
Unter Post-Market Surveillance (Überwachung nach der Inverkehrbringung) versteht man einen proaktiven und systematischen Prozess, um aus Informationen über Medizinprodukte, die bereits in Verkehr gebracht wurden, notwendige Korrektur- und Vorbeugemaßnahmen (CAPA, corrective and preventive action) abzuleiten.
Wir haben für Sie eine Checkliste zur Post-Market-Surveillance erstellt. Mit dieser Checkliste können Sie herausfinden, ob Sie die regulatorischen Anforderungen der MDR an die Post-Market Surveillance Ihrer Produkte erfüllen. Sie erhalten eine Übersicht über alle Aufgaben, die Sie erledigen müssen, um MDR-konform zu sein. Damit erhöhen Sie Ihre Sicherheit, im Audit und bei der Zulassung Ihrer Produkte erfolgreich zu sein.
Hier geht es zur Checkliste.
Beachten Sie auch die weiteren Artikel zur Post-Market-Phase.
1. Definition von Post-Market Surveillance
Sowohl die Medizinprodukteverordnung (MDR) und die Verordnung über In-vitro-Diagnostika (IVDR) als auch die FDA definieren den Begriff „Post-Market Surveillance“:
„all activities carried out by the manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from their devices placed on the market, made available or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions“
Quelle: MDR, IVDR
Die Definition der FDA ist vergleichbar:
„The active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device.“
Quelle: FDA 21 CFR part 822
Die Definition der FDA ist zwar angenehm kurz, doch die umständlicher zu lesende Darlegung der MDR erscheint hilfreicher. Denn sie beschreibt nicht nur die Tätigkeiten, sondern auch die Ziele der Post-Market-Aktivitäten.
2. Post-Market Surveillance: Ziele
Hersteller müssen die Risiken durch ihre Medizinprodukte minimieren und die Sicherheit der Patienten gewährleisten, bevor sie ihre Produkte in den Markt bringen. Behörden und Benannte Stellen überprüfen dies im Rahmen der Zulassung bzw. Konformitätsbewertung.
Allerdings offenbaren sich einige Risiken erst später im Laufe der Zeit, wenn die Anwender die Produkte täglich einsetzen.
Die Post-Market Surveillance hat zum Ziel,
- diese Risiken beim praktischen Gebrauch des Produkts systematisch zu identifizieren,
- die Leistungsfähigkeit der Produkte „im Feld“ zu überprüfen,
- Produktfehler und unentdeckt gebliebene Sicherheitsprobleme zu finden,
- die Nutzen-Risiko-Bewertung kontinuierlich zu aktualisieren und
- notwendige Maßnahmen wie Rückrufe schnell einzuleiten.
Nur durch eine kontinuierliche und systematische Überwachung nach der Inverkehrbringung (Post-Market Surveillance) können die Hersteller gewährleisten, dass die Medizinprodukte den Patienten den versprochenen Nutzen bieten und dass keine unbeherrschten Risiken existieren.
Welche Daten Sie bei der Post-Market Surveillance berücksichtigen können, lesen Sie weiter unten.
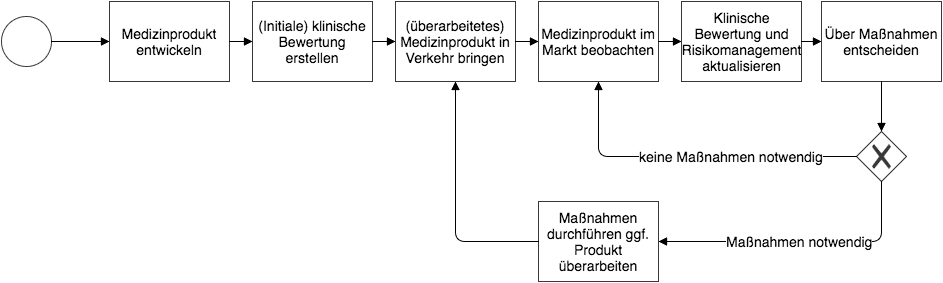
(zum Vergrößern bitte klicken)
3. Abgrenzungen
a) Post-Market Clinical Follow-up (PMCF)
Die Post-Market Surveillance verfolgt das Ziel, durch Beobachtung und Analyse des alltäglichen praktischen Gebrauchs den Nutzen von Medizinprodukten kontinuierlich zu belegen und bisher unbekannte Risiken zu identifizieren.
Eng verknüpft mit der Überwachung nach der Inverkehrbringung (Post-Market Surveillance) sind die Begriffe „Post-Market Clincial Follow-up“ (PMCF) bzw. – im Fall von IVD – „Post-Market Performance Follow-up“ (PMPF) und Vigilanz.
„Continuous process that updates the clinical evaluation referred to in Article 61 and Part A of this Annex“
Quelle: MDR
„Continuous process by which data are assessed and analysed to demonstrate the scientific validity, analytical performance and clinical performance of that device for its intended purpose as stated by the manufacturer“
Quelle: IVDR
Beim Post-Market Clinical Follow-up (PMCF) bzw. Post-Market Performance Follow-up (PMPF) geht es also um das systematische Sammeln klinischer Daten. Beim Post-Market Performance Follow-up (PMPF) werden proaktiv sowohl Daten zur Sicherheit und Leistung als auch wissenschaftliche Daten gesammelt.
Sowohl PMCF als auch PMPF im Fall von IVD haben zum Ziel, offen gebliebene wichtige Fragen zur Sicherheit oder Leistung des Medizinprodukts zu beantworten. Die Post-Market Surveillance beinhaltet das Sammeln aller Arten von bedeutsamen Information aus der Praxis, beispielsweise in Form von Serviceberichten, Anrufen bei der Hotline, Kundenbeschwerden usw.
Der PMCF hat zum Ziel, die klinische Bewertung zu aktualisieren; der PMPF soll die Leistungsbewertung von IVD erneuern. Die Post-Market Surveillance hat zum Ziel, über notwendige Maßnahmen zu entscheiden, um die Sicherheit der Patienten und Anwender zu gewährleisten. Bei dieser Entscheidung fließen die Ergebnisse der klinischen Bewertung bzw. Leistungsbewertung mit ein. Der PMCF bzw. PMPF ist somit eine Untermenge der Post-Market Surveillance.
