
Sowohl die FDA als auch die IEC 62304 kennen durch Dritte entwickelte Software. Sie sprechen von Off-the-Shelf Software (OTS) bzw. von Software of Unknown Provenance (SOUP).
Worin unterscheiden sich OTS und SOUP? Welche Gemeinsamkeiten haben beide? Welche gesetzlichen Anforderungen müssen sie erfüllen?
Dieser Artikel gibt Antworten.

1. SOUP und OTS: Definition und Abgrenzung
a) Definition SOUP
„Software-Komponente, die bereits entwickelt und allgemein verfügbar ist und die nicht entwickelt wurde, um in das Medizinprodukt integriert zu werden (auch bekannt als „Off-the-Shelf Software“), oder bereits fertig entwickelte Software, für die angemessene Aufzeichnungen zum Entwicklungs-PROZESS nicht verfügbar sind“
Quelle: IEC 62304
Die Definition des Begriffs SOUP hat die IEC 62304:2006+A1:2015 durch folgende Anmerkung ergänzt:
„A MEDICAL DEVICE SOFTWARE SYSTEM in itself cannot be claimed to be SOUP.“
IEC 62304:2006+A1:2015
Da scheinen die leidvollen Erfahrungen von Auditoren eingeflossen zu sein, die sich mit zweifelhaften Argumentationen ihrer Kunden auseinandersetzen mussten.
Die FDA verwendete den Begriff SOUP im vorherigen Guidance Dokument ebenfalls, das ist im neuen Guidance Dokument von 2023 aber entfallen und sorgt so (hoffentlich) für weniger Verwirrung:
„Some or all of the software contained in a Software Device may have been obtained by the submitter from a third party. […] Software for which adequate documentation may be difficult to obtain is referred to as Software of Unknown Pedigree or ‚SOUP‘.
Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices, 2005
b) Definition OTS
„a generally available software component, used by a medical device manufacturer for which the manufacturer can not claim complete software life cycle control (e.g., operating system, printer/display libraries).“
FDA Guidance Document „Content of Premarket Submissions for Device Software Functions“, 2023;
und identisch im FDA Guidance Document „Off-The-Shelf Software Use in Medical Devices“, 2023
Ein Sonderfall der OTS ist die COTS, die Commercial OTS-Software, also OTS eines kommerziellen Anbieters.
c) Gemeinsamkeiten und Unterschiede von SOUP und OTS
Überblick
Die Konzepte SOUP und OTS sind nicht ganz deckungsgleich:
- Es gibt SOUP, die auch OTS ist:
Alle allgemein verfügbaren Komponenten, die nicht dafür entwickelt wurden, Teil des zu entwickelnden Medizingerätes zu werden, und über dessen Entwicklungsprozess der Hersteller daher auch keine Kontrolle hat, sind sowohl SOUP als auch OTS. - Es gibt SOUP, die keine OTS ist:
Alle Software-Komponenten, die nicht allgemein verfügbar sind, etwa diejenigen, die der Medizinproduktehersteller zwar selbst, aber für einen anderen Zweck entwickelt hat und die er jetzt in das Medizingerät integrieren möchte, sind SOUP, aber keine OTS. - Es gibt OTS, die keine SOUP ist:
Ein typisches Beispiel ist jegliche Prozess-Software von anderen Herstellern, die unter den Begriff der OTS-Software fällt.
Erläuterungen
Zunächst fällt auf, dass sich die Definition der FDA nicht auf den Einsatz in Medizingeräten beschränkt („A generally available software component, used by a medical device manufacturer …“). Das Guidance-Dokument, in dem man die Definition findet, heißt Off-the-Shelf Software Use in Medical devices. Und wie der Titel schon sagt, beschäftigt sich das Dokument nur mit OTS-Software, die Teil eines Medizingerätes ist (bzw. als Laufzeitumgebung für die Software des Produkts vom Hersteller mit ausgeliefert wird).
Das verführt leicht dazu, anzunehmen, dass die Begriffe im Wesentlichen identisch sind. Das ist aber nicht der Fall! In dem betreffenden Dokument wird nur eine bestimmte Anwendung von OTS-Software behandelt.
Es gibt jedoch eine andere. Das wird beim intensiveren Lesen der General Principles on Software Validation klar. In Kapitel 6 geht es um „Validation of automated Process equipment and quality system software“. Dort heißt es: „Software tools are frequently used to design, build, and test the software that goes into an automated medical device. […] All of these applications are subject to the requirement of software validation […].“
Was hat das nun mit OTS-Software zu tun? Diesen Zusammenhang stellt das Kapitel 6.3 „Validation of the Off-the-Shelf Software“ her. All die oben erwähnte Software „to design, build and test the software“ fällt unter den Begriff OTS-Software, wenn sie von anderen Herstellern vertrieben wird. So gelangt man zum oben genannten Fazit.
2. Regulatorische Anforderungen
a) Regulatorische Anforderungen an SOUP
Anforderungen wie an jede andere Software-Komponente
Die IEC 62304 betrachtet SOUPs als Software-Komponenten. Die Hersteller müssen für SOUPs (wie für jede andere Komponente) die Anforderungen spezifizieren und deren Erfüllung testen. Zusätzlich zu „normalen“ Komponenten müssen die Hersteller auch die Voraussetzungen spezifizieren, von denen eine SOUP ausgehen kann, z. B. hinsichtlich der Ressourcen (RAM, CPU, …) oder des Betriebssystems.
