
Die korrekte und präzise Formulierung der Zweckbestimmung von Medizinprodukten und In-Vitro-Diagnostika (IVD) ist entscheidend für deren erfolgreiche Entwicklung und Zulassung. Doch bereits die Begriffsdefinitionen und der Zusammenhang von Zweckbestimmung und bestimmungsgemäßem Gebrauch erschweren die notwendige Klarheit und Präzision.
Dieser Artikel verrät, wie Sie eine Zweckbestimmung formulieren, um Schwierigkeiten bei Audits und Zulassungen zu vermeiden.
1. Relevanz der Zweckbestimmung
Mit der Zweckbestimmung drückt der Hersteller aus, zu welchem (medizinischen) Zweck sein Produkt angewendet werden soll. Diese Festlegung ist die Voraussetzung für viele Aktivitäten bei der Entwicklung und der Zulassung:
- Qualifikation des Produkts, d. h. die Entscheidung, ob es ein Medizinprodukt bzw. ein IVD ist
- Klassifizierung des Medizinprodukts/IVDs gemäß MDR oder IVDR
- Entscheidung, welche Normen bei der Produktentwicklung berücksichtigt werden müssen
- Spezifikation des Produkts und seiner Leistungsparameter
- Abgrenzung des normalen und abnormalen Gebrauchs (Ableitung der Use Specifications)
- Produktspezifisches Risikomanagement (Risikomanagementplan inkl. Herleitung der Risiko-Akzeptanz-Matrix sowie Risikoanalyse)
- Durchführen der klinischen Bewertung bzw. Leistungsbewertung, die nachweisen muss, dass der Zweck und der medizinische Nutzen erfüllt wurde
- Schreiben des Post-Market-Surveillance-Plans, der festlegt, wie das Erreichen der Zweckbestimmung im Feld überwacht wird.
Es ist die Aufgabe und die Freiheit des Herstellers, die Zweckbestimmung festzulegen.
2. Definitionen des Begriffs „Zweckbestimmung“
Bedauerlicherweise gibt es keine einheitliche Definition des Begriffs „Zweckbestimmung“. Zudem setzen viele Definitionen die „Zweckbestimmung“ (Intended Purpose) und den „bestimmungsgemäßen Gebrauch“ (Intended Use) gleich.
Dieses Kapitel stellt die relevanten Definitionen vor. Das nächste Kapitel grenzt die Zweckbestimmung und den bestimmungsgemäßen Gebrauch voneinander ab.
a) Definition gemäß MDR bzw. IVDR
’Zweckbestimmung‘ bezeichnet die Verwendung, für die ein Produkt entsprechend den Angaben des Herstellers auf der Kennzeichnung, in der Gebrauchsanweisung oder dem Werbe- oder Verkaufsmaterial bzw. den Werbe- oder Verkaufsangaben und seinen Angaben bei der klinischen Bewertung bestimmt ist;“ (MDR Artikel 2 (12))
‘Zweckbestimmung‘ bezeichnet die Verwendung, für die ein Produkt entsprechend den Angaben des Herstellers auf der Kennzeichnung, in der Gebrauchsanweisung oder dem Werbe- oder Verkaufsmaterial bzw. den Werbe- oder Verkaufsangaben oder seinen Angaben bei der Leistungsbewertung bestimmt ist;“ (IVDR Artikel 2 (12))
Auch die MDR/IVDR übersetzt den Begriff „Zweckbestimmung“ im Englischen als Intended Purpose.
Neben dem Begriff „Zweckbestimmung“ arbeitet die MDR/IVDR auch mit dem Begriff „bestimmungsgemäße Verwendung“ („Intended Use“). Beide Verordnungen definieren diesen Begriff aber nicht. Er entspricht dem „bestimmungsgemäßen Gebrauch“.
b) Definition in der IEC 60601-1
Die IEC 60601-1 definiert den Begriff „Zweckbestimmung“ als die „Anwendung eines Produkts, Prozesses oder einer Dienstleistung nach den vom Hersteller gelieferten Spezifikationen, Anweisungen und Informationen“ (IEC 60601-1:2005, 3.44).
c) Definition in der ISO 14971
Die ISO 14971 hat in ihrer dritten Ausgabe (ISO 14971:2019) die Definition des Begriffs überarbeitet. Sie setzt allerdings Intended Use und Intended Purpose gleich. Doch deutet die Anmerkung darauf hin, dass eher der medizinische Zweck gemeint ist.
„use for which a product, process or service is intended according to the specifications, instructions and information provided by the manufacturer
Note 1 to entry: The intended medical indication, patient population, part of the body or type of tissue interacted with, user profile, use environment, and operating principle are typical elements of the intended use“ (ISO 14971:2019, 3.6)
d) Definition der FDA
Die FDA definiert den Begriff „Intended Use“ im 21 CFR Part 801.4 wie folgt:
„The words intended uses […] refer to the objective intent of the persons legally responsible for the labeling of an article (or their representatives). The intent may be shown by such persons‘ expressions, the design or composition of the article, or by the circumstances surrounding the distribution of the article. This objective intent may, for example, be shown by labeling claims, advertising matter, or oral or written statements by such persons or their representatives. Objective intent may be shown, for example, by circumstances in which the article is, with the knowledge of such persons or their representatives, offered or used for a purpose for which it is neither labeled nor advertised; provided, however, that a firm would not be regarded as intending an unapproved new use for a device approved, cleared, granted marketing authorization, or exempted from premarket notification based solely on that firm´s knowledge that such device was being prescribed or used by health care providers for such use […]” (21 CFR Part 801.4).

Schreiben Sie eine Zweckbestimmung, die in Ihrem Unternehmen als Musterbeispiel dienen wird!
