
Die Validierung von IVD bestätigt den medizinischen Zweck des Produkts. Die Verifizierung von IVD hingegen weist nach, ob das IVD funktioniert wie beabsichtigt.
Mit diesem Artikel geben wir Ihnen einen Fahrplan an die Hand, wie Sie in jeweils 5 Schritten die Verifizierung und die Validierung Ihrer IVD-Produkte zielgerichtet und ohne unnötigen Aufwand durchführen. Zudem zeigen wir Ihnen die häufigsten Fehler und verraten, wie Sie diese vermeiden.
1. Begriffsdefinitionen und Abgrenzung
a) Begriffsdefinitionen
Bei einer Vielzahl an Begriffen wie „Verifizierung“, „Validierung“, „Leistungsbewertung“, „analytische Leistungsbewertung“, „klinische Leistungsstudien“ etc. ist es für IVD-Hersteller häufig schwierig, den Zusammenhang bzw. die Unterschiede der Begriffe zu verstehen. Wenn Ihnen das auch so geht, kann Ihnen der folgende Abschnitt einen raschen Überblick verschaffen.
Alle aufgeführten Begriffe haben gemeinsam, dass es um den Nachweis der Sicherheit und der Leistung eines IVDs geht. Ferner gelten sie für alle Arten von IVD gleichermaßen: sowohl für IVD-Geräte, IVD-Assays, Reagenzien und Kits als auch für IVD-Software.
Der Begriff „Validierung“ ist in der ISO 9000:2015 wie folgt definiert:
„Bestätigung durch Bereitstellung eines objektiven Nachweises, dass die Anforderungen für einen spezifischen beabsichtigten Gebrauch oder eine spezifische beabsichtigte Anwendung erfüllt worden sind“
Quelle: DIN EN ISO 9000:2015
Der in der ISO 9000:2015 allgemein gefasste „spezifische beabsichtigte Gebrauch“ wird bei einem IVD als „Zweckbestimmung“ bezeichnet. Anhand der Zweckbestimmung legen Hersteller fest, wofür ein IVD verwendet werden darf. Dies umfasst z. B. den medizinischen Zweck, die Art der bereitgestellten Informationen, den Probentyp, die Patientenpopulation, die vorgesehenen Nutzer und die Nutzungsumgebung. Die Validierung von IVD ist somit die Überprüfung, ob das Produkt die festgelegte Zweckbestimmung einschließlich der genannten Attribute erfüllt.
Die ISO 20916:2019, die sich auf die gute Studienpraxis bei klinischen Leistungsstudien für IVD bezieht, definiert „Validierung“ mit direktem Bezug zur Zweckbestimmung (Intended Use):
„verification that the specified requirements are adequate for an intended use“
Quelle: ISO 20916:2019
Hingegen bezieht sich die Verifizierung stets auf die Überprüfung einzelner Produktanforderungen und Produktdesign-Spezifikationen (Design Input Requirements, Design Specifications) und ist definiert als:
„Bestätigung durch Bereitstellung eines objektiven Nachweises, dass festgelegte Anforderungen erfüllt worden sind“
Quelle: DIN EN ISO 9000:2015
„confirmation by examination and provision of objective evidence that the specified requirements have been fulfilled“
Quelle: ISO 20916:2019
Der Begriff „Leistungsbewertung“ stammt aus der EU-Verordnung 2017/746 über In-vitro-Diagnostika (IVDR). Gemäß IVDR, Anhang XIII umfasst die Leistungsbewertung von IVD drei Elemente:
- Wissenschaftliche Validität des Analyten
- Analyseleistung
- Klinische Leistung
Die Analyseleistung bezieht sich auf die „Fähigkeit eines Produkts, einen bestimmten Analyten korrekt nachzuweisen oder zu messen“ (s. IVDR, Artikel 2 (40)). Die anwendbaren analytischen Leistungsparameter spezifizieren Hersteller als Produktanforderungen für das jeweilige IVD (s. IVDR, Anhang I, Abschn. 9.1.a)). Demnach kommt der Nachweis der Analyseleistung der Verifizierung des gesamten Produkts gleich (s. u.).