Dokumentation der SOUP
Um die Forderungen der IEC 62304 umzusetzen, empfehle ich Ihnen, die SOUPs anhand folgender Parameter z. B. tabellarisch zu dokumentieren:
- Name der SOUP
- Version (SOUPs unterliegen genau wie alle anderen Artefakte der Versionskontrolle.)
- SOUP-Hersteller
- Anforderungen an SOUP (ggf. Verweis auf getrenntes Dokument oder Software-Architektur)
- Anforderungen durch SOUP, z. B. an Speicher oder Hardware
- Webseite von Bug Reports und Release Notes
Festlegung der Sicherheitsklasse bei SOUPs
Weil SOUPs Software-Komponenten sind, erben sie die Sicherheitsklasse des umgebenden Software-Systems bzw. der umgebenden Software-Komponente. Diese Sicherheitsklasse regelt aber nur den Umfang der zu erstellenden Dokumentation. Forderungen, wie sie die FDA mit dem Lieferanten-Audit kennt, stellt die IEC 62304 nicht.
Spezifikation und Überprüfung der Anforderungen
Genau wie bei jeder anderen Software-Komponente müssen die Hersteller die Anforderungen an eine SOUP spezifizieren und die Erfüllung später prüfen.
Manche Hersteller fühlen sich verpflichtet, die komplette SOUP zu spezifizieren und zu prüfen, was etwa bei einem Betriebssystem kaum gelingen wird.
Vielmehr müssen die Hersteller die Anforderungen an die SOUP festlegen, die sie im Rahmen des Software-Systems benötigen. Bei einem Betriebssystem wären das beispielsweise Anforderungen an den Zugriff auf Festplatte und Netzwerke oder die Fähigkeit der parallelen Verarbeitung. Genau diese Anforderungen gilt es dann zu überprüfen.
Festlegung und Überprüfung der Voraussetzungen
Die IEC 62304 verpflichtet die Hersteller zu überprüfen, ob die Voraussetzungen für den Einsatz einer SOUP gegeben sind. Beispiele für solche Voraussetzungen sind:
- Verfügbarer Hauptspeicher (RAM)
- Typ und Version des zugrundeliegenden Betriebssystems
- Typ und Version von „Run-time environments“ wie die Java Run Time und die .NET-Laufzeitumgebung
- Versionen von Bibliotheken, mit denen die SOUP interagieren soll
b) Regulatorische Anforderungen an OTS
Festlegung des Levels of Concern bei OTS-Software (gültig bis August 2023)
Die Hersteller müssen/mussten den Level of Concern gemäß den Vorgaben des Guidance-Dokuments zur OTS der FDA bestimmen. Dabei stellen sich viele Hersteller die Frage, ob sie den Level of Concern vor oder nach den Maßnahmen bestimmen müssen.
Diese Frage ist nicht nur entscheidend für die einzureichende Dokumentation, sondern hat auch Einfluss auf die Entscheidung, ob die OTS überhaupt eingesetzt werden darf.
Im Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices verlangt die FDA, dass der Hersteller den Level of Concern zuerst „prior to mitigation of hazards“ bestimmt. Bei einer OTS-Software mit einem „major“ Level of Concern hätte das zur Folge, dass der Hersteller ein Audit beim OTS-Hersteller durchführen muss, was in der Regel nicht möglich ist.
Die gute Nachricht bezüglich OTS-Software besteht darin, dass es einen Level of Concern sowohl vor als auch nach den risikominimierenden Maßnahmen gibt.
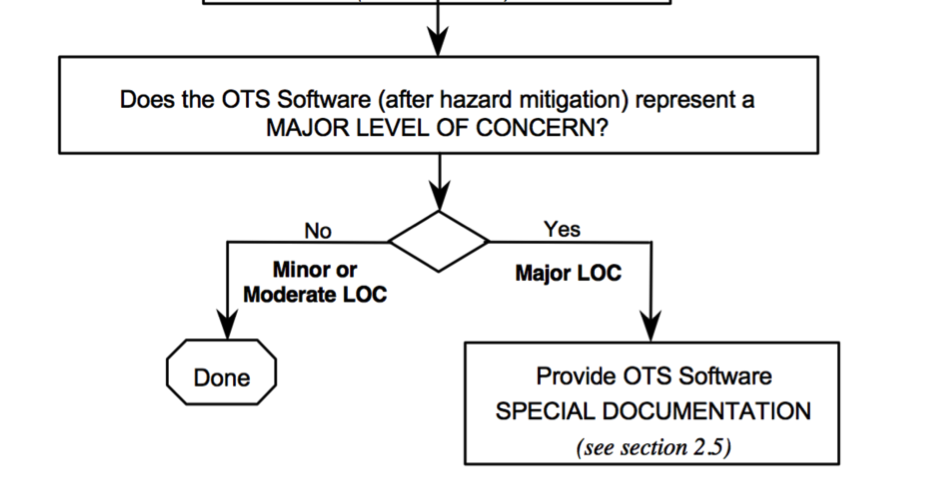
Nur wenn nach diesen Maßnahmen die OTS-Software noch einen „Major Level of Concern“ hat, wird die „Special Documentation“ notwendig, zu der das Audit beim OTS-Hersteller zählt.
Risikobasierter Ansatz
Obwohl die FDA den Begriff SOUP kennt, sind die Anforderungen eher unscharf:
„Therefore, we recommend that you explain the origin of the software and the circumstances surrounding the software documentation. Additionally, your Hazard Analysis should encompass the risks associated with the SOUP regarding missing or incomplete documentation or lack of documentation of prior testing. Nonetheless, the responsibility for adequate testing of the device and for providing appropriate documentation of software test plans and results remains with you.“
Man erkennt jedoch den risikobasierten Ansatz.