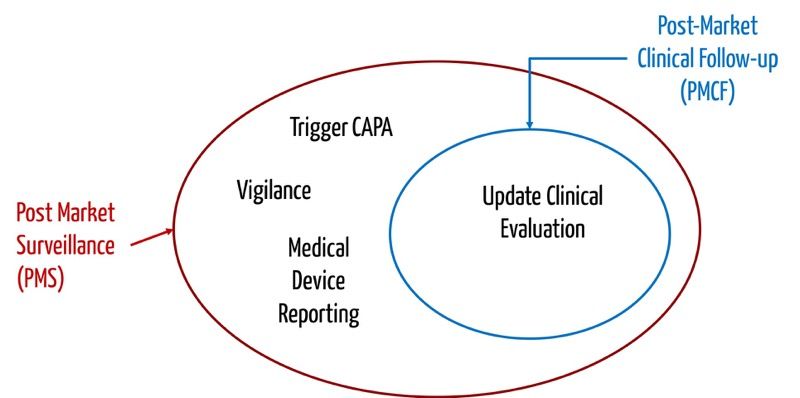
Dieser Ansatz spiegelt sich auch in den Anforderungen an den PMS-Plan in der MDR bwz. IVDR wider. Sie schreiben z. B.
The manufacturer shall undertake to institute and keep up to date a post-market surveillance plan, including a PMCF respectively a PMPF plan.
Im Gegensatz dazu unterscheidet die FDA die beiden Aspekte nicht so präzise: Sie verlangt beispielsweise, dass der PMS-Plan Angaben enthält, die man bei klinischen Studien festlegt, wie die Anzahl der Probanden, das Studienziel und Einverständniserklärungen.
Hier finden Sie Tipps für das Schreiben eines PMS-Plans und zu den Forderungen der ISO 20416.
b) Vigilanz
Unter einem Vigilanzsystem versteht man ein reaktives Meldesystem: Hersteller müssen im Rahmen der Vigilanz regeln, wie sie Vorkommnisse an die zuständigen Behörden melden. Gesetze und Verordnungen wie die MPAMIV lassen den Herstellern beim Festlegen des Meldesystems wenig Spielraum. Im Rahmen der proaktiven Post-Market Surveillance sollen Sie als Hersteller kontinuierlich auch die Daten aus Ihrem Vigilanzsystem auswerten.
Lesen Sie hier mehr zum Thema Vigilanz. Der Artikel geht auch auf die Abgrenzung zur Post-Market Surveillance ein.
c) Marktüberwachung
Nicht zu verwechseln mit der „Überwachung nach der Inverkehrbringung“ ist die „Marktüberwachung“. Letztere ist die Aufgabe der Behörden.
d) Fazit
Die Aktivitäten der Überwachung nach der Inverkehrbringung, des Meldewesens, des Post-Market Clinical Follow-ups bzw. Post-Market Performance Follow-ups und der Vigilanz sind teilweise überlappend. Daher werden die Begriffe häufig synonym verwendet – was sie aber nicht sind.
Die regulatorischen Anforderungen beziehen sich meist auf mehrere Aspekte.
4. Regulatorische Anforderungen
a) Medizinprodukte-Verordnung (MDR)
Die Medizinprodukte-Verordnung MDR beschreibt die Anforderungen an die Post-Market Surveillance wesentlich ausführlicher und konkreter als die zuvor gültige Medizinprodukterichtlinie (MDD, 93/42/EWG). Gleich vier Artikel und ein Anhang sind diesem Thema gewidmet.
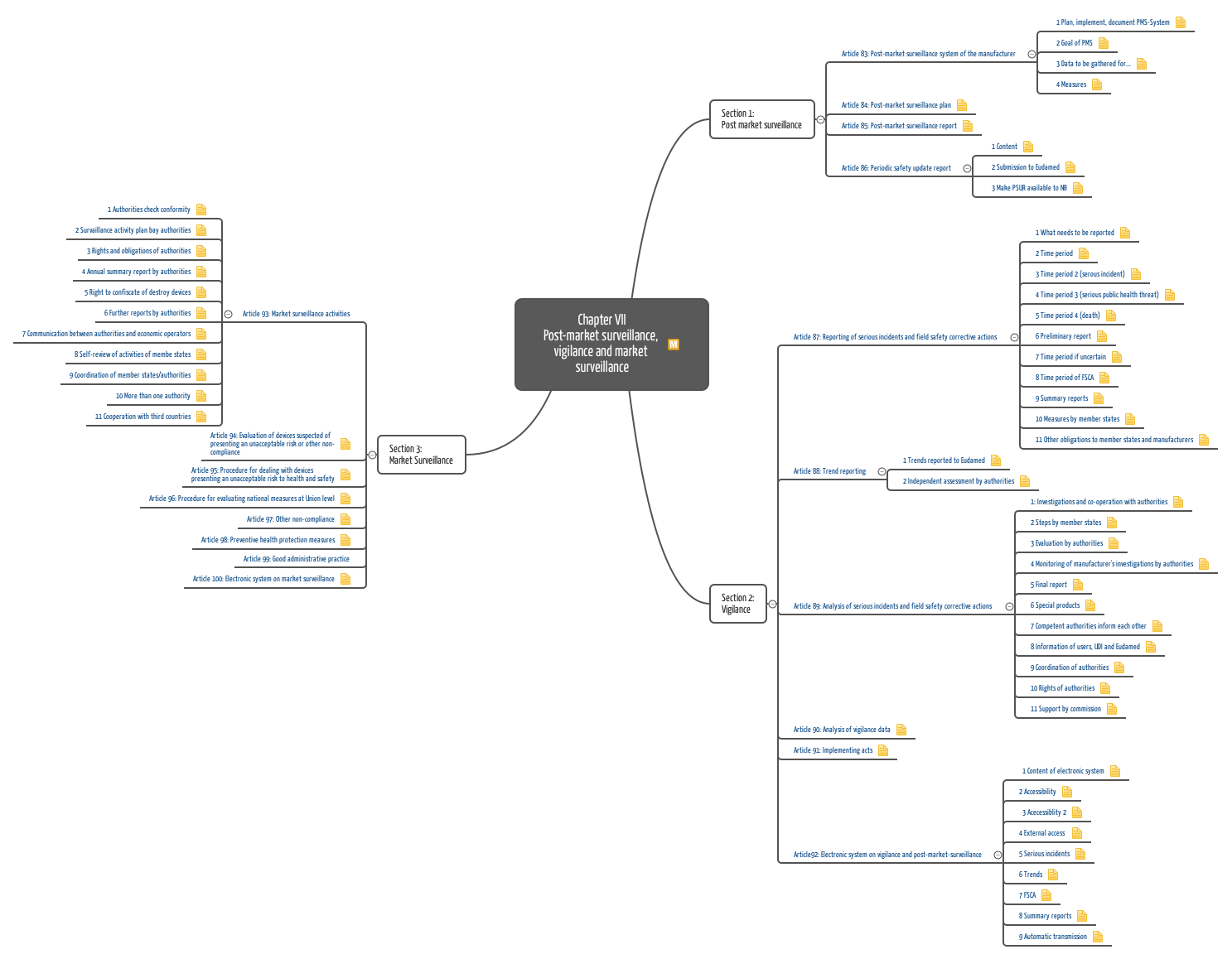
Wichtige Forderungen der MDR an die Post-Market Surveillance sind:
- Post-Market-Surveillance-Prozess definieren, planen und aufrechterhalten
- Kontinuierlich und systematisch Daten sammeln und bewerten
- Kontinuierlich auf Basis dieser Daten über Maßnahmen entscheiden wie
- CAPA initiieren (die kann das Produkt genauso betreffen wie den Hersteller und seine Prozesse)
- Behörden oder Anwender informieren
- Rückruf initiieren
- Klinische Bewertungsakte und/oder Risikomanagementakte aktualisieren (insbesondere als Ergebnis aus PMCF-Aktivitäten)
- Über Ergebnisse berichten (PMS-Bericht bzw. Periodic Safety Update Report PSUR)
Anhang III beinhaltet die Anforderungen an den PMS-Plan und somit, welche Quellen zu analysieren, wie diese zu bewerten und wie Maßnahmen zu ergreifen sind. Konkrete Anforderungen an den PMCF-Plan und die Aktivitäten sind in Anhang XIV, Teil B dargestellt.