Erstellen Sie mit unserem E-Learning-Kurs Ihre perfekte Zweckbestimmung. Nach Durchführen des Kurses sind Sie Expert:in für das Schreiben einer präzisen und schlanken Zweckbestimmung und halten ein freigegebenes und konformes Dokument für Ihr Produkt in der Hand.
3. Zweckbestimmung und bestimmungsgemäßer Gebrauch
Der Begriff Zweckbestimmung im weiteren Sinn umfasst bei Medizinprodukten und IVD mehrere Aspekte:
- Der eigentliche medizinische Zweck („medizinische Zweckbestimmung“ oder Zweckbestimmung im engeren Sinn) gibt an, welche Krankheit oder welche Verletzung diagnostiziert, therapiert oder überwacht werden soll.
- Die medizinische Anwendung bestimmt den medizinischen Kontext und damit die Bestimmungen, welche Anwender in welchem Nutzungskontext das Produkt für welche Patienten anwenden.
Der (sonstige) bestimmungsgemäße Gebrauch legt fest, wie insgesamt bzw. zusätzlich mit dem Produkt umgegangen werden soll. Das umfasst die Lagerung, den Transport, das Update oder die Reinigung des Produkts.
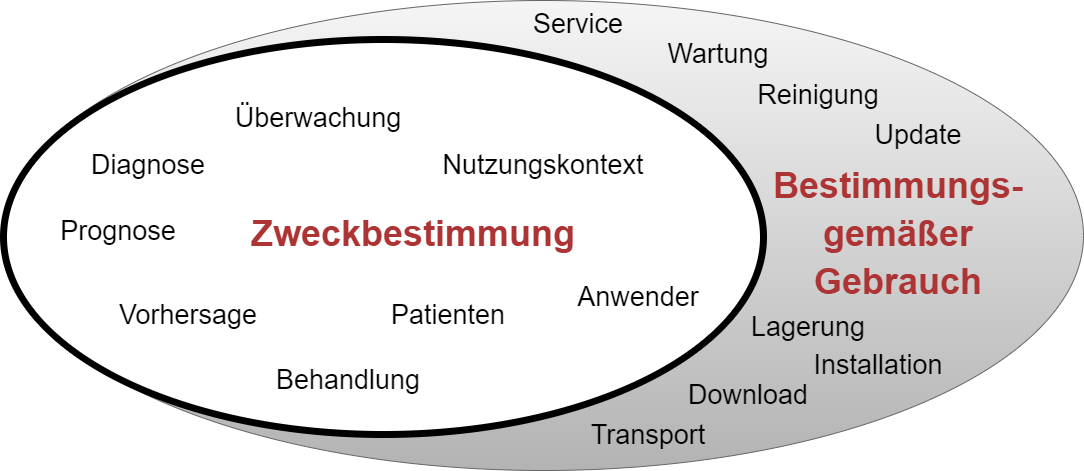
Die MDR bzw. die IVDR spricht statt vom „bestimmungsgemäßen Gebrauch“ von der bestimmungsgemäßen Verwendung. Beide Begriffe sind synonym zu verstehen.
4. Regulatorische Anforderungen
Es gibt mehrere Regularien, die Sie in diesem Kontext beachten sollten:
- Die MDR und die IVDR fordern die Festlegung der Zweckbestimmung als Teil der technischen Dokumentation (siehe Kapitel 5).
- Die ISO 13485 fordert in 7.3.7: „Eine Entwicklungsvalidierung muss […] sicherstellen, dass das resultierende Produkt in der Lage ist, die Anforderungen für die festgelegte Anwendung oder den bestimmungsgemäßen Gebrauch zu erfüllen.“ Das setzt voraus, dass diese bekannt sind.
- Die ISO 14971, die für das Risikomanagement harmonisierte Norm, setzt in Kapitel 5.2 die Zweckbestimmung für die weitere Risikoanalyse voraus.
- Die IEC 62366-1 verlangt in Kapitel 5.1 die Erstellung einer „Use Specification“, dessen Zusammenfassung von manchen Behörden als Zweckbestimmung bezeichnet wird.
- Die IEC 60601-1 setzt eine genaue Spezifikation der Anwendungsumgebung voraus, die beispielsweise physikalische Parameter wie Temperatur, Höhe/Luftdruck, Helligkeit ebenso umfasst wie die Versorgungsspannung(en) und den Verschmutzungsgrad.
5. Inhalte einer Zweckbestimmung
5.1 Zweckbestimmung eines Medizinprodukts
Adressieren Sie anlehnend an MDR Anhang I, Kapitel III, 23.4 b) sowie Anhang II 1.1 a) in Ihrer Zweckbestimmung folgende Aspekte:
- Medizinischer Zweck: Welche Krankheit oder welche Verletzung soll diagnostiziert, therapiert, überwacht, gelindert oder vorhergesagt werden?
- Medizinische Indikation und Kontraindikation
- Vorgesehene Patientengruppe(n)
- Vorgesehenes Körperteil
- Vorgesehene Anwender
- Vorgesehene Gebrauchsumgebung bzw. Nutzungsumgebung
- Physikalische Umgebung, z. B. Helligkeit, Lärm, Verschmutzung
- Soziale Umgebung, z. B. Stress, Schichtbetrieb
- Technische Umgebung, z. B. Werkzeuge, Software
- Klinische Umgebung, z. B. steril, Tragen von Schutzausrüstung
- Funktionsweise
5.2 Zweckbestimmung eines IVD
Die Inhalte der Zweckbestimmung eines IVD sind in Anhang I, Kapitel III, 20.4.1 c) sowie im Anhang II 1.1 c) der IVDR klar vorgegeben. Die Zweckbestimmung eines IVD umfasst:
- was nachgewiesen und/oder gemessen wird;
- seine Funktion (z. B. Screening, Überwachung, Diagnose oder Diagnosehilfe, Prognose, Vorhersage, therapiebegleitendes Diagnostikum);
- Hinweis: Bei der Festlegung der Funktion eines IVD helfen die Definitionen im Guidance Dokument GHTF/SG5/N8:2012 weiter.
