
Mit dem Post-Market-Surveillance-Plan (PMS-Plan) wollen Medizinproduktehersteller zwei (möglicherweise) gegensätzliche Ziele erreichen: Einerseits möchten und müssen sie die gesetzlichen Anforderungen erfüllen und die Sicherheit ihrer Produkte maximieren. Anderseits wollen sie sich nur soviel Arbeit machen, wie sie auf Dauer wirklich leisten können.
Doch kann dieser Spagat gelingen? Gibt die ISO 20416 eine Hilfestellung?
Lernen Sie die 7 häufigsten Fehler kennen und erfahren Sie, wie Medizinproduktehersteller diese vermeiden können. Denn diese führen schon jetzt regelmäßig zu Problemen in Audits.
1. Was Sie über den PMS-Plan wissen sollten
Bevor Sie weiterlesen, sollten Sie die Ziele der Post-Market Surveillance (PMS) und des PMS-Plans kennen.
a) Ziele der Post-Market Surveillance
Die Post-Market Surveillance (auf deutsch: Überwachung nach der Inverkehrbringung) ist ein produktspezifischer, systematischer Prozess mit folgenden Zielen:
- Frühzeitig unerwünschte Nebenwirkungen und Risiken Medizinprodukten erkennen
- Einflüsse der jeweiligen Anwendungsumgebung identifizieren
- Probleme der Anwender identifizieren
- Hinweise auf einen nicht-bestimmungsgemäßen Gebrauch erhalten
- Hinweise auf sonstige Produktfehler erhalten
Fazit: Bei der Post-Market Surveillance suchen Hersteller nach Informationen, um die Sicherheit und Leistung der Medizinprodukte zu erhalten oder sogar zu verbessern.
b) Ziele des PMS-Plans
Der PMS-Plan dient dazu, die Aktivitäten der Post-Market Surveillance zu koordinieren. Er muss somit produktspezifisch festlegen,
- welche Personen und Rollen,
- zu welchen Zeitpunkten und bei welchen Anlässen
- für welche Tätigkeiten verantwortlich sind,
- welche Informationen diese Personen zusammentragen und
- wie sie diese bewerten,
- an wen sie diese Bewertung kommunizieren und
- welche Maßnahmen oder Meldungen sie auslösen müssen.
Fazit: Der PMS-Plan hat zum Ziel, dass jeder Beteiligte weiß, was er wann und wie machen muss, um die Ziele der Post-Market Surveillance zu erreichen: die Sicherheit der Produkte und damit der Patienten sowie die Konformität der Firma.
2. Anforderungen der MDR an den PMS-Plan
Die MDR legt die Anforderungen an den PMS-Plan in Artikel 84 und im Anhang III Abschnitt 1 fest.
a) MDR Artikel 84
Der Artikel 84 der MDR besagt:
Das System zur Überwachung nach dem Inverkehrbringen gemäß Artikel 83 stützt sich auf einen Plan zur Überwachung nach dem Inverkehrbringen; die für diesen Plan geltenden Anforderungen sind in Anhang III Abschnitt 1 dargelegt…
b) MDR Anhang III Abschnitt 1
Dieser Anhang III, Abschnitt 1 listet im Abschnitt a) Anforderungen an die einzubeziehenden Informationsquellen:
- Informationen über schwerwiegende Vorkommnisse, einschließlich Informationen aus den Sicherheitsberichten, und Sicherheitskorrekturmaßnahmen im Feld,
- Aufzeichnungen über nicht schwerwiegende Vorkommnisse und Daten zu etwaigen unerwünschten Nebenwirkungen,
- Informationen über die Meldung von Trends,
- einschlägige Fachliteratur oder technische Literatur, Datenbanken und/oder Register,
- von Anwendern, Händlern und Importeuren übermittelte Informationen, einschließlich Rückmeldungen und Beschwerden und
- öffentlich zugängliche Informationen über ähnliche Medizinprodukte.