Der Artikel zur Trendanalyse bei der Post-Market Surveillance gibt Tipps, um häufige Schwierigkeiten zu überwinden.
Das im Artikel 83(4) verlangte Unterrichten von Behörden bzw. benannten Stellen über abgeleitete Maßnahmen durch den Hersteller bezieht sich dabei nur auf sicherheitsrelevante CAPA, wie im MPDG §85 spezifiziert wurde.
b) Verordnung für IVD (IVDR)
In der IVDR sind ebenfalls ausführliche Anforderungen an das Post-Market Surveillance System in vier Artikeln (Artikel 78-81) sowie in Annex III beschrieben. Diese sind mit den o. g. Forderungen der MDR nahezu identisch, unterscheiden sich aber bezüglich des Post-Market Performance Follow-ups zur Aktualisierung der Leistungsbewertung. Neben der kontinuierlichen Sicherstellung des Nutzen-Risiko-Verhältnisses steht auch die Gewährleistung des klinischen Nachweises im Mittelpunkt.
Die Hersteller sind aufgefordert, den aktuellen Stand der Technik zu bewerten, die wissenschaftliche Validität zu bestätigen, z. B. durch Screening neuester wissenschaftlicher Literatur, und die Kontinuität der analytischen und klinischen Leistung ihres IVD zu belegen, z. B. durch Teilnahme an Ringversuchen, epidemiologische Studien, Datenbank-Recherchen oder PMPF-Studien.
Teil B des Anhangs XIII beschreibt die Ziele des Post-Market Performance Follow-ups und die Anforderungen an die Planung, Auswertung und Dokumentation.
c) ISO 13485
Die ISO 13485 verpflichtet die Hersteller, die Wirksamkeit des QM-Systems und die Sicherheit der Medizinprodukte u. a. durch eine systematische Überwachung nach der Inverkehrbringung (Post-Market Surveillance) zu gewährleisten.
d) ISO 14971
Auch die Norm zum Risikomanagement stellt Anforderungen an die „nachgelagerte Phase“. Der Fokus dieser Norm ist allerdings nicht das Meldewesen. Hier geht es vielmehr darum, anhand von Informationen aus der Produktion oder danach (z. B. aus der Nutzung des Produkts) mehr zu lernen darüber, ob
- Wahrscheinlichkeiten und Schweregrade möglicher Schäden richtig geschätzt sind,
- die Risiken vollständig identifiziert sind,
- die vermuteten Risikoakzeptanzkriterien und Nutzen-Risikoverhältnisse gültig sind.
In der nachgelagerten Phase müssen Hersteller, die im Sinn der ISO 14971 handeln, nicht nur nach Problemen suchen. Alle Informationen sind bedeutsam, die helfen, die Korrektheit der eigenen Annahmen zu verifizieren oder zu falsifizieren.
Welche Daten Sie bei der Überwachung nach der Inverkehrbringung berücksichtigen können, lesen Sie weiter unten.
Der Risikomanagementbericht muss bestätigen, dass die geplanten Aktivitäten angemessen sind.
e) USA/FDA: 21 CFR part 822 und Guidance Document „Post-Market Surveillance“
Die FDA regelt im 21 CFR part 822 genau,
- wann eine Post-Market Surveillance notwendig ist,
- was die Hersteller bei der Planung der Überwachung nach der Inverkehrbringung (Post-Market Surveillance) beachten müssen,
- welche Dokumente die Hersteller vorweisen müssen und
- wie schnell die Hersteller im Problemfall reagieren müssen bzw. die FDA handelt.
Die FDA hat 2022 speziell zum 21 CFR part 822 ein Guidance Document veröffentlicht, das den Herstellern weitere Hilfestellungen geben soll.
Weitere Informationen zu den Forderungen der FDA finden Sie in diesem Artikel zum 21 CFR part 822 und im genannten Guidance Document.
f) ISO TR 20416
Der „Technical Report“ ISO TR 20416 beschreibt noch genauer, wie Hersteller die Post-Market Surveillance planen und durchführen können. Beachten Sie jedoch unbedingt, dass ein PMS-Plan nach ISO TR 20416 nicht alle von der MDR bzw. IVDR geforderten Elemente enthält.
In diesem Artikel zur ISO 20416 finden Sie eine Übersicht über die Norm und einen Vorschlag zur Kapitelstruktur von PMS-Plänen.
g) Fazit
Im Gegensatz zum Meldewesen (Vigilanz) waren die europäischen Vorgaben zur eigentlichen „Überwachung nach der Inverkehrbringung“ bisher nicht so handlungsleitend. Dies hat sich nun mit der neuen MDR und IVDR geändert, welche die Post-Market Surveillance wesentlich umfassender, konkreter und ausführlicher beschreiben. Die nachfolgenden detaillierten Tipps helfen Ihnen bei der Umsetzung.