- spezifische Informationen, die in folgenden Zusammenhängen bereitgestellt werden sollen:
- physiologischer oder pathologischer Zustand;
- kongenitale körperliche oder geistige Beeinträchtigungen;
- Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit;
- Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern;
- Voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf;
- Festlegung oder Überwachung therapeutischer Maßnahmen;
- ob es automatisch ist oder nicht;
- ob es qualitativ, semiquantitativ oder quantitativ ist;
- die Art der erforderlichen Probe(n);
- ggf. die zu testende Zielpopulation;
- der vorgesehene Anwender und
- bei therapiebegleitenden Diagnostika den internationalen Freinamen (INN) des dazugehörigen Arzneimittels.
Üblicherweise subsumieren Medizinprodukte- und In-Vitro-Diagnostika-Hersteller im Dokument zur Zweckbestimmung ebenfalls den bestimmungsgemäßen Gebrauch.
6. Stolpersteine vermeiden
a) Inkonsistente Formulierung der Zweckbestimmung
Die Zweckbestimmung eines Produkts ist der zentrale Ausgangspunkt für eine Vielzahl an nachfolgenden Prozessen, was die Auflistung in Kapitel 1 zeigt. Wir stellen bei Reviews von Technischen Dokumentationen immer wieder fest, dass die Zweckbestimmung in den verschiedenen Dokumenten inkonsistent formuliert ist. Das hat zur Folge, dass ggf. in der klinischen Bewertung bzw. Leistungsbewertung nicht alle erforderlichen Nachweise erbracht werden und die Produktakte nicht von der Benannten Stelle akzeptiert wird.
Wir empfehlen Ihnen, die Technische Dokumentation nicht dokumentenbasiert, sondern datenbasiert aufzustellen und somit Redundanzen und somit auch Inkonsistenzen zu vermeiden. Einen ersten Einblick liefert der Fachartikel 7 Tipps für die erfolgreiche digitale Transformation von Medizinprodukteherstellern.
Bitte beachten Sie zusätzlich, dass Angaben zur Zweckbestimmung neben der Technischen Dokumentation auch im dazugehörigen Marketingmaterial und auf der Webseite konsistent sein müssen.
b) Nicht nachgewiesene Claims
Mit der Zweckbestimmung „claimen“ Hersteller von Medizinprodukten und IVD den (medizinischen) Nutzen ihrer Produkte.
Die MDR definiert in Artikel 2 (52) die klinische Leistung als „Fähigkeit eines Produkts, die sich aufgrund seiner technischen oder funktionalen — einschließlich diagnostischen — Merkmale aus allen mittelbaren oder unmittelbaren medizinischen Auswirkungen ergibt, seine vom Hersteller angegebene Zweckbestimmung zu erfüllen, sodass bei bestimmungsgemäßer Verwendung nach Angabe des Herstellers ein klinischer Nutzen für Patienten erreicht wird“. Die IVDR definiert die „Leistung eines Produkts“ als „die Fähigkeit eines Produkts, seine vom Hersteller angegebene Zweckbestimmung zu erfüllen; sie besteht in der Analyseleistung und gegebenenfalls der klinischen Leistung zur Erfüllung dieser Zweckbestimmung“ (IVDR Artikel 2 (39)). Die „klinische Leistung“, als Bestandteil der Leistung eines Produkts, wird definiert als „die Fähigkeit eines Produkts, Ergebnisse zu liefern, die mit einem bestimmten klinischen Zustand oder physiologischen oder pathologischen Vorgang oder Zustand bei einer bestimmten Zielbevölkerung und bestimmten vorgesehenen Anwendern korrelieren“ (IVDR Artikel 2 (41)).
Das bedeutet, dass Hersteller den klinischen Nutzen ihres Produkts, den sie in der Zweckbestimmung angeben, durch die klinische Bewertung bzw. Leistungsbewertung nachweisen müssen. Dabei sollten Hersteller ebenfalls die definierte Patientenpopulation, die vorgesehenen Anwender und die vorgesehene Nutzungsumgebung berücksichtigen. Somit hat die Zweckbestimmung direkten Einfluss auf den Umfang der klinischen Bewertung bzw. Leistungsbewertung und den damit verbundenen Aufwand.
Ein Hersteller könnte z. B. die Zweckbestimmung zunächst auf eine bestimmte Patientenpopulation einschränken, um den Aufwand in der klinischen Bewertung bzw. Leistungsbewertung zu reduzieren. Nachdem sich das Produkt im Markt etabliert hat, kann der Hersteller die Zweckbestimmung mit den zugrundeliegenden klinischen Nachweisen erweitern.
7. Fazit
Die Zweckbestimmung stellt die Basis für die Entwicklung Ihres Medizinprodukts bzw. Ihres IVDs dar. Wenn diese fehlt, falsch, unvollständig, inkonsistent oder unverständlich ist, werden Sie wahrscheinlich im Laufe der Entwicklung nicht nur auf regulatorische Probleme stoßen. Üblicherweise lesen Auditoren und Inspektoren dieses Dokument gleich zu Beginn. Nehmen Sie sich also die Zeit, es sorgfältig zu erstellen!
Natürlich unterstützen wir Sie gern bei der Erstellung Ihrer produktspezifischen Zweckbestimmung. Nehmen Sie dazu einfach Kontakt zu uns auf.