Festlegung des Documentation Levels (gültig ab August 2023)
Im August 2023 hat die FDA die neue Version des Guidance-Dokuments „Off-the-shelf Software Use in Medical Devices“ publiziert.
Diese Gegenüberstellung hilft, die Guidance-Dokumente der FDA zur OTS-Software zu vergleichen.
Eine wesentliche Änderung betrifft den bisherigen Level of Concern. Die FDA passt die guidance damit an die Änderungen aus der FDA Guidance „Content of Premarket Submissions for Device Software Functions“ (2023) an und unterscheidet nicht mehr drei, sondern zwei Klassen. Sie spricht auch nicht mehr vom Level of Concern, sondern vom Basic bzw. Enhanced „Documentation Level“. Diese Documentation Level bestimmen (wie auch bereits die Level of Concern) die zur Einreichung bei der FDA zu liefernde Software-Dokumentation. Anders als bei der IEC 62304, bedeutet das aber nicht, dass man prinzipiell auf die Aktivitäten in der Entwicklung verzichten werden darf, so heißt es z.B. beim „Basic Documentation Level“:
„During premarket review, FDA may request additional information, if needed, to evaluate the safety and effectiveness of the device.„
FDA guidance „Off-The-Shelf Software Use in Medical Devices“, 2023
Auch wird die OTS nicht mehr eigenständig klassifiziert sondern erbt das „Documentation Level“ des Medizinprodukts, wie das Beispiel aus Appendix B (3) verdeutlicht:
„[…] since the device has an Enhanced Documentation Level, the OTS Software Documentation will follow the Enhanced Documentation Level in this guidance“
FDA guidance „Off-The-Shelf Software Use in Medical Devices“, 2023
Abhängig vom documenation level erwartet die FDA die folgenden Dokumentationen:
| Dokumentation | Basic | Enhanced |
| Beschreibung der OTS-Software | X | X |
| Risikobewertung der OTSS | X | X |
| Software-Testing | X | X |
| Sicherstellung der „Development Methodologies and Continued Maintenance“ | – | X |
Die letzte Anforderung für das „Enhanced Documentation Level“ bedeutet letztlich, dass man den Entwicklungsprozess und die angewendeten Methoden kennt. Damit wäre diese Software zwar noch OTS-Software, aber im Sinne der IEC 62304 nicht zwingend auch eine SOUP, denn die Lebenszyklusaufzeichungen müssten vorhanden sein.
Da das Documentation level für das gesamte Medizinprodukt gilt („[…] the documentation level reflects the device as a whole„) und die OTS das documentation level des Medizinprodukts erbt, führt das dazu, dass sehr viele OTS in das enhanced documentation level fallen werden.
Man bedenke die typische Sicherheits-Architektur kritischer Systeme aus Steuereinheit und Überwachungseinheit. Gemäß FDA hätte das Produkt als Ganzes ein enhanced documentation level. Alle OTS hätten ein enhanced documentation level, wo man aus Sicht der IEC 62304 die Steuereinheit ggf. herunterklassifiziert hätte bzw. bei der früheren OTS guidance andere Level of Concern vergeben hätte.
Diese enhanced documentation level Anforderungen lassen sich für viele OTS-Software nicht erfüllen. Damit stehen die Hersteller von Medizinprodukten vor den gleichen Herausforderungen wie früher beim „Major Level of Concern“. Noch nicht ganz klar ist, ob mit der Änderung von „Audit“ auf „Review“ der Entwicklungsdokumentation der OTS, die FDA bewusst die Anforderung etwas weicher formuliert und damit auf die Hersteller zugeht.
Glücklicherweise bietet auch die neue Guidance die Möglichkeit über das Risikomanagement nach Maßnahmen den Einsatz der OTS zu rechtfertigen, auch wenn man nicht den Entwicklungsprozess und die angewendeten Methoden des OTS-Lieferanten auditieren kann.
c) Vergleich der Anforderungen
Die Definitionen der beiden Begriffe sich ähneln sehr, doch es gibt folgenden regulatorischen Unterschied:
- Bei den SOUP hat die Sicherheitsklasse keinen direkten Einfluss auf die Zulassung bzw. Verwendbarkeit. Insbesondere fordert sie nicht unter Umständen ein Lieferantenaudit.
- Bei der OTS ist dies der Fall.
In beiden Fällen sind Risiken durch SOUP bzw. OTS zu analysieren und zu bewerten.
Lesen Sie hier mehr zur Abgrenzung von Legacy Software, SOUP und OTS.
Weitere Unterschiede sind:
- Die FDA nutzt einen risikobasierten Ansatz, um die Forderungen an OTS zu skalieren. Diese Forderungen können bis zum Verbot der OTS gehen. Die IEC 62304 nennt keine konkreten Forderungen, insbesondere gibt es keine Abhängigkeit von der Sicherheitsklasse.
- Die FDA hat eine konkrete Liste an Eigenschaften, die für jede OTS-Komponente zu dokumentieren ist. Die IEC 62304 ist unspezifischer (siehe Kapitel 5.3.3 und 5.3.4).