Bei der Post-Market Surveillance kann Sie auch der Post-Market Radar des Johner Instituts unterstützen.
5. SOP und Checkliste
Als Hersteller von Medizinprodukten sollten Sie Ihre Post-Market Surveillance anhand folgender Aspekte regeln – beispielsweise in einer entsprechenden Verfahrensanweisung.
- Auslöser von Aktivitäten
Es gibt generell zwei mögliche Trigger:- Basierend auf Zeitpunkt bzw. Zeitintervallen (z. B. einmal im Vierteljahr, jeden dritten Montag im Monat)
- Anlassbezogen (z. B. Kunde hat angerufen, neue Norm ist erschienen, tausendstes Gerät wurde ausgeliefert)
- Informationsquellen
Beispiele für Informationsquellen sind:- Kundenrückmeldungen einschließlich Kundenreklamationen
- Serviceberichte
- Ergebnisse bei Tests
- Beobachtungen von Mitarbeitern
- Behördendatenbanken mit Meldungen von Herstellern vergleichbarer Produkte, Technologien oder Verfahren über Probleme oder Maßnahmen
- Anrufe bei der Hotline
- Wissenschaftliche, technische und klinische Fachliteratur
- Ergebnisse von PMCF-Studien bzw. PMPF-Studien im Fall von IVD
- Messen und Konferenzen
- Tätigkeiten
Die Tätigkeiten betreffen typischerweise das- Sammeln der Daten,
- Auswerten der Daten,
- Bewerten der Daten,
- Ergreifen von Maßnahmen (z.B. Rückruf, Behördenmeldung, CAPA) oder das Begründen des Unterlassens.
- Verantwortlichkeiten
Die Verfahrensanweisungen sollten auch beschreiben, welche Rollen für welche Tätigkeit zuständig sind. Involviert sind beispielsweise die Medizinprodukteberater, die „für die Einhaltung der Regulierungsvorschriften verantwortliche Person“ PRRC, die Risikomanager, die Entwicklung, der Support, die Hotline, der Service und das Management. - Dokumentation und Werkzeuge
Da die Hersteller im Rahmen der Post-Market Surveillance umfangreiche Daten bewerten, empfehlen wir, diese Informationen mithilfe von Werkzeugen zu dokumentieren. Vergessen Sie nicht, diese Werkzeuge zu validieren. Die ISO 13485 fordert das.
Laden Sie sich hier unsere Checkliste als PDF-Dokument herunter. Sie listet detailliert alle Aufgaben, die Sie erledigen müssen, um einen MDR-konformen PMS-Prozess zu etablieren. So werden Sie im Audit bestehen und die Sicherheit Ihrer Produkte erhöhen.

Bestehen Sie Audits mühelos mit unserem Auditgarant
Sichern Sie sich eine stressfreie Audit-Erfahrung: Der Auditgarant bietet nicht nur Videotrainings zur Post-Market Surveillance, sondern auch erprobte Vorlagen, mit denen Sie schnell und kostengünstig Ihr PMS-System aufsetzen können.
Versionshistorie:
- 06.12.2022: Ergänzung der FDA-Guidance zu 21 CFR part 822 und allgemeines Update.



Sehr geehrter Herr Prof. Johner, Herr Geis von der Firma Pro Context hat mir empfohlen Sie zu kontaktieren bezüglich Post Market Surveillance.
Ich bin auf dem Gebiet noch ein greenhorn und möchte Sie fragen ob Sie Seminare für das „Post Market Surveillance“ anbieten?
Freundliche Grüsse
Mirko Gadza
Antwort per E-Mail folgt sofort 🙂
Sehr geehrter Herr Prof. Johner,
bei der Definition auf dieser Seite wird „Post-Market-Surveillance“ mit „Marktbeobachtung“ gleichgesetzt. In den neuen EU-Verordnungen werden doch aber unter „Marktbeobachtung“ die von den Behörden durchgeführten Tätigkeiten verstanden, während die Tätigkeiten des Herstellers „Überwachung nach dem Inverkehrbringen“ heißen. Falls ich hier richtig liege, mögen Sie Ihren Beitrag vielleicht überarbeiten.
Mit freundlichen Grüßen
Stephan Maihold
Da haben Sie Recht, lieber Herr Maihold! Wir werden den Beitrag aktualisieren.
Der PMCF war bei seiner Einführung 2007 eine merkwürdige, international einmalige Sache. Er war für viele KMU irritierend, weil sie begründen mussten, wenn sie keine PMCF durchführen wollten. Genau genommen war der PMCF eine klinische Studie ohne Ethikkommission und ohne Patienteneinverständniserklärung – eine Verletzung der Deklaration von Helsinki.
Hintergrund war nach meiner Erinnerung das Drängen von Herstellern von beispielsweise tierischem oder menschlichem Gewebe als Medizinprodukte. Da Langzeitschäden kaum innerhalb der üblichen wenigen Jahre klinischen Studien sicher auszuschließen waren, gab es die Furcht, eine Marktzulassung wirtschaftlich kaum überleben zu können. Also eröffnete die Kommission die Möglichkeit, klinische Studien alternativ nach Marktzulassung weiterzuführen. Der saubere Weg wäre gewesen, die Unsicherheit über langfristige Folgen als Restrisiko an Arzt und Patienten zu kommunizieren und die zuvor existierenden klinischen Studien längerfristig weiterzuführen.
Liest man die Kriterien für PMCF im MEDDEV 2.12/2 rev2, so sticht entsprechend hervor: „unanswered questions of long-term safety and performance “ Die anderen Punkte der Liste rechtfertigen keine PMCF. Die FDA ist hier ‚sauber‘ geblieben, vor allem ist es dort die Behörde, die die Notwendigkeit und die Kontrolle festlegt, und es ist ein sehr seltener Fall.
Schaut man sich nun die neue MDR an, so erscheint ein PMCF fast schon der Normalfall zu sein, eine Begründung, einen PMCF nicht durchzuführen, kaum möglich, insbesondere, weil es keine Kriterien gibt. Der Hersteller wird so zum Spielball der externen Gutachter. Die EU geht damit einen international völlig isolierten Weg, der zudem ethisch anrüchig ist: Klinische Studien müssen bei der Zulassung abgeschlossen sein. Wie soll ein PMCF brauchbare Daten liefern, wenn die Studie nicht engen Kriterien folgen kann und der Patient nicht eingeweiht ist? Es wäre mal interessant zu sehen, wie eine FDA reagiert, wenn ein in den USA und in der EU zugelassenes Produkt in der EU als in der klinischen Bewertung nicht abgeschlossen zu betrachten gilt.