Änderungshistorie
- 2025-10-06: Überarbeitung Kapitel 3 hinsichtlich des Zusammenhangs zwischen dem Begriff „Zweckbestimmung“ und „bestimmungsgemäßer Gebrauch“
- 2025-04-10: Am Ende von Kapitel 3 den Hinweis ergänzt, dass „bestimmungsgemäße Verwendung“ synonym zu „bestimmungsgemäßer Gebrauch“ zu verstehen ist
- 2024-08-12: Grundlegende Überarbeitung des Artikels; v. a. Einbindung IVD-relevanter Informationen
- 2021-03: Artikel völlig überarbeitet; v. a. die Einführung und Kapitel 3 (Definitionen) ergänzt und aktualisiert



Hallo,
eine umfassende Ausarbeitung – beeindruckend!
Ich habe (leider) einen formalen Hinweis zum letzten Absatz.
Aufgrund des letzten Satzes (Nehmen Sie sich die Zeit, es sorgfältig zu erstellen!) wirkt meine „Entdeckung eventuell ironisch, ist aber so nicht gemeint:
8. Fazit
Die Zweckbestimmung stellt die Basis für die Entwicklung Ihres Medizinprodukts dar. Fehlt es, ist es falsch…
Es müsste „Fehlt sie“ (die Zweckbestimmung) heißen
Üblicherweise lesen sich Auditoren und Inspektoren dieses Dokument auch zu allererst.
Entweder müsste „sich“ gelöscht werden, oder an das Satzende ein „durch“ gesetzt werden.
Weiterhin alles Gute und vielen Dank für die vielen verständlichen Erklärungen!
Sie haben absolut Recht, Frau Schulze-Beckinghausen!
Dank Ihre Hilfe konnte ich die grammatikalischen Schnitzer beheben. Vielen Dank!
Beste Grüße, Christian Johner
Hallo Herr Johner,
vielen Dank für diese tolle Ausarbeitung.
Ich habe diese Ausarbeitung gelesen, mit dem Ziel: etwas zur Definition zum „Abnormal use“ zu finden.
Gibt es diesen Begriff unter der MDR überhaupt noch? Gibt es eine allgemein gültige Definition hierfür?
In dem Template für die Vorkommnisbewertung wird der „abnormal use“ als Kriterium für das Nichtmelden von Ereignissen angegeben (im Entscheidungsbaum).
Vielleicht wäre es eine gute Idee den „nicht-bestimmungsgemäßen Gebrauch“ hier auch einzubauen?
Beste Grüße
Elena Keil
Sehr geehrte Frau Keil,
danke für Ihre Nachricht! Schauen Sie mal, ob der Beitrag zum vorhersehbaren Missbrauch u.a. in Abbildung die Antwort liefert? Ich bin nicht ganz sicher, auf welchen Entscheidungsbaum Sie sich beziehen.
Herzliche Grüße, Christian Johner
Guten Tag Herr Johner,
wir sind Händler von einem Urinsammelbecher. Ein Kunde hat bei uns angefragt, ob dieser Becher auch zum Sammeln von anderen Proben verwendet werden darf.
Nach Rückfrage beim Hersteller hat uns dieser eine allgemeine Gebrauchsanweisung für IVDs bereitgestellt (diese scheint für alle von ihnen hergestellten IVDs gültig zu sein). Z.B. wird als „Zweckbestimmung“ nur folgendes angegeben: For professional use only. Use only for laboratory analysis.
Meine Frage daher: ist das zulässig? Theoretisch könnten alle menschlichen und auch nicht-menschliche Proben darin gesammelt werden.
Beste Grüße,
Karl-Heinz
Sehr geehrter Herr Karl-Heinz,
der Hersteller hat die Hoheit über die Zweckbestimmung. Er darf die Anwendung, die Nutzer, die Nutzungsumgebung usw. festlegen.
Normalerweise machen das die Hersteller eher etwas breiter, um möglichst viele potenzielle Kunden zu haben. Wenn ein Hersteller aber aus anderen Gründen (z.B. um Medizinprodukt sein zu können oder Risiken zu minimieren) eine „enge“ Zweckbestimmung wählt, ist das absolut rechtlich okay.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
neben der ISO 14971:2019 setzt auch die MDCG 2020-6 (S. 6) den Intended Purpose mit dem Intended Use gleich:
“‘intended use’: The MDR defines ‘intended purpose’, but not ‘intended use’. ‘intended use’ should be considered to have the same meaning as ‘intended purpose’.”
Die IEC 62366-1:2015 definiert als den umfassenderen „bestimmungsgemäßen Gebrauch“:
„NORMAL USE: operation, including routine inspection and adjustments by any USER, and stand-by, according to the instructions for use or in accordance with generally accepted practice for those MEDICAL DEVICES provided without instructions for use
Note 1 to entry: NORMAL USE should not be confused with INTENDED USE. While both include the concept of use as intended by the MANUFACTURER, INTENDED USE focuses on the medical purpose while NORMAL USE incorporates not only the medical purpose, but maintenance, transport, etc. as well.“
Das Gegenstück ist der „Abnormal Use“.
Eine Vereinheitlichung der Definitionen, wie Sie sie fordern, wäre absolut wünschenswert.
Mit besten Grüßen
Michael Lang
Eine sehr wertvolle Ergänzung, Herr Lang! Ich werde den Beitrag noch entsprechend ergänzen.
Vielen Dank!
Beste Grüße, Christian Johner
Sehr geehrter Hr. Prof. Johner!
Wo kann die Gültigkeitsdauer einer CE- Kennzeichnung bei einem Medizinprodukt (z.B. Hochfrequenzchirurgie) gefunden werden?
Ist die CE für Medizinprodukte (Gerätschaften) einmal ausgestellt immer gültig?
