Hersteller allerdings Produkte anmeldet, wird man auch gefragt, ob es bei diesen Produkten einen (anderen) Hersteller gibt. Und den trägt man dort ein. Mit anderen Worten: Sie als Lieferant eines IVD-Herstellers müssen sich nicht als solcher in der EUDAMED registrieren.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Professor Johner,
Wir sind ein Importeur und würden gerne die vom Hersteller in Verkehr gebrachten Medizinprodukte in eigene Kartons mit eigenem Design, Logo etc. um-verpacken. Natürlich sollen weiterhin alle relevanten Informationen des Herstellers ebenso auf der Verpackung erscheinen.
Ich habe eine Frage zu Punkt C) MDR: Alternativen zum OEM-PLM-Konstrukt? 2. Variante: Händler statt PLM
„Er benötigt ein Qualitätsmanagementsystem (Artikel 16(3)), das durch die benannte Stelle begutachtet sein muss. Derzeit ist noch unklar, ob dies eine Zertifizierung bedingt und ob die Begutachtung sich nur auf die entsprechenden Prozesse des „Re-Labelings“ bezieht oder auf alle Prozesse, die eine ISO 13485 vorschreibt. Ebenfalls bleibt unklar, ob diese Bewertung einmalig oder fortlaufend erfolgen muss.“
In dem genannten Artikel 16(3) finde ich nichts zur Vorgabe, dass das QM eines Händlers/Importeurs durch die benannte Stelle begutachtet sein muss. Woher entnehme ich diese Pflicht einer Begutachtung? Weiß man zwischenzeitlich ob dies außerdem eine Zertifizierung bedingt?
Vielen Dank im Voraus für Ihre Antwort.
Mit freundlichen Grüßen,
Maximilian Koglek
Lieber Herr Koglek,
im Artikel 16 (4) der MDR steht: „Der Händler oder Importeur legt der zuständigen Behörde im selben Zeitraum von 28 Tagen eine Bescheinigung vor, ausgestellt von einer Benannten Stelle, die für die Art der Produkte benannt ist, auf die sich die in Absatz 2 Buchstaben a und b genannten Tätigkeiten erstrecken, in der bescheinigt wird, dass das Qualitätsmanagementsystem des Händlers oder Importeurs den in Absatz 3 festgelegten Anforderungen entspricht.“
Die Benannten Stellen scheinen hier bisher unterschiedliche Vorgehensweisen zu haben. Einige erwarten ein vollständiges Qualitätsmanagement-System, das der ISO 13485 entspricht. Unser Tipp: Sprechen Sie die verfügbaren Benannten Stellen an und holen Sie sich Informationen zu deren Vorgehensweisen und mögliche Kosten ein.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Johner
Ich hätte eine Frage zu folgender Aussage:
„Einige Tätigkeiten und Aktivitäten lassen sich nicht strikt zwischen OEM und PLM aufteilen. Dazu zählen insbesondere das Risikomanagement und die Bewertung der Gebrauchstauglichkeit. Dies wird zum Problem, da regelmäßig nur die PLMs über ein ausreichendes Verständnis des Nutzungskontexts verfügen.“
Wäre es in diesem Fall ausreichend, wenn der OEM allein ein Risikomanagement implementiert und aufrechterhält, oder müssen beide Parteien ein eigenes Risikomanagement führen und regelmäßig aktualisieren? Wie bereits erwähnt, hat der PLM wahrscheinlich mehr Daten zu diesem Thema als der OEM. Eine doppelte Durchführung erscheint mir daher wenig sinnvoll. Eine gemeinsame Lösung wäre zwar sinnvoll, könnte jedoch mit erheblichem Zeit- und Arbeitsaufwand verbunden sein und müsste von beiden Parteien gewünscht werden.
Welche Methode halten Sie in diesem Zusammenhang für die einfachste und dennoch konforme?
Vielen Dank für Ihre Antwort!
Freundliche Grüße
Flavia Ingold
Liebe Frau Ingold,
vielen Dank für Ihre spannende Frage! Wie im Beitrag beschrieben, ist das Konstrukt OEM/PLM unter der MDR nicht mehr so möglich. Wir haben als Alternative zum Beispiel aufgeführt: PLM wird zum Händler oder PLM wird zum Hersteller. Auch in diesen Fällen kann nicht einer alleine das Risikomanagement machen und braucht zumindest Informationen vom anderen.
Grundsätzlich ist also zunächst zu klären, wer der Hauptverantwortliche für das Risikomanagement nach ISO 14971 ist und das ist in meinen Augen in der primären Verantwortung der legale Hersteller. Er kann Teile der operativen Erstellung der Akte aber an seine(n) Lieferanten auslagern (z.B. Produktions-FMEA, Design-FMEA) bzw. die Händler auffordern, Rückmeldungen aus dem Feld reaktiv oder proaktiv einzusammeln und an ihn weiterzuleiten. Auf diese Weise stellt das Risikomanagement die zentrale Drehscheibe zwischen dem Hersteller des Medizinproduktes, seinen Lieferanten und seinem Kundennetzwerk dar.
Wir schauen uns die Situation in Ihrem Fall gerne spezifisch mit Ihnen in einem workshop an. Kommen Sie gerne jederzeit auf uns zu.
Herzliche Grüße
Christian Rosenzweig