Guten Tag,
muss ich als Händler eines nicht-EU Herstellers ebenfalls eine PMS durchführen? Es handelt sich hierbei um ein Klasse I Produkt
Vielen Dank
Die Aufgabe der PMS liegt beim Hersteller, nicht beim Händler. Allerdings kann der Hersteller die Händler verpflichten, Daten für die PMS bereitzustellen. Beispielsweise ist der Händler meist auch Medizinprodukteberater, der vom Gesetz verpflichtet ist, Informationen zur Sicherheit der Produkte an den Hersteller zu liefern. Das zählt auch zur PMS.
Sehr geehrter Herr Professor Johner,
Ich bin Student der TUM und interessiere mich für die neuen Zulassungs Richtlinien von Medizin Produkten. Mit der eintretenden MDR, welche Risikoklassen müssen die PMS durchführen und was passiert, wenn diese nicht durchgeführt werden. Verliert das Produkt dann seine Zulassung?
Vielen Dank für ihre Hilfe im Voraus.
Sehr geehrter Herr Amm,
danke für Ihre Nachricht!
Alle Produkte müssen im Markt überwacht werden. Nur die Anforderungen an die Berichte unterscheiden sich.
Wenn die PMS nicht durchgeführt wird, kann es zwei Konsequenzen haben:
1. Die Behörden merken und ergreifen dann eine Maßnahme. Das kann bis zur Stilllegung reichen.
2. Die benannte Stelle bemerkt es und kann dann das ISO 13485 Zertifikat aussetzen. Das kann ebenfalls zur Folge haben, dass keine Produkte mehr vermarktet werden dürfen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner
Wir sind Zulieferer von fertigen Medizinprodukten, wir liefern keine OEM Produkte noch bringen wir Produkte unter eigenem Namen in Verkehr.
In diesem Fall benötigen wir doch weder Regelungen für Post-Market Surveillance, der Überwachung nach der Inverkehrbringung, des Meldewesens, des Post-Market Clinical Follow-up und der Vigilanz oder eine Qualified Person?
Sehr geehrtere Herr Hirschauer,
in diesem Fall sind Sie zu nichts verpflichtet — von der üblichen Produkthaftung abgesehen.
Für die genannten Themen ist der Hersteller verantwortlich. Er kann Sie über eine QSV natürlich zu etwas verpflichten.
Viele Grüße, Christian Johner
Eine prima Übersicht mit sauberer Abgrenzung der ähnlichen und sich teils überschneidenen Begriffe und Aufgaben der Überwachung nach der Inverkehrbringung (PMS), des Post-market clinical follow-up (PMCF) und des Meldesystems über Vorkommnisse (Vigilanzsystem).
Danke, lieber Herr Dudzinski! Ich freue mich über Ihre Rückmeldung! Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
mich würde interessieren wie lange die Post-Market Surveillance (incl Aktualisierung des Clinical Evaluation Reports und PMS bzw PSUR Reports) nach dem Market Exit erfolgen muss? Gibt es da einen Richtwert (voraussichtliche /empfohlene Lebensdauer + x bzw Mindesthaltbarkeitsdatum +x); Meist wird von 10 Jahren nach Market Exit ausgegangen aufgrund der Aufbewahrungsfristen der Daten, aber gibt es für die PMS verbindliche Richtwerte?
Herzlichen Dank
Sehr geehrte Frau Dr. Bernek,
die MDR fordert in Artikel 61, dass die klinische Bewertung über den kompletten Lebenszyklus erfolgen muss. D.h. wenn Sie die PMCF vermeiden wollen, müssen Sie den Einsatz des Produkt beenden. Diese Anforderungen gilt unabhängig von den Aufbewahrungsfristen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
im Zuge der Umstellung auf die MDR bin ich in meinem Unternehmen mit dem Thema PMS beauftragt worden.
Hierzu stellt sich mir eine Frage: Gibt es ein „mindest“ Datum, an dem mit der Überwachung nach dem Inverkehrbringen von Medizinprodukten begonnen werden muss?
Mein Unternehmen hat 2010 ein neues Warenwirtschaftssystem eingeführt. Deshalb kann nur auf die Daten der letzten 9 Jahre zugegriffen werden.
Ist dieser Zeitraum für den PMS-Bericht ausreichend?
Vielen Dank im Voraus für Ihre Hilfe!
Freundliche Grüße,
M. Korf
Sehr geehrte/r Herr/Frau Korf,
danke für Ihre Frage!
Die PMS startet im Moment der Inverkehrbringung beginnt sofort nach der Inverkehrbringung. Allerdings wird keiner ein Problem machen, wenn man „nur“ auf die Daten der letzten 9 Jahre zurückgreifen kann. Je älter die Daten sind, umso weniger relevant. Es geht v.a. darum, *zeitnah* zu reagieren.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich hätte eine Frage bezüglich Vigilanz und Reporting unter PMS. Die Definiton von „Icidence“ hat sich im MDR geändert und die Interpretation dieser ist eher unklar in meine Augen.
„Any malfunction or deterioration in the characteristics or performance of a device made
available on the market, including use-error due to ergonomic features, as well as any
inadequacy in the information supplied by the manufacturer and any undesirable side-effect“
Würde dies dedeuten, dass ein „Incident“ auch einfach „any undesirable side-effect“ sein kann, oder muss dies in Kombination mit einer „malfunction“ stehen. Due deutsche Übersetzung dieser Definition hat anstatt dem „und“ ein „oder“, was zu noch mehr Unklarheit führt.
Besten Dank
Julia
Sehr geehrte Julia,
nein, es ist nicht „any undesirable side-effect“. Sonst müssten wir viel melden. Undesirable side-effects können in der RM-Akte sogar als akzeptabel bewertet sein. Es geht hier um die Kombination mit „malfunction or deterioration in the characteristics or performance of a device“.
Sie haben Recht, die deutsche Übersetzung ist nicht gut, weshalb wir in den Schulungen immer den englischen Text nutzen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner
Artikel 61 der MDR fordert, dass die klinische Evaluation über den gesamten Lebenszyklus eines Medizinproduktes aktuell gehalten werden muss. Falls ein Unternehmen sich entscheiden ein Medizinprodukt nach Mai 2020 nicht mehr auf den Markt zu bringen, muss dann folglich trotzdem weiterhin eine PMS durchgeführt werden? Solange wie die Lebensdauer der Produkte ist?