Im Abschnitt b) legt die MDR die Anforderungen an die Methoden fest, mit denen die Hersteller diese Informationsquellen auswerten müssen, z.B.
- wirksame und geeignete Methoden und Prozesse zur Bewertung der erhobenen Daten,
- wirksame und geeignete Methoden und Instrumente zur Prüfung von Beschwerden und Analyse von marktbezogenen Erfahrungen, die im Feld erhoben wurden…
Die Anforderungen der IVDR an den PMS-Plan sind im Wesentlichen buchstabenidentisch mit denen der MDR.
Wir haben für Sie eine Checkliste zur Post-Market- Surveillance erstellt. Diese Checkliste wird Ihnen helfen herauszufinden, ob Sie die regulatorischen Anforderungen an die Post-Market Surveillance Ihrer Produkte erfüllen. Mit dieser Liste bekommen Sie eine Übersicht über alle Aufgaben, die Sie erledigen müssen, um MDR-konform zu sein. Damit erhöhen Sie Ihre Sicherheit, im Audit und bei der Zulassung ihrer Produkte erfolgreich zu sein. Laden Sie sich hier unsere Checkliste als PDF-Dokument herunter. Diese listet detailliert alle Aufgaben, die Sie erledigen müssen um einen MDR-konformen PMS-Prozess zu etablieren. So werden Sie im Audit bestehen und die Sicherheit Ihrer Produkte erhöhen.
3. ISO TR 20416: Eine nützliche Hilfestellung?
Der Technical Report ISO TR 20416 trägt den Titel Medical devices — Post-market surveillance for manufacturers. Diese internationale Norm widmet sich auch dem PMS-Plan.
a) Überblick über die ISO 20416
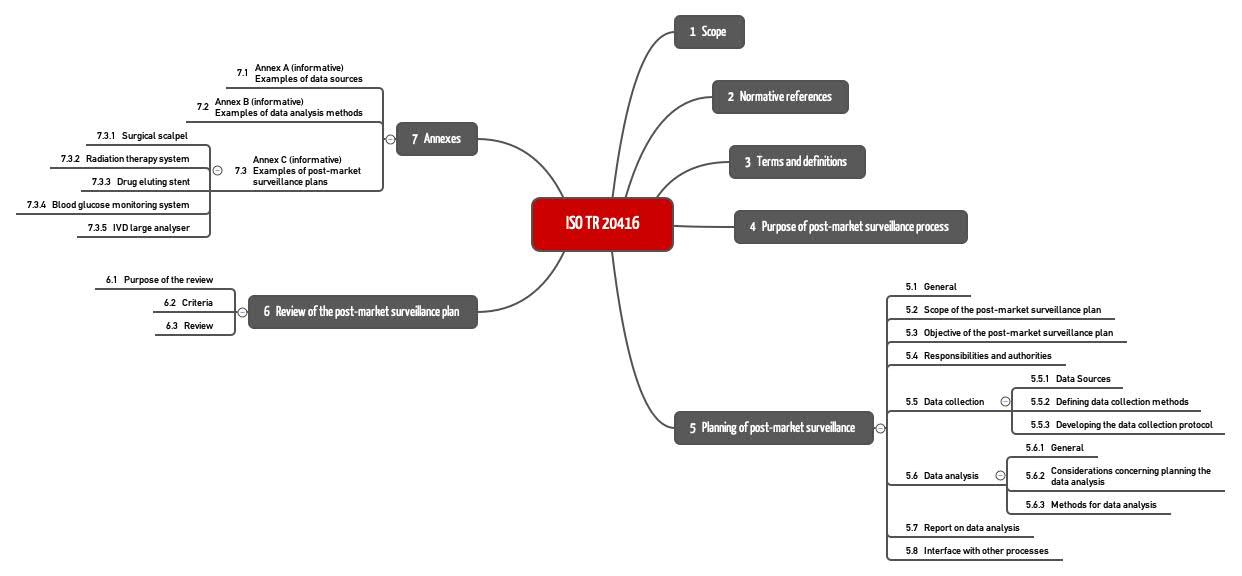
Die ISO 20416 nennt in Kapitel 4 die Ziele der Post-Market Surveillance und stellt in Kapitel 5 Anforderungen an den PMS-Plan. Kapitel 6 beschreibt das Review dieses Plans (s. Abb. 1). Bemerkenswert sind die Beispiele im Anhang C.
b) Zusammenspiel von ISO 20416, ISO 13485 und ISO 14971
Die ISO 20416 beschreibt auch das Zusammenspiel mit der ISO 13485 und der ISO 14971 (s. Abb. 2).