- Die FDA erlaubt es nicht, eigene Software, für die keine adäquaten Aufzeichnungen vorhanden sind, als OTSS zu behandeln.
d) „Indirekte“ regulatorische Anforderungen an SOUP & OTS
Versionskontrolle
Wie alle anderen müssen auch die SOUPs der Versionskontrolle unterliegen. Im Gegensatz zum Quellcode würde man aber nicht etwa den kompilierten Code (z. B. die DLL- oder die JAR-Datei, das Executable) im Versionsverwaltungssystem einchecken.
Zusätzlich unterliegt die auszuliefernde Software, also das Ergebnis des Build-Prozesses, der Konfigurationskontrolle – und nicht nur die Artefakte, die als Input dessen dienen.
Post-Market Surveillance
Im Rahmen der Post-Market Surveillance und der nachgelagerten Phase des Risikomanagements müssen die Hersteller Informationen aktiv nachverfolgen, die Rückschlüsse auf Bugs oder auf durch SOUP verursachte Risiken erlauben. Zu den Informationsquellen zählen:
- Kundenrückmeldungen (Beschwerden, Fehlermeldungen, Beobachtungen)
- Meldungen der Hersteller, z. B. in Form von Release Notes oder Bug Reports
- Meldungen anderer Hersteller zu Problemen mit der SOUP, z. B. beim BfArM oder der MAUDE-Datenbank der FDA
- Hinweise zu Problemen mit der IT-Sicherheit, z. B. in der NIST-Datenbank
Wie bei jedem potenziellen Problem muss der Hersteller das Problem analysieren und entsprechende Maßnahmen ergreifen. Dazu gehören:
- Austausch der SOUP durch neue Version
- Ersatz durch eigene oder andere Komponente
- Entfernen und ggf. deaktivieren von Funktionalität
- Verbot, das Produkt weiterhin zu nutzen
- Hinweise zum Umgang mit dem Produkt bzw. dem Fehler
Wie Hersteller am besten Meldungen über Schwachstellen kommunizieren, beschreibt der Artikel zur ISO 29147. Ein weiterer Artikel beschreibt die regulatorischen Anforderungen an die IT-Sicherheit.
3. Praxistipps zum Umgang mit SOUP und OTS
a) Kriterien zur Auswahl von SOUPs und OTS
Die folgenden Kriterien helfen bei der Auswahl eines SOUP-Herstellers bzw. einer SOUP:
- Kann man den SOUP-Hersteller auditieren? => z. B. ein Lieferantenaudit des Entwicklungsprozesses und des Wartungsprozesses
- Bietet die SOUP die Funktionalität, die wir benötigen? Wie vollständig erfüllt sie unsere Anforderungen?
- Können wir der SOUP die von ihr notwendigen Laufzeitbedingungen wie Hardware, RAM, Betriebssystem usw. bereitstellen?
- Wie oft ist die SOUP im Einsatz? => Abschätzen der Fehlerwahrscheinlichkeit
- Veröffentlicht der Hersteller Buglisten? => Input für die Risikoanalyse
- Ist eine technische Dokumentation der SOUP verfügbar, etwa eine Architektur? Gibt es Testberichte?
- Sind gar Testergebnisse einsehbar, wie beispielsweise bei den Apache-Projekten?
- Ist möglicherweise sogar der Source-Code erhältlich?
- Gibt es Berichte aus vorangegangenen Audits?
- Ist ein QM-System beim SOUP-Hersteller vorhanden? => z. B. nach Normen ISO 9001, ISO 15504, ISO 13485 etc.
- Ist die SOUP in anderen Medizinprodukten bereits im Einsatz?
- Was kostet sie?
- Wie gut ist der Support? Wie schnell reagiert er? Zu welchen Zeiten ist er verfügbar?
Erstellen Sie eine Nutzerwertanalyse, mit der Sie die einzelnen Kriterien für Ihren konkreten Anwendungsfall gewichten.
b) Umgang mit SOUP bzw. OTS in Hardware-Bauteilen
Wie geht man mit Software um, die bereits in Bauteilen enthalten ist, die Hersteller von Dritten beziehen, z. B.
- Software in einem Modem- oder GSM-Chip
- Software in einem USB-Baustein
- Software in Display-Controllern
- Software in Ethernet-Chips und sonstigen Mikrocontrollern

Die Frage, ob Software in Hardware-Bauteilen als SOUP gilt, stellen v. a. Hersteller, die die Gesamtheit aller Software in einem Medizinprodukt als das Software-System definieren. Davon rät das Johner Institut ab.
Definieren Sie als Software-System die Gesamtheit der Software innerhalb eines Prozessoren- bzw. Speicherbereichs.
In einem Medizingerät kann es somit mehrere Software-Systeme geben, zumindest eines pro PESS (programmierbares elektrisches Subsystem). Weshalb Sie dies so aufteilen sollten, erfahren Sie in den Videotrainings im Auditgarant.
Das Johner Institut empfiehlt, diese Software aus folgenden Gründen nicht als SOUP zu behandeln:
- Für den Hersteller ist es nicht nachvollziehbar, welcher Teil der Funktionalität einer gekauften Hardware in Software implementiert ist. Damit kann der Hersteller auch keine Anforderungen an diese Software formulieren, wie von der IEC 62304 formuliert.
- Diese „embedded“ Software fällt nicht unter die Definition des Begriffs SOUP.
- Ein Auditor wird Sie wahrscheinlich nicht auf diese Software ansprechen, sondern auf das ganze Bauteil. Machen Sie die Sache nicht unnötig kompliziert.