Herzlichen Dank!
Anja
Die Forderung nach PMS gab es schon immer – unabhängig von der MDR. Die Forderungen PMCF (> Aktualisierung der klinischen Bewertung) gibt es auch schon vor der MDR (u.a. MEDDEV). Daher ist die Antwort ja.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Johner,
gibt es in der MDR eine Festlegung in welchen Abständen die klinische Bewertung und der PMCF-Report aktualisiert werden müssen? Oder ist es den Herstellern überlassen einen angemessenen Zeitraum zu definieren. Bis jetzt haben wir uns bei der klinischen Bewertung an der MEDDEV 2.7.1 orientiert.
Danke im Voraus!
Beste Grüße, K.H. Suddera
Sehr geehrter Herr Suddera,
meine Expertin für klinische Bewertungen, Daniela Penn schreibt mir dazu:
In der Hoffnung, geholfen zu haben, und mit herzlichen Grüßen
Christian Johner
Vielen Dank für die erneute schnelle Hilfe! Die Antwort war sehr hilfreich!
Schöne Grüße, K.H. Suddera
Sehr geehrter Prof. Johner,
Frau Penn schreibt: „Die Aktualisierung hängt vom Ergebnis der letzten klinischen Bewertung bzw. deren Update ab. Die MDR macht keine genauen Vorgaben bis auf Klasse IIb und III Produkte. Hier liegt die Frist bei 1 Jahr.“
Wo genau finde ich diese Frist(en) in der MDR wieder?
Grüße
Kai Markus
Sehr geehrter Herr Markus,
hier die Antwort von Frau Penn:
Artikel 61, Abschnitt 11
The clinical evaluation and its documentation shall be updated throughout the life cycle of the device concerned with clinical data obtained from the implementation of the manufacturer’s PMCF plan in accordance with Part B of Annex XIV and the post-market surveillance plan referred to in Article 84.
For class III devices and implantable devices, the PMCF evaluation report and, if indicated, the summary of safety and clinical performance referred to in Article 32 shall be updated at least annually with such data.
Sehr geehrter Prof. Johner,
bei regulatorische Anforderungen a) hat sich der Fehlerteufel eingeschlichen, die klinische Bewertung steht im Anhang X und nicht IX.
Viele Grüße
Sie haben absolut Recht, lieber Herr Baudis!
Ich habe das sofort behoben.
Danke für den Hinweis, der mir geholfen hat, den Beitrag zu korrigieren.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner
In der MEDDEV 2.12-2 wird die ISO14155:2011 referenziert. Für PMCF Studien sind jedoch nicht alle Anforderungen der ISO umsetzbar. Wie bindend ist die ISO für PMCF Studien generell?
Vielen Dank und herzliche Grüsse
Anna
Ich vermute, dass diese Frage der anderen entspricht. Bitte verziehen Sie, wenn wir einen Tag zum Antworten benötigen.
Sehr geehrter Herr Prof. Johner
In der MEDDEV 2.12-2 wird die ISO14155 referenziert. Für PMCF Studien sind jedoch nicht alle Punkte vollumfänglich umsetzbar. Ist die ISO für solche Studien bindend oder stellt sie lediglich eine Empfehlung dar?
Vielen Dank im Voraus
Liebe Grüsse
Anna
Sehr geehrte Frau Rüegg,
danke für Ihre spannende Frage. Meine Kollegin, Daniela Penn, die bei uns die klinischen Studien betreut, ließ mich dazu Folgendes wissen:
Beantwortet das Ihre Frage? Falls nicht, wenden Sie sich gerne direkt an [email protected].
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
Genau, die Frage hatte ich erneut gepostet, da es für mich zuerst so aussah, als wäre sie am Tag zuvor nicht gespeichert worden – Entschuldigung.
Vielen Dank für die ausführliche Antwort, das hilft mir sehr weiter!
Herzliche Grüsse
Anna
Sehr geehrter Herr Prof. Johner,
in Artikel 83 „System des Herstellers für die Überwachung nach dem Inverkehrbringen“ der MDR bin ich über folgenden Passus gestolpert:
„(4) Zeigt sich im Verlauf der Überwachung nach dem Inverkehrbringen, dass *Präventiv-* oder Korrekturmaßnahmen oder beides erforderlich sind, so ergreift der Hersteller die geeigneten Maßnahmen und unterrichtet die zuständigen Behörden und gegebenenfalls die Benannte Stelle.“
In Ihrem Artikel „Rückruf: Ein gefährliches Missverständnis“ erklären Sie was man unter dem Begriff Rückruf zu verstehen hat und weisen auf die Meldepflicht von Rückrufen hin. In dem Artikel und den Kommentaren erklären Sie jedoch auch, dass Rückrufe korrektive Maßnahmen und nicht vorbeugende Maßnahmen betrifft.
Müssen durch die MDR (s.o.) nun auch jede vorbeugende/präventive Maßnahme gemeldet werden?
Wie soll man „im Verlauf der Überwachung nach dem Inverkehrbringen“ verstehen? Wird damit die halbjährliche PMS gemeint, in welcher man einen längeren Zeitraum von Rückmeldungen betrachtet und ggfs. Trends entdeckt? Wie sieht es bei der Meldepflicht von Rückmeldungen aus, welche sofort zu vorbeugende/präventive Maßnahmen führen und nicht erst bei der halbjährlichen PMS?
Mit freundlichen Grüßen
Dr. Martin Hartig
Lieber Dr. Hartig,
das ist eine ausgezeichnete Frage! Der Artikel 83(4) liest sich in der Tat so, als müsse jede Vorbeuge- und Korrekturmaßnahme gemeldet werden. Ich hatte deshalb bereits bei einer Benannten Stelle angefragt gehabt, die meinte, das sei ein Fehler, der korrigiert werden müsste. Das ist zwar nicht erfolgt. Allerdings enthält defacto das MDCG diese „Abschwächung“:
Hier schränkt man das auf die sicherheitsrelevanten CAPAs ein. Das ergibt meines Erachtens mehr Sinn.
Nochmals besten Dank!
Viele Grüße, Christian Johner
guten tag herr johner,
ich bin gerade über denselben text gestoplert, dass any preventive/corrective action, die isch aus der PMS ergibt, gemeldet werden müsse.
wissen sie, ob dieser fehler mittlerweile in form eines guidance doks adressiert wird? in der MDR ist der wortlaut ja noch immer gleich…
ansonsten darf ich mich an die vorgaben meiner competent authority halten, in meinem fall das BASG, das klar sagt dass jedes schwerwiegende vorkommnis sowie jede sicherheitskorrekturmassnahme im feld gemeldet werden müssen und damit preventive/corrective actions, die keine FSCAs sind, nicht gemeldet werden müssen?
danke für die hilfe!