Vielen Dank und LG
Sehr geehrter Herr,
die CE-Kennzeichnung enthält keine Gültigkeitsdauer. Die Kennzeichnung generell kann die Haltbarkeitsdauer enthalten.
Oder sprechen Sie von der Konformitätserklärung? Die kann muss aber keine zeitliche Beschränkung enthalten. Mehr dazu finden Sie im Artikel zur Konformitätserklärung.
Viele Grüße, Christian Johner
Hallo Herr Prof. Dr. Johner!
In der Zweckbestimmung wird ja auch das Anwenderprofil beschrieben.
Soll oder muss man hier auch das technische Servicepersonal mit berücksichtigen, welches je nach Vorgaben des Herstellers Wartungs- und Reparaturarbeiten durchführen darf?
Ich denke, dass es gerade im Hinblick auf die Risikoanalyse wichtig sein kann, bereits in der Zweckbestimmung Anforderungen/Einschränkungen beim Servicepersonal formuliert zu haben.
Wie ist Ihre Meinung dazu?
Vielen Dank schon einmal und Grüße an den Bodensee.
Ralf Philipp
Lieber Herr Philipp,
absolute Zustimmung zu dem was Sie schreiben. Zum bestimmungsgemäßen Gebrauch gehört auch das Anwendungsszenario „Service“. Also wie Sie sagen: Tätigkeiten wie Wartung und Reparaturarbeiten am System, von wem sie durchgeführt werden und ob damit Risiken verbunden sind. Aus der Zweckbestimmung lassen sich Gefährdungen, die mit diesen Benutzungsszenarien verbunden sind ableiten, die Sie in der Risikoanalyse untersuchen und ggf. mit einer Gebrauchstauglichkeitsprüfung absichern. Die Einschränkung des Service-Personals zum Beispiel auf Hersteller-eigene Mitarbeitende kann ein Ergebnis Ihrer Risikobeurteilung sein und damit eine Risiko-Kontrollmaßnahme. Die konkrete Beschreibung der User und deren Einschränkung kann in der Zweckbestimmung festgelegt werden. Jedoch eignet sich bei umfangreichen Beschreibungen die Gebrauchstauglichkeits-Akte besser. Hier finden Sie mehr Informationen dazu: https://www.johner-institut.de/blog/category/iec-62366-usability/
Herzliche Grüße,
Sebastian Grömminger
Vielen Dank für Ihren Kommentar, Herr Grömminger!
Sehr gerne geschehen lieber Herr Philipp!
Sehr geehrter Herr Johner,
wird sind gerade am Gruppieren unserer Medizinprodukte für die Basis-UDI-DI. Diese sollte ja nur Produkte mit gleicher Zweckbestimmung gruppieren. Jetzt stellt sich für uns die Frage, wie eng man diese Zweckbestimmung sieht (Medizinischer Zweck oder zusätzlich Medizinische Anwendung?).
Dürften zum Beispiele ein Medizinprodukt was für Erwachsene bestimmt ist und ein Medizinprodukt, dass nur für Kleinkinder bestimmt ist unter einer Basic-UDI-DI gruppiert werden, wenn alle anderen Merkmale der Zweckbestimmung identisch sind?
Vielen Dank im Voraus!
Mit freundlichen Grüßen,
Marius Berthel
Lieber Herr Berthel,
Die Patientenzielgruppe fällt unter die medizinische Anwendung. Wenn das Produkt z.B. mit geänderter Konfiguration angewandt wird, der eigentliche medizinische Zweck bei den unterschiedlichen Patientengruppen hingegen derselbe ist, können Sie eine Basic-UDI-DI für beide Produkte verwenden. Entscheidend ist zudem, dass es sich bei den Anpassungen für die Patientengruppe (hier Kleinkinder) um nicht-essentielle Charakteristika handelt. So zumindest der Entscheidungsbaum der Medtech Europe: https://www.medtecheurope.org/wp-content/uploads/2020/06/200602_MTE-Basic-UDI-DI-guidance-v1.1_final.pdf.
Herzliche Grüße,
Sebastian Grömminger
Hallo liebes Johner-Team,
ich habe eine frage, die ihr mir vielleicht beantworten könnt.
ist es möglich, ein medizinprodukt einmal mit breitem intended use und einmal mit eingeschränktem intended use unter dem selben zertifikat laufen zu lassen?
wir haben aktuell zwei medizinprodukte die, bis auf die zweckbestimmung, ident sind. eines der beiden produkte hat eine breite zweckbestimmung (katheterisierung, intubierung, koloskopie), das zweite wird (aus marketingtechnischen gründen) rein zur intubierung verkauft. wäre es aus ihrer sicht möglich, das unter einer TD laufen zu lassen und trotzdem marketingtechnisch als zwei individuelle produkte zu verkaufen oder sehen sie hier stolpersteine?
danke für die hilfe,
mfg
eva albertini
Liebe Frau Albertini,
prinzipiell ist es möglich, dass Sie beide Produkte auf einem Zertifikat listen. Beide Produkte hätten dann dieselbe Basis UDI-DI mit einer übergeordneten Zweckbestimmung, welche für beide Produkte identisch ist. Die beiden Produkte würden sich dann hinsichtlich der Indikationen unterscheiden.
Auch eine Dokumentation beider Produkte in einer Technischen Dokumentation ist möglich. Allerdings sollten Sie in diesem Fall darauf achten, dass sich z.B. Aspekte hinsichtlich des Klinischen Nutzens, die Zuordnung einzelner Nachweise, oder Risiken in der Risikotabelle ggf. zwischen den beiden Produkten unterscheiden. Es muss immer klar erkenntlich sein, welche Informationen innerhalb der Technischen Dokumentation zu welchem Produkt gehören.