- Die Risiken, dass diese Software Fehler enthält, können und sollten Sie nicht auf Ebene dieser Software diskutieren und beherrschen, sondern auf Ebene des Bausteins bzw. der System-Architektur.
c) Umgang mit Linkern
Wie gehen Auditoren mit den Linkern um? Hierzu gibt es leider unterschiedliche Antworten:
Ein Auditor vertrat die Meinung, ein Linker würde Software-Teile für den Kompiliervorgang in das Programm rein-linken. Insofern müsste dieser in die Software gelinkte Teil der Software als SOUP betrachtet werden.
Diese Meinung teilen andere Auditoren und das Johner Institut nicht: Ein Linker selbst ist keine SOUP. Vielmehr ist er wie der Compiler eine Prozess-Software. Man könnte eher diskutieren, ob der Linker deshalb zu validieren ist.
Eine SOUP ist, nochmals zu Erinnerung,
„eine Software-Komponente, die bereits entwickelt wurde, und allgemein verfügbar ist, und die nicht entwickelt wurde, um in das Medizinprodukt integriert zu werden, oder bereits fertig entwickelte Software, für die angemessene Aufzeichnungen zum Entwicklungsprozess nicht verfügbar sind.”
IEC 62304
Ein Linker erstellt aber gerade keine Komponenten, außerdem war das, was der Linker erstellt, keinesfalls vorher vorhanden und allgemein verfügbar.
d) Vorsicht mit der Behauptung, eine SOUP sei „zertifiziert“
Oft stellt sich die Frage: Wäre es nicht besser, eine zertifizierte SOUP zu nehmen? Es gibt ja Hersteller von Betriebssystemen, die so etwas anbieten (Beispiel).
Es ist immer das Ziel, einen Hersteller auszuwählen, der Software professionell entwickelt. Grundsätzlich braucht aber kein SOUP-Hersteller die Regulierungen zu Medizinprodukten (z. B. die MDR oder IEC 62304) zu beachten, da er kein Inverkehrbringer ist.
Dass es IEC-62304-zertifizierte Produkte geben soll, erscheint fragwürdig, denn die IEC 62304 ist eine Prozessnorm und keine Produktnorm. Das heißt: Man kann sich nur zertifizieren lassen, dass man konform mit den (geringen) Anforderungen dieser Norm entwickelt. Über die Güte des Produkts sagt das aber nichts aus.
Diese Form der Werbung oder Kommunikation ist zumindest missverständlich.
4. Fazit und Zusammenfassung
In Medizinprodukten wächst kontinuierlich der Anteil der Software, die die Hersteller nicht mehr selbst schreiben, sondern etwa in Form von Bibliotheken übernehmen. Damit wird es immer wichtiger, dass Hersteller die Konformität ihrer Produkte auch dann nachweisen können, wenn sie die Software Dritter verwenden. Das Log4J-Desaster hat dieses Problem sehr ins Bewusstsein gerückt.
Die IEC 62304 und die FDA führen dazu die nicht ganz deckungsgleichen Konzepte der SOUP und der OTS-Software ein. Hersteller müssen diese Konzepte und die damit verbundenen regulatorischen Anforderungen kennen und beachten, um Schwierigkeiten bei Zulassungen und Audits sowie v. a. Probleme mit der Sicherheit ihrer Produkte zu vermeiden.
Das Johner Institut unterstützt Medizinproduktehersteller bei der normen- und gesetzeskonformen Entwicklung von Software. Wir stellen sicher, dass Sie schnell und verlässlich die „Zulassungen“ Ihrer Produkte erreichen, unnötige Aufwände vermeiden und sichere Medizinprodukte entwickeln.
Melden Sie sich, wenn Sie Unterstützung wünschen, sei es beim Erstellen oder Prüfen der Software-Akte oder für Pentests der Produkte.
Änderungshistorie
- 2023-11-15: kleinere Korrekturen, Konkretisierung für bessere Verständlichkeit
- 2023-10-30: Im Kapitel 2.b) Ergänzungen zum neuen 2023 Guidance-Dokument hinzugefügt
- 2023-09-05: Im Kapitel 2.b) Unterkapitel zum neuen Guidance-Document der FDA ergänzt
- 2023-03-13: Artikel völlig überarbeitet und aktualisiert und mit dem bisherigen Schlagwortbeitrag zusammengeführt
- 2017-09-13: Erste Version des Artikels



Ich habe den Verdacht, dass die regulatorischen Vorgaben bisweilen die Sicht auf das Wesentliche verstellen.
Unter der hoffentlich allgemein anerkannten Prämisse das d. Medizin-SW so sicher wie möglich sein sollte, ist der History-Track der SW wohl d. aussagekräftigste Faktor. Wenn ich einen langen und ereignisarmen History-Track habe (vielleicht sogar mit Anwendung im Med.bereich) sind meine Risiken auch bei näherer Betrachtung wohl eher klein. UNIX/ Linux oder Legacy-Software sind sicher gute Beispiele dafür.
Wenn ich einen schlechten History-Track habe sollte wohl schon von Anfang an eine bessere Alternative suchen.
Wenn ich hingegen keinen oder nur einen sehr kurzen History Track habe, DANN wird die Prozessbeherrschung à la 62304 ein Thema (neben der Validierung). Das würde auch wieder das Thema der verschiedenen WIndows-Versionen beleuchten (von XP bis 8).
Ist so eine Argumentationslinie bei Pre-Market-Approvals und Audits belastbar?