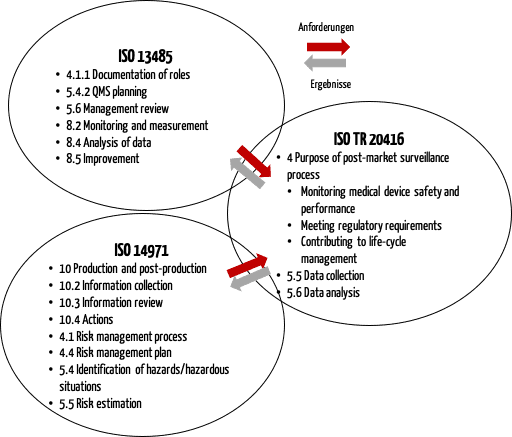
Diese Abbildung verdeutlicht, dass der PMS-Prozess mit anderen Prozessen im Qualitätsmanagement verknüpft werden muss, insbesondere mit dem Risikomanagement.
Aus der ISO 20416 ergeben sich konkrete Anforderungen die Hersteller:
- Kompetenzen der verantwortlichen Rollen im PMS-Prozess sicherstellen
- Aktuelle regulatorischen Anforderungen erheben (Regulatory Update)
- Bei der Managementbewertung den geforderten „Input“ einbeziehen und z.B. Kennzahlen und sinnvolle Zeiträumen festlegen
- Methoden zur Auswertung festlegen, z.B. Verfahren zur Kategorisierung oder Priorisierung von Kundenrückmeldungen
- Risiken und risikominimierende Maßnahmen fortlaufend bewerten und kontrollieren
4. Kapitelstruktur des PMS-Plans (ISO 20416)
a) Übersicht
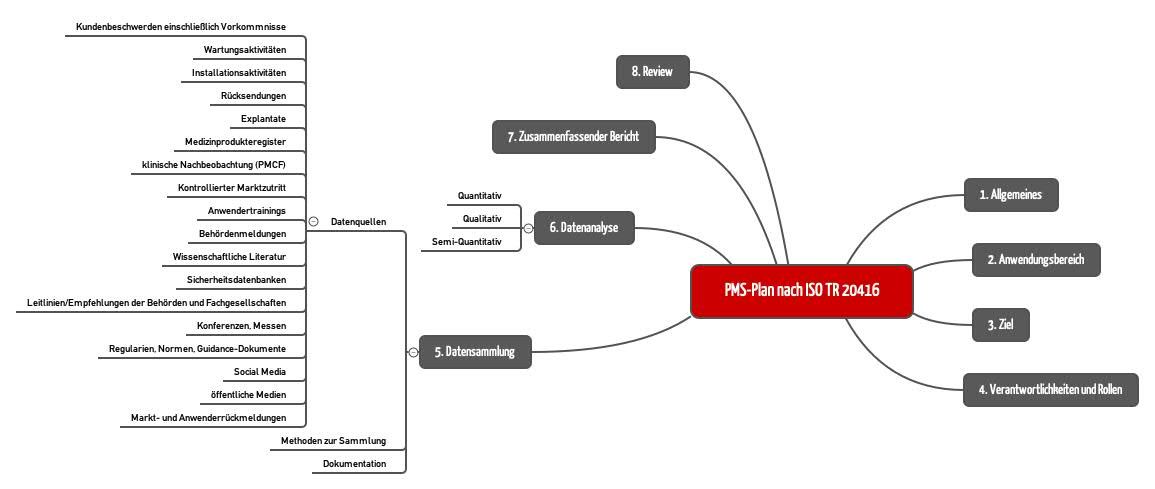
Die ISO TR 20416 schlägt eine konkrete Kapitelstruktur für den PMS-Plan vor (s. Abb. 3).
b) Hinweise der Norm zu den Kapiteln des PMS-Plans
1. Allgemeines
Hierzu gibt die Norm keine spezifischen Hinweise.