Herzliche Grüße,
Juliane Havlicek
Guten Tag, ich hätte folgende konkrete Fragen zum Thema Zweckbestimmung – aber aus Sicht eines Importeurs/Distributors:
Ist es zulässig ein Device, das vom Hersteller nicht für einen Medizinischen Zweck deklariert ist, für einen medizinischen Zweck zu importieren/vertreiben ohne es als MD handzuhaben (Transport und Lagerung)? Falls nein – übernimmt der Distributor dann im Grunde die Rolle des Legal Manufacturers, weil er dem Device eine neue Zweckbestimmung gibt?
Beispiele:
– Ein Kühlbeutel, der nicht als MD deklariert ist, wird zur Kühlung zB bei Sportverletzungen abgegeben.
– Ein Messbecher, der nicht als MD deklariert ist, wird zur Dosierung eines Arzneimittels abgegeben.
Ist es zulässig ein Device, das vom Hersteller nicht als MD deklariert ist, aber für einen medizinischen Zweck bestimmt ist, als nicht-MD zu vertreiben oder ist man da in der Verantwortung die Deklaration des Herstellers zu hinterfragen?
Beispiel:
– Ein Pflaster, das nicht als MD ausgewiesen ist.
Lieber Daniel,
vielen Dank für Ihre spannenden Fragen.
Sie haben die Situation in Ihrer ersten Frage bereits selbst richtig eingeschätzt. Da der Importeur/Distributor dem Produkt einen medizinischen Zweck zuweist, ändert er die Zweckbestimmung des Produktes. Aufgrund des medizinischen Zwecks, fällt das Produkt nach Art. 2 (1) unter die MDR und es sind die Anforderungen der MDR zu erfüllen.
Für Ihre zweite Frage können wir einen Blick in den Leitfaden für die Umsetzung der Produktvorschriften der EU 2022 („Blue Guide“) werfen. In Kapitel 2.8 „Vernünftigerweise vorhersehbarer und vorgesehener Verwendungszweck/Fehlanwendung“ steht „der vorgesehene Verwendungszweck bezeichnet dabei die Nutzung, die für ein Produkt entsprechend den Informationen seitens des inverkehrbringenden Herstellers (oder Einführers) vorgesehen ist, oder die übliche Nutzung entsprechend dem Entwurf und die Bauweise des Produkts“. Bei einem Pflaster fällt es schwer, dem Produkt einen medizinischen Zweck abzusprechen.
Des Weiteren heißt es im „Blue Guide“ im Kapitel 3.3. „Einführer“: „Der Einführer muss sicherstellen, dass der Hersteller seinen Verpflichtungen ordnungsgemäß nachgekommen ist. Er ist kein bloßer Wiederverkäufer von Produkten, sondern spielt bei der Gewährleistung der Konformität der eingeführten Erzeugnisse eine sehr wichtige Rolle“. Somit ist der Importeur in der Verantwortung, die Angaben des Herstellers zu hinterfragen.
Herzliche Grüße,
Juliane Havlicek
Vielen lieben Dank, Frau Havlicek
Guten Tag,
die Definition der Zweckbestimmung in Ihrem Artikel lässt mich hier mit folgenden Fragen zurück:
– Wieso benennt Annex I chapter III 23.4 (b) zusätzlich zur Zweckbestimmung (intended purpose) nochmal gesondert die indications, contra-indications, patient target group(s) und intended users, wenn diese default Bestandteil der Zweckbestimmung sind?
– Wieso benennt Annex II 1.1 (a) noch zusätzlich den intended user, wenn dieser default Bestandteil der Zweckbestimmung ist?
– Annex XII chapter I 4(a) besagt, dass die Zweckbestimmung (intended purpose) auf die Zertifikate zu drucken ist. Wenn all die oben genannten Bestandteile der Zweckbestimmung auf die Zertifikate zu drucken ist, dann könnte man jetzt extrapolieren, wie groß jene wären, wenn man mehrere hundert Produkte dort abbildet (was real der Fall ist).
Es wäre hier zielführender, die Zweckbestimmung auf die Definition der MDR einzuschränken denn es ist dort ziemlich klar als „use for which a device is intended“ abgebildet. Vice versa ist der claim, den der Hersteller mit der beabsichtigten An/Verwendung im Rahmen der Formulierung „The is intended for…“ die Formulierung des med. Zwecks. Alle zusätzlichen Attribute, die die Erreichung des Intended Purpose entsprechend der vom Hersteller validierten Anwendungsfälle und -szenarien beschreiben in der IfU sind Intended Use. Deshalb werden sie in weiteren Punkten des Annex I 23.4. auch gesondert gelistet und nicht unter dem Intended Purpose subsummiert. Gleiches gilt für die Inhalte von Annex II1.1(c), die oben als Teil des Intended Purpose erscheinen, jedoch in der grundlegenden Gesetzgebung dort nicht mit in den operativen Abschnitten drunter geführt werden.
All diese Inhalte können sich ohne Veränderung des eigentlichen Intended Purpose des Produkts (wofür ist es gedacht) während der Zeit verändern – sei es durch Erprobung neuer Anwendungsfälle und folge dessen Erweiterung dieser (Anwendungsumgebung, neue Schnittstellen, interoperative System, …) oder auch durch PMS-Aktivitäten, die bspw. zu Kontraindikationen führen. All das erweitert oder begrenzt die beabsichtigte Anwendung des Produktes, ändert am eigentlichen Zweck des Produktes jedoch zunächst nichts.