Die Prüfung der klinischen Leistung ist hingegen Teil der Validierung des IVDs. Sie ist definiert als die „Fähigkeit eines Produkts, Ergebnisse zu liefern, die mit einem bestimmten klinischen Zustand oder physiologischen oder pathologischen Vorgang oder Zustand bei einer bestimmten Zielbevölkerung und bestimmten vorgesehenen Anwendern korrelieren“ und belegt somit die Zweckbestimmung des IVDs.
Hersteller sollten beachten, dass sich die Leistungsbewertung eines IVDs wesentlich von der klinischen Bewertung eines Medizinprodukts unterscheidet.
Lesen Sie unseren ausführlichen Artikel über die Leistungsbewertung von IVD.
Falls Sie mehr über die Klinische Bewertung von Medizinprodukten erfahren wollen, finden Sie in diesem Artikel einen Überblick.
Haben Sie eine IVD-Software? In diesem Artikel haben wir für Sie die speziellen Anforderungen an die klinische Bewertung von Medizinprodukte-Software und die Leistungsbewertung von IVD-Software zusammengefasst.
b) Abgrenzung zur Qualifizierung von Anlagen und Geräten (DQ, IQ, OQ, PQ)
Die Medizinprodukte-Regularien kennen die Unterscheidung in DQ, IQ, OQ und PQ nicht. Es ist jedoch ratsam und erforderlich, dass sich Hersteller mit der Qualifizierung von Geräten und Anlagen auseinandersetzen.
Um Verifizierungstests und Validierungsstudien durchzuführen, benötigen Hersteller u. U. neben dem zu überprüfenden IVD weitere Geräte und Messmittel. Dabei müssen sie sich auf die Funktionalität und Eignung dieser Geräte für den vorgesehenen Zweck (z. B. Messung eines pH-Werts, Bestimmung einer Nukleinsäure-Konzentration, Extraktion von DNA aus Blutproben) verlassen können. Um die Eignung zu prüfen, führen sie eine Qualifizierung der erforderlichen Geräte und Messmittel durch. Auch die Anlagen und Geräte, die für die Produktion eines IVDs eingesetzt werden, sind zunächst für ihren Einsatzzweck zu qualifizieren.
Es werden folgende Qualifizierungsaktivitäten unterschieden:
- Design Qualification – DQ
Wurde das Produkt wie (in der Systemarchitektur) spezifiziert entwickelt und das System aus diesen Komponenten zusammengesetzt? - Installation Qualification – IQ
Ist das Gerät bzw. die Anlage vollständig und lässt es sich wie beschrieben in Betrieb nehmen? - Operation Qualification – OQ
Stehen alle Funktionen wie spezifiziert zur Verfügung? Funktioniert das Gerät bzw. die Anlage unter den festgelegten Umgebungsbedingungen? - Performance Qualification – PQ
Erfüllt das Gerät bzw. die Anlage auch an den Leistungsgrenzen und bei Fehlgebrauch seine Funktion mit der spezifizierten Genauigkeit?
Auch medizinische Labore – die „Anwender“ von In-vitro-Diagnostika – müssen eine Qualifizierung ihrer „Laboratoriumsausrüstung“ vor der ersten Nutzung durchführen. Dies fordert sowohl die ISO 15189 als auch die Richtlinie der Bundesärztekammer (RiLiBÄK). Das Laborpersonal qualifiziert ein Gerät, um sicherzustellen, dass es für die vorgesehenen Arbeitsschritte (z. B. Probenprozessierung, Qualitätskontrolle) geeignet ist und die erforderliche Leistung erbringt. Die ISO 15189 erläutert weiter, dass der Begriff Laboratoriumsausrüstung sowohl „Hardware und Software für Apparate, Messsysteme und Laboratoriums-IT-Systeme“ umfasst. Damit ist auch das Thema Computer System Validation (CSV) relevant.
ISO 15189:2023-03, Kapitel 6.4
c) Abgrenzung zur Prozessvalidierung
Während der Validierung eines Prozesses soll nachgewiesen werden, dass der Prozess stets zu reproduzierbaren Ergebnissen bzw. Produkten gemäß zuvor spezifizierter Qualitätsmerkmale führt.