2. Anwendungsbereich des PMS-Plans
Hierzu zählen:
- Medizinprodukt, Produktfamilie und Zubehör und Anwendergruppe
- Klassifizierung und Märkte
- Erwartete Lebensdauer, erwartete Nutzungszahlen
- Verfügbare klinische Daten
- Aussage zum Innovationsgrad des Produkts
3. Ziel des PMS-Plans
Um neben den allgemeinen Zielen (Erkennung von neuen Risiken, Erfüllung der regulatorischen Anforderungen) die wirklich produktspezifischen Ziele abzuleiten, schlägt die ISO TR 20416 einen Fragenkatalog vor und nennt konkrete Beispiele:
- Überwachung der Sicherheit und Leistung in einem engen Intervall für eine begrenzte Zeitspanne (neues Produkt)
- Sicherstellen, dass die Verbindungen zwischen der klinischen Bewertung, den präklinischen Studien und den Risikomanagementprozessen robust und transparent sind (neues Produkt)
- Beschaffung von Informationen über die langfristige Sicherheit und Leistung des Medizinprodukts, einschließlich des klinischen Nutzens (Produkt mit CE nach klinischer Studie)
- Bestätigung der Prävalenz bekannter oder vermuteter unerwünschter Ereignisse (Produkt mit CE nach klinischer Studie)
- Überwachung der anhaltenden Zufriedenheit der Anwender mit dem Medizinprodukt und der Entwicklung des Stands der Technik (etabliertes Produkt)
- Einholen von Feedback für Verbesserungen, die nicht notwendigerweise mit Sicherheits- und Leistungsfragen zusammenhängen (etabliertes Produkt)
4. Verantwortlichkeiten und Rollen
Jeder Aktivität im PMS-Prozess muss eine für diese Aufgabe kompetente Person zugewiesen werden.
5. Datensammlung
Die Norm fordert die Nennung der produktspezifischen Informationsquellen sowie die Festlegung der Methoden zur Datensammlung. Die Sammlung der Daten muss entsprechend dokumentiert werden.
6. Datenanalyse
Hier unterscheidet die Norm in
- quantitative Methoden (z.B. deskriptive Statistik, inferentielle Statistik),
- qualitative Methoden (z.B. Kontextanalyse, narrative Analyse),
- semiquantitative Methoden.
7. Zusammenfassender Bericht zur Datenanalyse
Der Plan soll einen Hinweis auf den zu erstellenden Report geben. Das schließt Vorgaben für den Zeitraum, ein zu nutzendes Template u.ä. ein.
8. Review des PMS-Plans
Der Plan muss regelmäßig geprüft und ggfs. angepasst werden. Auch hierzu sollen Verantwortlichkeiten, Zeiten und Kriterien festgelegt werden.
c) Fazit
Die von der ISO TR 20416 vorgeschlagene Kapitelstruktur für einen PMS-Plan ist nachvollziehbar und sinnvoll.
Die Norm macht deutlich, dass es nicht mehr ausreicht, festzulegen, dass die Supportdaten und die Behördenmeldungen einmal im Jahr auf Hinweise überprüft werden. Sie beschreibt zahlreiche weitere Informationsquellen und deren Nutzen.
Was die Norm leider nicht verrät, ist, wie man einzelne Funde sinnvoll und objektiv bewerten kann. Und das führt zum nächsten Kapitel.