Unter dem Intended Purpose so viel zu subsummieren birgt folglich nicht nur eine administrative Last (bspw. Zertifikate) sondern auch eine strategische Unflexibilität in der Weiterentwicklung von Produkten (Revalidierung des gesamten Scopes bei Erweiterung), da es Merkmale des Intended Use vereinnahmt, die an anderen Stellen der Gesetzgebung mit Grund nicht darunter inkludiert sind.
Deutlich einfacher macht es die Auslegung, dass der Intended Purpose den eigentlich Zweck (… is intended for …) beschreibt und der Intended Use sämtliche Beschreibung, wie dieser nachweislich sicher und leistungsfähig erreicht werden kann. Das ist erprobt in vielen Zulassungen und kompatibel mit den versch. Gesetzes- und Normungstexten. Vielleicht einfach mal diesen modularen Ansatz ausprobieren und selbst sehen, dass das gehen kann.
Liebe Frau Dr. Richter,
herzlichen Dank für Ihre wertvollen Gedanken zum Thema Zweckbestimmung. Wir haben diese mit großer Aufmerksamkeit im Team diskutiert und möchten Ihnen unsere Betrachtung dazu schildern.
Die MDR definiert in Art. 2 (12) den Intended Purpose als „use for which the device is intended according to the data supplied by the manufacture on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacture in the clinical evaluation”. Diese Definition umfasst keine detaillierten Aspekte. Daher erweitert die MDR in den Anhängen I und II die Aspekte, welche der Intended Purpose enthalten soll. Als Beispiel ist es für die klinische Bewertung oder das Risikomanagement wesentlich, dass Sie bestimmen, für welche Patientenzielgruppe Ihr Produkt gedacht ist oder wer das Produkt anwendet (health care professionals oder Laienanwender).
Bitte beachten Sie in diesem Zusammenhang ebenfalls das MDCG 2020-3, welches im Kapitel 4.3.2.2 significant changes des Intended Purpose auflistet (z.B. neue Indikationen, neue Targetpopulationen, neue Nutzer usw.). Diese Aspekte sind daher ebenfalls Teil des Intended Purpose.
Es ist tatsächlich durchaus machbar, die Aspekte, welche die MDR fordert, in ein bis zwei kurzen Sätzen zu definieren. Diese Executive Summary können Sie dann in den unterschiedlichen Dokumenten angeben (Risikomanagementakte, klinische Bewertung, Usability Engineering File, oder auch auf der Konformitätserklärung).
Andere Aspekte, die zur bestimmungsgemäßen Verwendung Ihres Produktes relevant sind (z.B. Service, Wartung, Transport, Lagerung usw.), stellen den bestimmungsgemäßen Gebrauch dar. Wir empfehlen, diese Aspekte zusätzlich zur Executive Summary in dem Dokument zur Zweckbestimmung zu beschreiben.
Auf diese Weise können Sie sicherstellen, dass Sie im Intended Purpose alle Aspekte für nachfolgende Prozesse berücksichtigen, ohne dass die Ausführungen zu umfangreich werden.
Wir hoffen, Ihnen mit unseren Ausführungen weitergeholfen zu haben.
Herzliche Grüße,
Juliane Havlicek
Guten Tag,
herzlichen Dank für Ihre wertvollen Hinweise.
Ich habe eine Frage zur nachgelagerten, praktischen Verwendung der (korrekten und vollständigen) Zweckbestimmung als Teil der „vom Hersteller bereitzustellenden Informationen“ gemäß MDR.
Die MDR beschreibt ja die Zweckbestimmung wie folgt:
…
„Zweckbestimmung“ bezeichnet die Verwendung, für die ein Produkt entsprechend den Angaben des Herstellers auf der Kennzeichnung, in der Gebrauchsanweisung oder dem Werbe- oder Verkaufsmaterial bzw. den Werbe- oder Verkaufsangaben und seinen Angaben bei der klinischen Bewertung bestimmt ist;
…
Dies erweckt den Eindruck, dass auf der Kennzeichnung (z.B. Etiketten) stets die Zweckbestimmung in Gänze zu nennen wäre.
Ich interpretiere es eher so, dass die Angaben auf der Produktkennzeichnung und in den Werbematerialien der Zweckbestimmung zumindest in keiner Weise widersprechen dürfen.
Weiter steht in der MDR, 23.2 „Angaben auf der Kennzeichnung“ jedoch:
Die Kennzeichnung enthält alle folgenden Angaben:
…
alle unbedingt erforderlichen Angaben, aus denen der Anwender ersehen kann, worum es sich bei dem Produkt, dem Packungsinhalt sowie der Zweckbestimmung eines Produkts, sofern diese für den Anwender nicht offensichtlich ist, handelt;
…
In der Praxis scheint es dennoch aus nachvollziehbaren Gründen kaum Hersteller zu geben, welche die komplette Zweckbestimmung tatsächlich auf ein MDR-Produktetikett bringen.
Ist davon auszugehen, dass ein (exakt) beschreibender Produktname auf dem Etikett, z.B. „Spritzenpumpe“ in Verbindung mit dem Symbol „Gebrauchsanweisung beachten“, in welcher die Zweckbestimmung sowie die Anwendergruppe enthalten ist, in jedem Fall die regulatorischen Anforderungen der MDR erfüllt?
Herzlichen Dank für Ihre Einschätzung.
Beste Grüße
Johannes Hoffmann
Lieber Herr Hoffmann,
vielen Dank für Ihre Frage.
Sie haben die Situation bereits richtig eingeschätzt. Die Zweckbestimmung auf der Kennzeichnung anzugeben wäre nicht zielführend und oft nicht realisierbar. Es sollte klar erkennbar sein, worum es sich bei dem Produkt handelt; hier reicht der Name an sich nicht aus. In der Praxis wird häufig eine Zweckbestimmung in Kurzform (z.B. „Spritzenpumpe“, „Zentralvenöser Katheter“, „COVID-19 Test Kit“ etc.) aufgebracht. Aus unserer Sicht steht Ihrer vorgeschlagenen Vorgehensweise nichts entgegen.