Sehr geehrte Frau Albertini,
das MDCG 2022-21 zum PSUR geht in Kapitel 2.1.2 auf die in Artikel 83(4) MDR genannten Präventiv- oder Korrekturmaßnahmen ein und erläutert im Detail, dass es sich dabei nur um CAPAs handelt, die einen direkten Einfluss auf die Sicherheit, Leistung oder Qualität des Produkts haben.
Viele Grüße.
Sehr geehrter Herr Johner,
Die bisherige klinische Bewertung konnte sich ja bis jetzt auf ähnliche Produkte am Markt stützen, was sich mit der MDR ja erübrigt hat, da ja mehr oder weniger nur mehr idente Produkte für die klinische Bewertung herangezogen werden können.
Für viele Produkte, die bereits auf dem Markt sind, müssen nun klinische Bewertungen erstellt werden, die sich strikt auf das eigene Produkt beziehen, was sich hinsichtlich der Datenlage schwierig darstellt, da speziell für Niedrig- Risko Produkte der Klasse I klinische Studien finanziell nicht durchführbar (und gesetzlich auch nicht gefordert) sind und in speziellen Fällen (Schienen, Bandagen, etc) weder wissenschaftlich einen Erkentnissgewinn haben bzw. ethisch kaum vertretbar sind (Stichwort: Vergleich mit Nichtbehandlung?).
Wie sehen sie in diesem Zusammenhang Erhebungen, bei denen nicht interventionell über Fragebögen Sicherheits- und Leistungsmerkmale erhoben werden, um Daten für die PMCF und die Aktualisierung der klinischen Bewertung zu erheben? Nach welchen Kriterien müssten diese Erhebungen erfolgen?
Bei den zu erhebenden Merkmalen für genannte Klasse 1 Produkte handelt es sich um „weiche Kriterien“, die der Patient selbst beantworten kann.
Vielen Dank und liebe Grüsse
Sehr geehrter Herr S.,
vielen Dank für Ihren Kommentar. Es gibt meines Wissens keine formalen Anforderungen an Fragebögen, die Sie im Rahmen von PMCF-Aktivitäten bei Medizinprodukten einsetzen. Sie sollten natürlich die gängigen Kriterien bzw. den Stand der Technik für die Erstellung von Fragebögen berücksichtigen. Außerdem sollte es eine Planung, Protokollierung der Durchführung und einen Bericht geben. Unter Umständen kann es sein, dass Sie auch einzelne Kapitel der ISO 14155 berücksichtigen müssen, da auch nicht-interventionellen klinische Prüfungen im Scope dieses Standards sind. Da kommt es ganz auf die Zielstellung dieser PMCF-Maßnahme an. Auf jeden Fall würde ich empfehlen eine Nichtanwendbarkeit zu begründen.
Sehr geehrter Herr Professor Johner,
ist es in einer PMS zulässig das Medizinprodukt unentgeltlich zur Verfügung zu stellen?
Oder ist dies hinsichtlich der Anerkennung der erhobenen Daten problematisch?
Vielen Dank im Voraus und freundliche Grüße
T. Wohnlich
Sehr geehrte Frau Wohnlich,
die MDR und IVDR unterscheiden nicht, ob Produkte entgeltlich oder unentgeltlich bereitgestellt werden. Sie schreiben sogar explizit, dass beides einer Inverkehrbringung entspricht.
Somit gibt es auch keine anderen Pflichten an die PMS. Sie können durchaus Produkte kostenfrei bereitstellen. Die Anwendung dieser Produkte sollte aber repräsentativ sein. Auch sollten Sie keinen Verdacht erwecken, dass die Art der Informationen durch diese kostenfreie Bereitstellung einen „Bias“ bekäme.
Beste Grüße, Christian Johner
Hallo Herr Prof. Johner,
ich habe eine Frage bzgl. der PMS in Verbindung mit Drittländern.
Ist es für mich als Hersteller in der EU unerheblich, ob meine Ware an einen Kunden in der EU oder Drittland geht bzw. kann/ muss ich mit meinem Händler im Drittland (über welchen ich die Produkte vertreibe) die PMS-Vereinbarung ebenfalls abschließen?
Besten Dank vorab und VG
Elia M.
Vielen Dank für Ihre Nachfrage. Der PMS-Prozess schließt alle Wirtschaftsakteure mit ein. Die ist z.B. erkennbar aus der Definition in der MDR: „Überwachung nach dem Inverkehrbringen“ bezeichnet alle Tätigkeiten, die Hersteller in Zusammenarbeit mit anderen Wirtschaftsakteuren durchführen…“ oder auch im Punkt 1.1a) des Anhang III zum Inhalt des PMS-Plans. Auch aus der Zielstellung des PMS-Prozesses zur Identifizierung jeglicher notwendiger Korrektur- und Vorbeugemaßnahmen um die Sicherheit und Leistung des Produkts zu überwachen ergibt sich für mich, dass alle verfügbaren Daten, also auch aus Drittländern, gesammelt werden müssen. Wenn Sie hier Ausnahmen vornehmen, müssten Sie zumindest die Angemessenheit begründen können (z.B. Ergebnisse nicht übertragbar auf europäischen Raum). Die Pflicht zur Datenübermittlung sollte in die entsprechenden Vereinbarungen aufgenommen werden.
Ich hoffe, ich konnte Ihre Frage beantworten. Beste Grüße, Andrea Seeck
Hallo Herr Prof. Johner,
leider musste ich eine Ungereimtheit in der IVDR zumThema Field Safefty Notice (Art. 84, Punkt 8, Abs. 2) feststellen.
In der EN Variante steht: „field safety notice shall allow the correct identification of the device or devices involved, in particular by including the relevant UDIs, …“
In der DE Variante steht: „Die Sicherheitsanweisung im Feld ermöglicht die korrekte Identifizierung des Produkts bzw. der Produkte, insbesondere durch Aufnahme der Basis-UDI-DI und gegebenenfalls anderer UDI, …
Auf der EC Webseite (https://ec.europa.eu/docsroom/documents/32521) findet man ein Template von der EU (Stand 2018), das die UDI-DI vorschreibt.
Es stehen also untersch. Angaben im Raum, die hinsichtlich UDI in einer FSN gemacht werden sollen. Welchen Ansatz empfehlen Sie hier?