5. Die 7 häufigsten Fehler beim PMS Plan
Das Johner Institut übernimmt nicht nur die Post-Market Surveillance für Dutzende Medizinproduktehersteller (mehr dazu hier). Es bereitet auch auf Audits durch Benannte Stellen vor, indem es Hersteller auditiert und die Pläne und Ergebnisse der Post-Market Surveillance einem Review unterzieht. Dabei fallen die folgenden Fehler immer wieder auf:
Fehler 1: Suchstrategie ist nicht ausreichend dokumentiert
Wenn der PMS-Plan die Suchstrategie nicht genau festlegt, kann der Hersteller nicht sicher sein, dass zwei Bewerter zu den gleichen Suchergebnissen gelangen.
Der Plan sollte also genau vorschreiben, mit welchen Suchbegriffen, Suchkriterien und Suchfiltern die Bewerter in welchen Datenbanken und anderen Informationsquellen recherchieren.
Mithilfe eines Templates kann sichergestellt werden, dass die PMS-Pläne aller Produkte diese präzisen Festlegungen enthalten.
Fehler 2: Bewertung der Funde ist subjektiv, intransparent und nicht dokumentiert
Selbst wenn zwei Bewerter dieselben Suchergebnisse erhalten, ist noch nicht sichergestellt, dass beide auch zum gleichen Ergebnis bei der Bewertung kommen.
Damit eine objektive Analyse gelingt, ist eine konkrete Bewertungsstrategie bzw. ein präziser Fragen- und Kriterienkatalog erforderlich.
Zudem müssen die Hersteller die Transparenz und Nachvollziehbarkeit der Bewertung gewährleisten. Dazu sollten sie
- die bewerteten Daten sowie die Ergebnisse und Zwischenergebnisse der Bewertung dokumentieren,
- ein Template nutzen, das eine einheitliche Struktur und Verständlichkeit schafft, und
- auf die Kriterien verweisen, mit der sie zum Bewertungsergebnis kamen.
Der PMS-Plan sollte auch diese Bewertungskriterien enthalten.
Diese Bewertungskriterien in einer übergeordneten SOP festzulegen ergibt (meist nur) dann Sinn, wenn der Hersteller vergleichbare Produkte überwacht und Informationen bewertet.
Fehler 3: PMS-Pläne sind nicht produktspezifisch
Um relevante Ergebnisse zu erhalten, ist ein produktspezifischer Plan unumgänglich. Es ist z.B. nicht sinnvoll, in den FDA-Datenbanken zu recherchieren, wenn das Produkt in den USA gar nicht als Medizinprodukt reguliert wird.
Wer hingegen OTS-Software wie Betriebssysteme oder Datenbanken verwendet, wird um die US-amerikanische NIST-Datenbank nicht herumkommen, auch wenn er das Produkt nicht in den USA vertreibt.
Die PMS-Pläne müssen spezifisch sein für beispielsweise:
- Produkttyp, Produktkategorie
- Medizinische Indikation und Zweckbestimmung
- Vorgesehene Nutzer, vorgesehene Nutzungsumgebung
- Verwendete Technologien und Verfahren, z.B. Produktionsverfahren
- Märkte
- Lebensdauer, Verkaufsvolumen, Anwendungshäufigkeit
Fehler 4: PMS-Pläne sind nicht mit anderen Dokumenten abgestimmt
Hersteller dürfen die PMS-Pläne nicht als isolierte Dokumente verstehen. Vielmehr müssen sie abgestimmt sein mit z.B.
- der Zweckbestimmung,
- der klinischen Bewertung,
- der Risikomanagementakte.
Die klinische Bewertung empfiehlt PMS-Maßnahmen und PMCF-Maßnahmen: Finden sich diese im PMS-Plan wieder?
Der Risikomanagementplan legt die Überwachungstätigkeiten in der nachgelagerten Phase fest. Enthält der PMS-Plan diese Tätigkeiten?