Lieber Herr Beisswenger,
eine solche Argumentation ist aus Sicht der IEC 62304 bedingt möglich, aus der der ISO 14971 eher. Der „History Track“ wie Sie es nennen, gibt Anhaltspunkte zur Wahrscheinlichkeit von Fehlern. Diese möchte die 62304 nicht wirklich diskutiert haben.
Für mich und auch einen Auditor sind diese Überlegungen aber absolut zielführend und unter einem ISO 13485 Blick sogar fast zwingend.
Viele Grüße
Christian Johner
Hallo Herr Johner,
Wie ist es mit „bereits fertig entwickelte Software, für die angemessene Aufzeichnungen zum Entwicklungs-PROZESS _nicht_ verfügbar sind“, die aber _nicht_ allgemein erhältlich ist?
Diese fällt zwar unter die IEC Definition von SOUP, aber nicht unter die FDA Definition von OTSS. Soweit, so gut. Aber es kann der FDA doch nicht recht sein, diese Komponente ohne die für OTSS gültigen Maßnahmen einfach zu integrieren? Muss man in diesem Fall nicht trotzdem die „Guidance […] on Off-The-Shelf Software Use in Medical Devices anwenden (wenn man im USA Markt verkaufen möchte)?
MfG, C. Beck
Absolut, Herr Beck. Es ist genau wie Sie sagen: OTS ist hier nicht gleich SOUP.
Es wäre der FDA nicht Recht, die Alt-Sünden einfach einzubauen. Hier hilft nur der Weg, diese Sünden zu „heilen“, indem man entsprechende Aufzeichnungen aus dem Software-Lebenszyklus nachholt oder gemäß „risk based approach“ diese als vertretbar bewertet. Diese Diskussion muss aber geführt sein.
Hallo Herr Johner,
macht es in der Betrachtung einen Unterschied, ob ich eine OTSS als binäre, nicht veränderbare Komponente einbaue oder den ggf. verfügbaren Source Code benutze und die Komponente selber im Zuge meines Medizinproduktes baue?
Für den Fall, dass es möglich ist, die Komponenten selber zu bauen: Wie verhält es sich, wenn ich frei verfügbare Software, wie z.B. das DCMTK nutze und Änderungen am Source Code vornehmen um z.B. einen mir bekannt gewordenen Bug zu fixen oder Funktionalität anzupassen oder zu erweitern? Reicht es aus, wenn ich dann nur die angepasste Komponente gemäß meines QM Systems einer Qualitätssicherung unterziehe oder muss sich diese dann auf die gesamte OTSS erstrecken?
Viele Grüße
Anna Henke
Sehr geehrte Frau Henke,
wenn Sie über OTSS sprechen, sind Sie im FDA-Kontext.
Falls Sie über den Source-Code verfügen, haben Sie die Möglichkeit, den Code der eigenen Qualitätssicherung zu unterwerfen und damit den Code gar nicht mehr als OTSS zu betrachten. Das geht natürlich bei rein binären Artefakten nicht.
Wenn Sie eine OTSS-Komponente ändern, weil Sie z.B. einen Fehler beheben oder Funktionalität hinzufügen, wäre das nicht mehr im Sinn der FDA. Die Idee besteht gerade darin, dass man eine gewisse Güte erwartet, weil die Software allgemein verfügbar und damit in gewisser Weise bewährt ist.
Bei SOUPs wäre das anders. Aber danach haben Sie ja nicht gefragt.
Viele Grüße
Christian Johner
Sehr geehrter Herr Johner,
mich würde Ihre Einschätzung zu folgendem Sonderfall sehr interessieren:
Es handelt sich um eine Software die mithilfe eines Algorithmus anhand von US-Bildern exakt den Körperfettanteil bestimmen kann. Die SW ist erfolgreich im Einsatz, es existieren auch wissenschaftliche Studien dazu. Die SW soll jetzt aber auch als eine SaMD(Risikoklasse A) zum Einsatz kommen. Allerdings wurde diese Software mehr im Zuge eines „wissenschaftlichen Projektes“ entwickelt. Es existieren demnach keine dokumentierten Tests, kein Entwicklungsplan oder generell Software-Lebenszyklus Prozesse nach EN 62304 und auch kein QM-System nach ISO 13485. Lediglich die Bestätigung anhand von zahlreichen Praxisanwendungen, dass sie die erwarteten Ergebnisse liefert.
Meiner Meinung nach wäre dann das ganze Software-System eine SOUP, was gemäß EN 62304 ja nicht möglich ist.
Wie würde hier die Strategie für eine erfolgreiche Zulassung aussehen, falls dies überhaupt möglich ist?
Die für RK A geforderten Software-Systemtests können ja nachgeholt und dokumentiert werden. Die Entwicklung innerhalb eines QM-Systems und nach einem definierten Entwicklungsplan aber, ist nicht mehr möglich.
Vielen Dank für Ihre wertvollen Beiträge.
Liebe Grüße
Sehr geehrter Herr Boser,
danke für die spannende Frage!
Wenn Sie das Produkt unter der MDR in den Verkehr bringen wollen (die MDD läuft ja bald ab), dann fällt das wahrscheinlich gemäß Regel 11 in die Klasse IIa. Damit wäre sogar ein zertifiziertes QM-System notwendig.
Aber auch bei einem Klasse-I-Produkt müssen Sie die Anforderungen nach Lebenszyklusprozesse einhalten, die bereits seit der MDD gelten.
