Die Validierung wird von vielen Gesetzen und Normen gefordert. Allerdings verstehen diese Regularien den Begriff in den verschiedenen Kontexten unterschiedlich.
1. Grundlagen
a) Definition
Die ISO 9000 definiert den Begriff Validierung wie folgt:
Definition (1)
die Bestätigung durch objektiven Nachweis erbracht, dass die Anforderungen für eine bestimmte Anwendung oder eine bestimmte Zweckbestimmung erfüllt sind
Quelle: ISO 9000
Da der Begriff Anforderung unterschiedlich interpretiert wird, hilft diese Definition:
Definition (2)
Prüfung mit objektiven Mitteln, ob spezifizierte Nutzer im spezifizierten Nutzungskontext die spezifizierten Nutzungsziele erreichen
Quelle: Johner Institut, angelehnt an IEC 62366-1
b) Was validiert werden kann
Die Definitionen legen nicht fest, welche Objekte validiert werden. Im Kontext von Medizinprodukten sind das beispielsweise:
- Medizinprodukte
- Komponenten dieser Produkte
- Prozesse zum Entwickeln, Produzieren und Überwachen dieser Produkte
- Werkzeuge und computerisierte Systeme
2. Validierung von Medizinprodukten
a) Eine Aktivität im Entwicklungsprozess
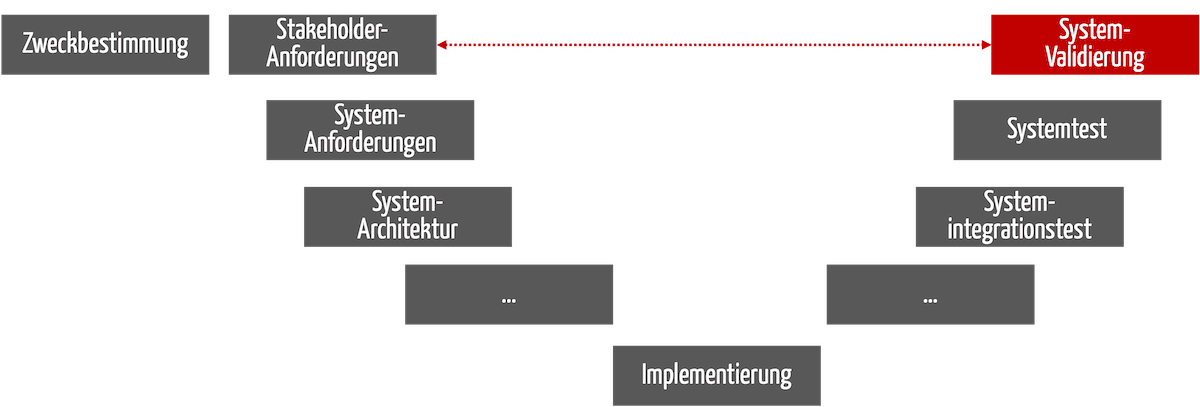
Bei Medizinprodukten erfolgt die Validierung am Ende der Entwicklung (s. Abb. 1). Das gilt nicht nur beim V-Modell, sondern bei jeden Entwicklungsprozess.
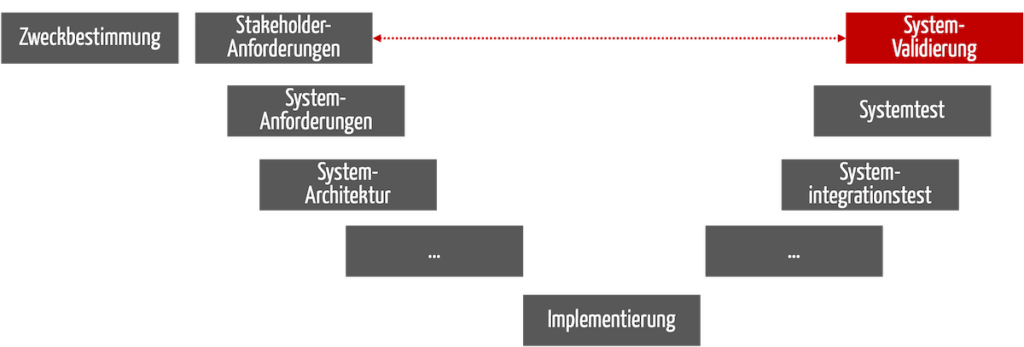
Abb. 1: Die Validierung findet am Ende des Entwicklungsprozesses statt.
Im Englischen spricht man von Design Validation.
Sie muss sicherstellen, dass die Stakeholder-Anforderungen erfüllt sind und die Anwender damit die Zweckbestimmung tatsächlich erreichen (s. gestrichelter roter Pfeil in Abb. 1).
b) Methoden
Die wichtigsten Methoden bei der Validierung von Medizinprodukten sind die klinische Bewertung und die Usability Validation, z. B. in Form einer summativen Evaluation bzw. von Usability-Tests.
Bei der V&V von IVD entspricht die Leistungsbewertung der klinischen Bewertung.
Mache Hersteller zählen auch Tests in Prüflaboren wie Prüfungen der Biokompatibilität (z. B. Zytotoxitätstests) und Prüfungen der elektrischen Sicherheit zur Validation. Aber das sind eher Verifizierungsaktivitäten.
Vorsicht!
Der Begriff „Validation“ wird auch im Kontext von Komponenten genutzt, beispielsweise bei Machine Learning Libraries. Allerdings sind das im regulatorischen Sinne eher Verifizierungsaktivitäten.
c) Regulatorische Anforderungen
Alle wichtigen Normen und Gesetze fordern die Validierung der Medizinprodukte. Dazu zählen:
Die ISO 13485 zählt die klinische Bewertung als Teil der Validierung.
3. Software-Validierung und Computerised Systems Validation
Die ISO 13485 fordert gleich mehrfach, dass die Hersteller ihre Infrastruktur und Werkzeuge validieren. Beispielsweise verpflichtet das Kapitel 4.1.6 die Hersteller zur Computerized Systems Validation (CSV). Normen wie die ISO 80002-2 und der AMII TIR 36 geben dabei Hilfestellung.
Dieser Artikel beschreibt die genauen Anforderungen der Norm an die Werkzeug-Validierung.
Der Begriff Software-Validierung ist mehrfach belegt, wie dieser Artikel zeigt.
4. Validierung von Prozessen
Gesetze und Normen wie MDR, IVDR und ISO 13485 verpflichten die Hersteller auch zur Prozessvalidierung. Validiert werden müssen alle Prozesse, die eine Auswirkung auf die Konformität der Produkte haben. Das sind nicht nur Entwicklungs- und Produktionsprozesse, sondern auch Prozesse zum Umgang mit Kundenrückmeldungen und zur Post-Market Surveillance.