Fehler 5: Statistische Signifikanz ist nicht (sinnvoll) festgelegt
Zugegeben, die Forderung der MDR ist schwer zu verstehen und zu erfüllen:
„… Methoden und Protokolle, die zur Feststellung jedes statistisch signifikanten Anstiegs der Häufigkeit oder des Schweregrades dieser Vorkommnisse anzuwenden sind, …“
Anhang III, Abschnitt 1b, MDR
Besonders bei geringen Stückzahlen ist die statistische Signifikanz häufig ein Problem. Hier kann oft nur eine Einzelfallbewertung vorgenommen werden. Die ISO TR 20416 unterstützt so einen Ansatz explizit.
Fehler 6: Verantwortlichkeiten sind nicht (ausreichend) festgelegt, Ressourcen fehlen
Die Post-Market Surveillance ist mit vielen Prozessen im Unternehmen verknüpft und fordert den Input zahlreicher Akteure. Daher müssen viele Rollen in die PMS eingebunden und entsprechend im PMS-Plan benannt sein, u.a.:
- Risikomanager
- Sicherheitsbeauftragter und/oder PRRC
- Support
- Service
- Produktion
- Clinical Affairs
- Anwender
- Händler
- Zulieferer
- Management
Alle Rollen und Personen sollten wissen, dass sie für die Post-Market Surveillance eingeplant sind; sie sollten ausreichende Kompetenzen und Kapazitäten haben und erhalten.
Fehler 7: Zeitintervalle zur Prüfung der Informationsquellen sind nicht angemessen
Viele Hersteller gehen davon aus, dass eine jährliche Überprüfung der Informationsquellen ausreicht. Doch das kann eine Fehleinschätzung sein.
- Neue Produkte
Häufig sind bei neuen Produkten viele Fragen zur Sicherheit, zur Leistungsfähigkeit und zum klinischen Nutzen noch nicht ausreichend beantwortet. - Produkte mit sich rasch ändernden Technologien und Regularien
Beispielsweise zum Machine-Learning erscheinen monatlich neue Publikationen zu neuen Verfahren und zur „Interpretability“.
Auch die Frequenz, in der neue Regularien publiziert werden, lässt eine nur jährliche Überprüfung als nicht adäquat erscheinen. - Produkte mit OTS-Software
IT-Security-Datenbanken publizieren manchmal Hunderte von Meldungen zu Schwachstellen pro Tag!
Insbesondere bei den genannten Produkten müssen die Hersteller eine engmaschigere Überwachung im Markt durchführen.
Tipp
Die PMS-Experten des Johner Instituts
- beantworten Fragen zu PMS-Plänen,
- unterziehen vorhandene PMS-Pläne einem schnellen Review
- und unterstützen bei der Erstellung neuer PMS-Pläne.
So erlangen Sie regulatorische Sicherheit und vermeiden nicht nur Ärger in Audits, sondern auch die Aufwände für unnötige Überwachungstätigkeiten. Das hilft Ihnen zudem, die Sicherheit Ihrer Produkte zu gewährleisten.
Melden Sie sich einfach bei uns. Die Expertinnen und Experten des Johner Instituts freuen sich schon auf Sie!
6. Fazit, Empfehlung
Die Post-Market-Surveillance kann und muss einen wesentlichen Beitrag dazu leisten, die Sicherheit und Leistung von Medizinprodukten zu gewährleisten und sogar zu verbessern.
a) Bedeutung des PMS-Plan, Unterstützung durch die ISO 20416
Voraussetzungen für eine wirkungsvolle PMS sind ein produktspezifischer PMS-Plan und die Bereitstellung der notwendigen Ressourcen, um alle Informationsquellen zu erfassen und zu bewerten.
Die ISO 20416 hilft mit einem Vorschlag für der Kapitelstruktur eines solchen Plans. Hingegen lässt die Norm die Hersteller bei der Bewertung der Meldungen alleine. Im Anhang B gibt sie Tipps, wie die Hersteller die von der MDR geforderten Trends identifizieren und Schwellwerte festlegen können.
b) Das dicke Ende kommt noch. Zum Glück gibt es Lösungen
Die Datenmenge, die Hersteller bei der Post-Market Surveillance erfassen, aufbereiten und bewerten müssen, nimmt stetig zu. Die Frequenz für diese Überwachung wird von jährlich auf kontinuierlich sinken. Insbesondere PMS-Pläne softwarebasierte Produkte müssen das ensprechend berücksichtigen.