Herzlichen Dank vorab.
Viele Grüße,
Corinna Bausch
Liebe Frau Bausch, vielen Dank für Ihre Anfrage. Die von Ihnen genannte Passage ist der letzten konsolidierten Fassung der IVDR korrigiert wurden in „Die Sicherheitsanweisung im Feld ermöglicht die korrekte Identifizierung des Produkts bzw. der Produkte, insbesondere durch Aufnahme der relevanten UDI,…“ (Artikel 84, Abschnitt 8, Current consolidated version: 05/05/2017). In diesem Fall macht nur die UDI-DI Sinn, die Basis UDI-DI ermöglicht keine eindeutige Identifizierung z.B. bei verschiedenen Produktvarianten.
Hallo Frau Dr. Seeck,
vielen Dank für Ihre Antwort.
Dann korrigiere ich meine Frage:
Die IVDR besagt, dass die FSN die UDI (UDI-DI + UDI-PI) als korrekte Identifizierung aufführen soll.
Das Template von der EU führt lediglich die UDI-DI auf.
In einer Feldaktion kann dies einen beträchtlichen Unterschied machen.
Nehme ich richtig an, dass je nach vorliegender Ursache für die Feldaktion entweder das komplette Produkt (UDI-DI) oder die betroffenen Chargen (UDI-DI+UDI-PI) in die FSN eingetragen werden sollten?
Viele Grüße,
Corinna Bausch
Liebe Frau Bausch,
ich würde das genauso wie Sie interpretieren, dass je nach Ursache und Relevanz die entsprechend sinnvollere UDI eingetragen werden sollen. Die Bewertung dazu würde ich dokumentieren.
Beste Grüße,
Andrea Seeck
Hallo Herr Prof. Johner,
ich habe mir die Frage gestellt, ob Systeme gemäß Artikel 22 der MDR auch eine Post Market Surveillance benötigen. Werden die mögliche Risiken durch die PMS der einzelnen Komponenten abgedeckt, sodass eine PMS bei den neu entstandenen System gemäß Artikel 22 nicht nötig ist?
Mit freundlichen Grüßen
Kristin S.
Vielen Dank. Das ist eine sehr spannende Frage. Ich würde mich hier dazu am Abschnitt 3 orientieren. Dort heißt es: „Die Anwendung dieser Verfahren und die Beteiligung der Benannten Stelle sind auf die Aspekte des Sterilisationsverfahrens beschränkt, die der Gewährleistung der Sterilität des Produkts bis zur Öffnung oder Beschädigung der Verpackung dienen.“ Auf diese Aspekte würde ich auch einen PMS-Prozess ausrichten, wenn anwendbar. Außerdem würde ich sicherstellen, das mögliche Informationen zu den Einzelkomponenten, die bei mir als Systemersteller eingehen, an die jeweiligen Hersteller weitergeleitet werden. Hilft Ihnen das weiter?
Beste Grüße,
Andrea Seeck
Hallo Frau Dr. Seeck,
vielen Dank für Ihre Ausführungen.
Ich habe zwei Fragen. 🙂
MDCG-2021
1) MDR-Artikel 83(4) – meldungspflichtige CAPAs:
Sie haben das oben ausgeführt.
Wenn wir eine CAPA in Bezug auf das Produkt haben, die sicherheitsrelevant ist, dann muss man diese CAPA BfArM melden. Ob man die benannte Stelle informieren muss, hängt vom Vertrag mit der BS ab.
QM-relevante CAPAs sind in den Auditberichten aufgeführt und müssen nicht gemeldet werden.
„The scope of the various types of CAPAs under Article 83(4) does not cover quality management system related CAPA’s unless these could have a direct impact on product safety, performance or quality.” (MDCG 2022-21)
Meine Frage: Ich habe noch Schwierigkeiten mit dem Konzept von meldungspflichtigen CAPAs. Gäbe es eine Situation, bei der ich eine CAPA ohne FSCA und umgekehrt eine FSCA ohne CAPA hätte?
Worauf ich hinaus möchte: auf die separate Erwähnung von meldungspflichtigen CAPAs. Ich gehe momentan davon aus, dass es bei einem FSCA eh eine CAPA gibt. Daher wird die CAPA automatisch mit gemeldet, oder? Wo ist bei mir der Denkfehler?
2) Leading Device:
Man soll in einem Sammel-PSUR ein Leading Device nennen.
„Leading device: The “leading device” in a group of devices covered by the same PSUR corresponds to the highest risk class device. In the case when there are several devices with the same risk classification, the manufacturer should assign a leading device.“ (MDCG 2022-21)
Ein Leading Device wird benötigt, wenn im Sammel-PSUR Produktgruppen unterschiedlicher Risikoklassen behandelt werden. So verstehe ich das.
Beispiel: HF-Instrumente (Klasse IIb) und HF-Kabel (Klasse I).
Wenn in einem Sammel-PSUR Produktgruppen gleicher Risikoklasse behandelt werden, benötigt man dennoch ein Leading Device?
Beispiel: elevators, spatulas, speculum. Das sind alles Ir-Produkte. Somit gibt es keinen PSUR, aber einen PMSR. Dennoch nur als Beispiel. Gehen wir mal davon aus, diese Produkte seien Klasse 2. For the sake of argument … Benötigt man dennoch ein Leading Device?
Vielen Dank.
Grüße
Ayfer Bektas
Guten Tag Herr Bektas,
vielen Dank für Ihre Fragen.
Zur ersten Frage fallen mir spontan die Trends ein, die einen meldepflichtigen CAPA, aber ggfs. ohne FSCA, auslösen könnten.
Ihre zweite Frage kann ich auf Basis des MDCG-Dokuments nur mit Ja beantworten. Mir ist keine andere Regelung bekannt.
Beste Grüße,
Andrea Seeck
Guten Tag,
sie schreiben „Die FDA hat am 16. Mai 2016 speziell zum 21 CFR part 822 ein Guidance Document veröffentlicht, das den Herstellern weitere Hilfestellungen geben soll. Für dieses Guidance-Dokument wurde 2021 ein neuer Draft publiziert.“
Nur eine kleine Anmerkung meinerseits > der Draft ist mittlerweile finalisiert worden.
Mit freundlichen Grüßen
Lilli Tietjens
Liebe Frau Tietjens,
da haben Sie natürlich völlig recht, ich habe die Anpassung direkt vorgenommen.
Beste Grüße,
Andrea Seeck