„Process validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications“
Quelle: FDA 21 CFR 820.3
Im Sinne der guten Herstellungspraxis (Good Manufacturing Practice, GMP) spielt die Prozessvalidierung für die Produktion eines IVDs eine wichtige Rolle. Hat der Hersteller den Herstellprozess etabliert und anhand von Herstellvorschriften und Herstellanweisungen dokumentiert, untersucht er die einzelnen Produktionsschritte auf mögliche Risiken. Anhand dieser Risikoanalyse leitet der Hersteller ab, welche Prozessschritte kritisch sind und ggf. mit Qualitätskontrollen abgesichert und validiert werden müssen.
Auch die Prozesse zur Sterilisation eines IVDs müssen validiert werden.
2. Regulatorische Anforderungen
Die Verifizierung und Validierung von IVD soll den Nutzen eines IVDs nachweisen. Was sind jedoch die gesetzlichen Anforderungen, an die sich IVD-Hersteller bei ihren Verifizierungs- und Validierungsaktivitäten halten müssen?
Artikel 5 der IVDR gibt vor, welche Anforderungen IVD-Hersteller erfüllen müssen, bevor sie ein IVD in Verkehr bringen dürfen bzw. ein IVD durch den Anwender in Betrieb genommen werden kann: So sollen Hersteller zunächst die für ein Produkt relevanten grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der IVDR spezifizieren, indem sie die Zweckbestimmung des IVDs berücksichtigen. Dass das Produkt die spezifizierten Anforderungen an die Sicherheit und Leistung (s. Definitionen) erfüllt, müssen die Hersteller nachweisen. Dafür sollen sie u. a. eine Leistungsbewertung gemäß Artikel 56 durchführen, also eine Verifizierung und Validierung.
„Freiheit von unvertretbaren Risiken“
Quelle:ISO 20916:2019
„bezeichnet die Fähigkeit eines Produkts, seine vom Hersteller angegebene Zweckbestimmung zu erfüllen; sie besteht in der Analyseleistung und gegebenenfalls der klinischen Leistung zur Erfüllung dieser Zweckbestimmung“
Quelle: IVDR, Artikel 2 (39)
Die folgenden Normen beschreiben weitere Kriterien für die Verifizierung und Validierung eines IVDs.
- Die ISO 13485:2016 fordert in den Kapiteln 7.3.6 und 7.3.7 die Durchführung und Dokumentation einer „Entwicklungsverifizierung“ und einer „Entwicklungsvalidierung“.
- Für IVD-Software gibt die IEC 62304:2006 + A1:2015 in den Kapiteln 5.5. bis 5.7. als Verifizierungsaktivitäten die Unit-Tests, die Integrationstests und die Prüfung des Software-Systems vor.
- Auf die Durchführung und Dokumentation einer Validierung von Standalone-Software für Medizinprodukte geht die IEC 82304-1:2016 in den Kapiteln 6.1. bis 6.3. ein. Sie ist für Standalone-Software für IVD anwendbar.
- Die bereits erwähnte Norm ISO 20916:2019 spezifiziert die Anforderungen an die Durchführung und Dokumentation von Leistungsbewertungsstudien für IVD und wird in der IVDR referenziert.
3. In 5 Schritten zur Verifizierung von IVD
Schritt 1: Requirements und Spezifikationen festlegen
Die Verifizierung eines IVDs hat zur Aufgabe, die spezifizierten Produktanforderungen zu überprüfen. Diese müssen Hersteller folglich zuvor festgelegt haben.
Schritt 2: Wahl der Verifizierungstätigkeit
Hersteller können sich für die Verifizierung einer Vielzahl von unterschiedlichen Tests, Experimenten und Studien bedienen. Die Wahl der Verifizierungsaktivität hängt von der Art des IVDs (z. B. Assay, Gerät, Software) und der zu überprüfenden Produkt- bzw. Software-Anforderung ab. Sie können die Verifizierung unter Verwendung von humanen Patientenproben, einer synthetischen Probenmatrix oder in silico durchführen. Bei der Verifizierung von IVD-Software können Hersteller sowohl echte Patientendaten zum Testen verwenden als ggf. auch simulierte Daten bzw. Computermodelle.