D.h. Sie müssten u.a. konform u.a. mit der IEC 62304 nachdokumentieren. Dazu zählt nicht nur die Dokumentation fürs Produkt, sondern auch für das QMS. Denn die IEC 62304 und die MDR fordern bei jeder Klasse ein QM-System.
Unter der MDR müsste man erwägen, ob eine Dokumentation nach Sicherheitsklasse A überhaupt die Anforderungen an die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt, da z.B. keine SW-Architektur vorliegt.
Kurze Version der Antwort: Sieht nach „risikobasierter Nachdokumentation“ aus.
Danke auch für Ihr wertschätzendes Feedback, über das ich mich freue!
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner
Ich beziehe mich auf Ihre Antwort auf die Frage von Frau Henke vom Juni 2017.
Wie sieht die Situation denn für SOUP aus? Was ist, wenn die Software sowohl SOUP als auch OTS ist?
Ich kann die Argumentation bei OTS nicht recht nachvollziehen. Wenn ich OTS wähle, damit sie Software eine hohe Qualität und wenige Fehler hat, und ich Fehler darin korrigiere und den veränderten Code teste, dann ist doch die Qualität noch höher als vorher, was im Sinne der FDA sein müsste?
Beste Grüsse
Andreas Schätti
Sehr geehrter Herr Schätti,
ich bin nicht ganz sicher, ob ich Ihre Frage wirklich verstehe. Eine SW kann durchaus SOUP und OTS sein. Das ist bei uns Herstellern sogar der Normalfall.
Die Annahme bei OTS besteht darin, dass die SW durch die Tatsache, dass sie allgemein verfügbar ist, eine gewisse (Mindest-)Qualität habe.
Wenn Sie diese Software ändern, kollabiert diese Vermutung, denn man kann ja gerade keine Annahme mehr bezüglich der Prozesse der ändernden Organisation treffen. D.h. Ihr Schluss, dass eine Software nach einer Änderung eine höhere Qualität habe, wird für Ihre Organisation stimmen. Ich halte diese aber in dieser Allgemeingültigkeit für gerade nicht zutreffend.
Um diese Vermutung aufrecht zu halten, würde man eine qualitätsgesicherte Entwicklung benötigen. Hätte ein Hersteller diese, könnte er je nach Art der (OTS-) Komponente auch den konsequenten Schritt gehen und die Software zu „internalisieren“, d.h. die fehlende Evidenz einer Konformität zu heilen z.B. durch eine entsprechende Dokumentation. Dazu zähen beispielsweise die von Ihnen erwähnte Dokumentation der Tests.
Beste Grüße, Christian Johner
Ich habe Anmerkungen bzw. Anregungen zu drei Textstellen in diesem hilfreichen Artikel:
„Die FDA verwendet den Begriff SOUP ebenfalls, und zwar im Guidance-Dokument.“: Das Zitat ist aus dem alten Guidance von 2005. Im neuen Guidance von 2023 ist diese Definition von SOUP erfreulicherweise entfernt worden.
„Und wie der Titel schon sagt, beschäftigt sich das Dokument nur mit OTS-Software, die Teil eines Medizingerätes ist.“: Das ist etwas irreführend, denn das OTS Software Guidance Dokument der FDA bezieht sich auch auf OTS-Software, die nicht Bestandteil des Medizingerätes ist, die es aber in der Anwendungsumgebung benötigt und vom Betreiber bereitgestellt wird (z.B. das Betriebssystem oder der Browser auf dem Computer, wo eine SaMD installiert wird). Das wäre auch noch ein gutes Beispiel für eine OTS-Software, die keine SOUP im Sinne der IEC 62304 ist (sondern Supported bzw. Required Software im Sinne der IEC 81001-5-1).
„Abhängig vom documenation level erwartet die FDA die folgenden Dokumentationen“: Das ist so weit richtig, aber es wäre gut, hinzuzufügen, dass die FDA beim Basic Level die letztgenannte Dokumentation zwar nicht in der Einreichung haben möchte, wohl aber soll der Hersteller es im DHF ablegen, und die FDA behält sich vor, es nachzufordern. Damit ist diese Unterscheidung bei der Entwicklung irrelevant. Beim Basic Level fällt es lediglich leichter, über das Risikomanagement zu argumentieren, dass trotz der meist nicht verfügbaren Details zur Entwicklungsmethodik des OTS-Software-Herstellers die Qualität der OTS-Software als gut genug angenommen werden kann.
Sehr geehrter Herr Pesara,
vielen Dank für die ergänzenden Hinweise!
Viele Grüße
Daniel Reinsch
Sehr geehrter Herr Johner,
vielen Dank für diesen sehr hilfreichen Artikel zum Thema SOUP / OTS. Ich habe eine spezielle Frage in diesem Zusammenhang.
Wenn wir innerhalb des Unternehmens eine eigene Abteilung haben, die „Off-the-Shelf Software“ entwickelt für Nicht-Medizinprodukte des Unternehmens und Medizinprodukte des Unternehmens (deren intended use sie aber nicht kennt und die teilweise noch gar nicht existieren), muss dann diese Abteilung zwangsläufig in einem QM System arbeiten oder ist das in diesem Fall nicht nötig? Die genannte Abteilung folgt dem ISO 62304 Gedanken wie auch der FDA Guidance zum Thema OTS Software.
Danke und viele Grüße
Frederik Panse
Sehr geehrter Herr Panse,
die Intention der Norm und der Guidance ist es sicher nicht, dass medizinische Software-Entwicklung ausgelagert wird, um sie dann als SOUP/OTS wieder reinzuholen. Genau das könnte einem hier unterstellt werden.