Herzliche Grüße,
Juliane Havlicek
Werte Frau Dr. Havlicek,
vielen Dank für die theoretische Nachhilfe, wenngleich diese Abschnitte sehr wohl bekannt sind.
In diesem Zusammenhang sind jedoch einige weitere Abschnitte aus der MDR und Normen zu betrachten, die hier die Trennung von Intended Purpose und den von Ihnen darunter gefassten Inhalten formulieren bzw. in welchen der Intended Purpose konkatenierend mit anderen nutzungsrelevanten Inhalten benannt ist sodass er eben diese nicht vereinnahmt. Beispielsweise würde hier Art. 32 2(b) einer Antwort in Ihrer Interpretation bedürfen:
„2. The summary of safety and clinical performance shall include at least the following aspects:
(a) […]
(b)
the intended purpose of the device and any indications, contraindications and target populations; […].“
Oder auch Annex X:
„5. Changes to the type
5.1. The applicant shall inform the notified body which issued the EU type-examination certificate of any planned change to the approved type or of its intended purpose and conditions of use.
5.2. Changes to the approved device including limitations of its intended purpose and conditions of use shall require approval from the notified body which issued the EU type-examination certificate where such changes may affect conformity with the general safety and performance requirements or with the conditions prescribed for use of the product. The notified body shall examine the planned changes, notify the manufacturer of its decision and provide him with a supplement to the EU type-examination report. The approval of any change to the approved type shall take the form of a supplement to the EU type-examination certificate.
5.3. Changes to the intended purpose and conditions of use of the approved device, with the exception of limitations of the intended purpose and conditions of use, shall necessitate a new application for a conformity assessment.“
um hier nur ein paar ausgewählte zu nennen. Wenn die nachgenannten Inhalte aus Ihrer Sichtweise immer Teil des Intended Purpose sind, warum muss das hier gesondert zusätzlich geführt werden?
Es ist ja üblich, dass es verschiedene Sichtweisen auf einen Text gibt und in Regulatory Affairs essentiell hier im Austausch zu sein, um einen common sense zu entwickeln. Diese Diskussion ist jedoch leider schon so polarisierend geworden, dass es nur noch Lager gibt, die einander nicht verstehen. Vielleicht auch weil es doch Unterschiede in den Randbedingungen von Medizinprodukten und IVD`s gibt oder weil es viele in einer rein akademischen Betrachtung lassen und sich daran auch normativ geübt abarbeiten, wohingegen andere die globale Integration von Zweckbestimmungen für ein Produkt schon lange durchführen, was fast immer nur mit modularer Herangehensweise gelingt.
Ich kann Ihnen nur sagen, dass die distinkte, modulare Handhabung von Intended Purpose (als zentraler Benefit-Claim am Produkt) und den weiteren use-relevanten Attributen eines Produktes (die Sie auch unter Intended Purpose fassen würden) schon länger als die MDR gebräuchlich ist in globalen Settings und auch Produkte von vielen Unternehmen so zugelassen wurden und werden. Wie soll das funktioniert haben, wenn es so gar nicht der Rechtsauffassung entspricht?
Mir fiel jetzt noch ein wesentlicher Beispiel-Abschnitt ein, den man auch in Ihrer Sichtweise nochmal erläutern müsste, wie das zu handhaben wäre: Annex I, 10.2. „Devices shall be designed, manufactured and packaged in such a way as to minimise the risk posed by contaminants and residues to patients, taking account of the intended purpose of the device, and to the persons involved in the transport, storage and use of the devices.“ – Hier wird der Intended Purpose inhärent mit dem Nutzen für den Patienten verknüpft. Die Use- relevanten Attribute für Transport, Lagerung und „Use of the devices“ sind zusätzlich geführt für die entsprechenden Anwendergruppen. Entsprechend der anwendbaren Normen ist jedoch die Kennzeichnung von bspw. Entsorgungshinweisen etc. (nehmen wir mal kontaminierte Produkte nach Gebrauch als Beispiel) auch nutzungsrelevant, sind jedoch für den Patienten selbst nicht relevant oder von Nutzen.
Das würde dann bei Ihnen also mit unter Intended Purpose fallen?
Besten Dank an Herrn Hoffmann auch nochmal für den Praxischeck. Mich würde interessieren, wie die Zweckbestimmung in Kurzform denn spezifiziert ist? Wo kann man sicher ableiten, was da drin enthalten ist? Wäre für die entsprechenden vorgesehenen Ziele, auf die diese anwendbar ist (IfU, DoC, Zertifikate).
Ich bin auf Ihrer Antwort gespannt und verbleibe mit vielen Grüßen!
Sehr geehrte Frau Dr. Richter,
herzlichen Dank für Ihr Feedback zu unserem Fachartikel.
Wir schätzen es sehr, dass Sie Ihre Gedanken mit uns teilen. Die im Artikel dargelegten Perspektiven basieren auf der fundierten Expertise des Teams des Johner Instituts, welche die Facetten der Branche widerspiegeln. Selbstverständlich ist uns bewusst, dass die regulatorischen Vorgaben individuell und produktspezifisch betrachtet und umgesetzt werden müssen.
Um sicherzustellen, dass wir Ihnen möglichst effektiv und zielgerichtet helfen können, konzentrieren wir uns am besten auf reale und aktuelle Anliegen. Sollten Sie spezifische Unterstützung benötigen, können Sie uns gern unter [email protected] kontaktieren. Wir würden uns freuen, Ihnen zu helfen.
Mit herzlichen Grüßen,
Juliane Havlicek