Hersteller werden diese sowohl quantitativ als auch qualitativ steigende Belastung nur durch Automatisierung bewältigen können.
Daher hat das Johner Institut bereits begonnen, diese Prozesse zu automatisieren. Es bietet Herstellern einen Service an, der in nennenswertem Umfang Zeit und Kosten für repetitive Arbeiten erspart. Lesen Sie hier, wie Sie das Johner Institut unterstützt:



Sehr geehrter Hr. Johner,
die Formulierung bei Fehler 3 könnte man so auffassen, dass es nicht sinnvoll sei, die FDA-Veröffentlichungen mit in das PMS einzubeziehen, wenn man nicht in den USA vertreibt.
Ich bin der Auffassung, dass es generell sinnvoll ist, die FDA-Datenbanken als Quelle für das PMS seiner Produkte heranzuziehen, solange es eine entsprechende Produktkategorie in den USA gibt, was in den allermeisten Fällen der Fall sein dürfte.
Damit hat man die Chance auf Schwachstellen seiner Produktkategorie aufmerksam zu werden, die natürlich in Bezug auf die Auswirkung auf das eigene gewählte Design bewertet werden müssen.
Beste Grüße, Uwe Zisterer
Ich stimme Ihnen zu, Herr Zisterer!
Danke für den wertvollen Hinweis!
Sehr geehrter Herr Johner,
Wie setzt ein Importuer einen PMS-Prozess um?
Welche Verpflichtungen und oder im welchen Umgfang muss es sein?
Muss er die PMS, PMCFund PSUR Prozesse haben?
Beste Grüße
Sara Schmidt
Sehr geehrte Frau Schmidt,
danke für Ihre spannende Frage!
Die PMS-/PMCF-/PSUR-Pflichten gelten für die Hersteller. Allerdings verpflichtet der Artikel 13 der MDR die Importeure dazu, bei diesen Post-Market-Aktivitäten mitzuwirken. Diese Anforderungen benennen v.a. die Absätze 5ff.
Beste Grüße, Christian Johner
Sehr geehrte Damen und Herren,
ist es ihrer Ansicht nach möglich mehrere Produkte / Produktgruppen / Produktfamilien in einen einzigen PMS-Plan unterzubringen? Die EU 2017/745 MDR sowie MDCG 2022-21 und MDCG 2025-10 verbieten dies nicht konkret. MDCG 2025-10: „Each device must be covered by a PMS plan. A plan can cover a single device or a group of devices. For example, devices with the same manufacturing process, design and intended purpose, or in the same device family can be covered by the same PMS plan. The plan should clearly state which devices are included within its scope.“ Laut dieser Guidance scheint ein Grouping allein auf einer nachvollziehbaren Begründung des Herstellers zu basieren, wobei die aufgeführten Beispiele wie „same manufacturing process“ lediglich als Orientierung dienen.
Vielen Dank vorab und VG
Manuel H.
Sehr geehrter Herr H.,
jedes Medizinprodukt muss durch einen PMS-Plan abgedeckt sein. Es ist jedoch nicht zwingend erforderlich, für jedes einzelne Gerät einen eigenen Plan zu erstellen. Ein Plan kann mehrere Geräte umfassen, sofern sie einem gemeinsamen Abdeckungsgrundsatz folgen, beispielsweise Zweckbestimmung, generische Produktgruppe oder Technologie. Nutzen Sie klare Gruppierungskriterien und definieren Sie Scope sowie Leading Device. Diese Vorgehensweise wird in MDCG 2025-10 und verwandten Leitlinien (MDCG 2022-21) bestätigt.
Viele Grüße, N. Jurrmann.