Prinzipiell sieht die Definition von SOUP vor, dass die Software-Komponente möglicherweise sogar im eigenen Unternehmen entwickelt wurde, aber keine geeigneten Aufzeichnungen vorhanden sind. Es geht aber immer nur um Komponenten der Software. Die Software als Ganzes darf nicht als SOUP betrachtet werden.
Die Definition von OTS sieht vor, dass es eine allgemein verfügbare Software oder Software-Komponente ist, über dessen Entwicklungsprozess man keine Kontrolle hat. Das stimmt bei einer Abteilung innerhalb eines Unternehmens eher nicht. Dort kann man den Entwicklungsprozess kontrollieren.
Sofern man also zeigen kann, dass die SOUP vor Projektstart des Medizinprodukts entwickelt wurde und es nur eine Komponente der Medizinprodukte-Software ist, kann man diese als SOUP integrieren.
Bei der FDA gehe ich davon aus, dass die Inspektoren das merkwürdig finden und die Betrachtung als OTS hier als nicht anwendbar sehen.
Nicht ganz klar ist mir die Aussage „Die genannte Abteilung folgt dem ISO 62304 Gedanken wie auch der FDA Guidance zum Thema OTS Software.“ Wenn die Abteilung nach IEC 62304 entwickelt, dann muss/kann man die SW-Komponenten ja gar nicht als SOUP/OTS behandeln, denn dann sind ja Aufzeichnungen zum Medizinprodukte-Software-Lebenszyklus vorhanden.
Viele Grüße
Daniel Reinsch
Sehr geehrter Herr Johner, sehr geehrter Herr Reinsch,
ich habe eine kurze Frage zum Punkt 3b: Sie empfehlen, Software in separaten Hardware-Bauteilen nicht als SOUP zu deklarieren. Im Abschnitt vor dieser Empfehlung sprechen Sie von mehreren Software Systemen.
Was ist denn Ihre Empfehlung für solche Software? Nach meinem Verständnis wäre es noch schwieriger, sie als separates Software System zu behandeln denn als SOUP. Kann/soll solche „embedded“ Software in der Dokumentation ignoriert werden mit dem Verweis auf nicht-veränderbare embedded Software innerhalb einer abgeschlossenen Hardware?
Vielen Dank und mit freundlichen Grüßen
– Oliver Rath
Sehr geehrter Herr Rath,
vielen herzlichen Dank für Ihre spannende Nachfrage.
Der Bezug auf die Software-Systeme rührt daher, dass das Medizinprodukt in seiner Gesamtheit in mehrere Software-Systeme unterteilt werden kann. Diese Trennung erfolgt typischerweise anhand von Prozessor- oder Speicherbereichsgrenzen. Dadurch können in einem Medizinprodukt Software-Systeme unterschiedlicher Sicherheitsklassen existieren. Innerhalb dieser Software-Systeme können Sie mit diversen hardwarenahen Firm- bzw. Software(s) konfrontiert sein. Unsere Empfehlung ist, diese hardwarenahe Firmware nicht als SOUP zu deklarieren, sondern die korrekte Funktionalität der „embedded Software“ implizit über die Verifizierung des Hardware-Bauteils zu testen. Dies hat unter anderem den Hintergrund, dass etwaige Tests der Firmware oder embedded Software zumeist nur in Kombination mit dem Bauteil stattfinden können. Ignoriert wird die embedded Software dadurch nicht.
Herzliche Grüße
Alexander Wassel
Sehr geehrte Frau Delor,
danke für Ihre Frage. Ich gehe im Folgenden davon aus, dass Sie mit BSP ein Board Support Package meinen, also Software wie Treiber, Bootloader usw.
Die kurze Antwort auf ihre Frage, ob Sie das BSP als SOUP behandeln dürfen, ist ja. Daher sind Ihre Maßnahmen wie die Konfigurationskontrolle und das Monitoring von Vulnerabilities absolut zielführend. Beachten Sie aber, dass die IEC 62304 auch verlangt, dass Sie die Anforderungen an die SOUP bestimmen und die Erfüllung der Anforderungen auch prüfen. Die Anforderung, dass Sie die Voraussetzungen z.B. an die Hardware sicherstellen müssen, dürften implizit erfüllt sein, wenn das BSP vom Hersteller mitgeliefert wird.
Ob man ein Qt dem BSP zuschlagen sollte, weiß ich nicht. Aber wahrscheinlich meinten Sie das auch gar nicht.
Beste Grüße, Christian Johner
Sehr geehrte Frau Delor,
Sie haben die Freiheit, selbst festzulegen, was Sie als SOUP definieren. Eine SOUP kann sowohl ausschließlich eigener Code sein, ausschließlich Code von Dritten als auch jede Mischform.
Sie haben auch die Freiheit, über die Granularität der SOUP-Komponenten zu entscheiden. Es müssen nur Komponenten im Sinn von identifizierbaren Teilen der Software sein.
Natürlich wollen Sie diese Freiheiten weise nutzen, um dabei berücksichtigen:
– Risiken
– Wartbarkeit
– Lebenszyklus der Komponenten
– Wiederverwendung der Komponenten
– Testbarkeit
– Aufwand zum Formulieren der Anforderungen und zum Identifizieren der Komponenten
Das können wir uns gerne zusammensetzen, um die besten Entscheidungen für Sie zu treffen.
Herzliche Grüße
Christian Johner