Die grundsätzliche Teststrategie sollten Hersteller bereits im produktspezifischen Entwicklungsplan dokumentieren. Detaillierte Testpläne beschreiben schließlich die genaue Vorgehensweise, um Experimente bzw. Teste durchzuführen.
Schritt 3: Bottom-up-Testen der einzelnen Komponenten
In der Regel überprüfen Hersteller zunächst die einzelnen Bestandteile/Komponenten ihres Produkts und testen, ob diese die spezifizierten Anforderungen erfüllen. Das kann z. B. ein Reagenz eines Kits sein, dessen tatsächliche Enzymaktivität gemessen wird, oder Unit-Tests bei einer IVD-Software.
Mit den Komponenten- bzw. Unit-Tests stellen die IVD-Hersteller sicher, dass die einzelnen Bestandteile ihres Produkts funktionieren wie beabsichtigt. Sollten den Testern Fehler in einer Produktkomponente auffallen, können die Entwickler diese korrigieren.
Je nach Art des IVDs führen Hersteller unterschiedliche Aktivitäten durch:
IVD-Assay
Bei einem IVD-Assay umfassen die Tests zur Komponenten-Verifizierung z. B.:
- Messungen zur Prüfung der Konzentration einer Substanz im Reagenz
- Analysen zur Überprüfung der Länge und Sequenz eines Primers
- Testung adäquater Primer- und Sondenkonzentrationen in einem PCR-Kit
- Optimale Antikörperkonzentrationen in einem ELISA-Kit
IVD-Gerät
Während der Verifizierung von IVD-Geräten wird geprüft:
- Die Funktion einzelner Geräte-Komponenten und ihr Zusammenspiel
- Elektromechanische Sicherheit gemäß EN 61010-2-101
- Elektromagnetische Verträglichkeit gemäß EN 61326-2-6
- Steuerungssoftware u. v. m.
IVD-Software
Die Verifizierung von IVD-Software – also das Software-Testing – umfasst u. a.:
- Unit-Tests
- Intergrationstests
Die Validierung der ggf. zum Testen verwendeten Tools unterliegt der Computer System Validation (CSV).
Lesen Sie mehr zur Anwendung von künstlicher Intelligenz bei Medizinprodukten und den speziellen Anforderungen an AI-basierte Medizinprodukte-Software sowie die Validierung von Machine Learning Libraries.
Schritt 4: Verifizierung des Produkts – Systemtests
Erst wenn die einzelnen Komponenten eines Produkts ihre spezifizierten Anforderungen erfüllen, sollten Hersteller das gesamte Produkt prüfen – also das finale IVD-Kit, das Analysegerät bzw. die Standalone-Software. Bei IVD-Software spricht man von Systemtests. Die Tester prüfen das Zusammenspiel der einzelnen Produkt- und Softwarekomponenten im Endprodukt.
Die Verifizierung des gesamten Produkts umfasst u. a. die analytische Leistungsbewertung, um die Analyseleistung des IVDs zu bestätigen (s. IVDR, Anhang I, Abschnitt 9.1.a). Dabei verifizieren Hersteller stets das fertig entwickelte Endprodukt, um beispielsweise folgende Fragen zu beantworten:
- Wie konstant sind die Ergebnisse bei konstanten Bedingungen (Wiederholbarkeit)?
- Beeinflussen die Umgebungsbedingungen die Ergebnisse (Reproduzierbarkeit)?
- Wie genau können Extremwerte bestimmt werden (Nachweisgrenze, Quantifizierungsgrenze)?
- Welche Probeneigenschaften inklusive pathologischer Veränderungen und Medikamente in der Probe haben welchen Einfluss auf die analytische Spezifität und Sensitivität des IVDs (Interferenzen, Kreuzreaktionen)?
- Unterscheiden sich die Ergebnisse abhängig vom Benutzer (Reproduzierbarkeit)?
- Wie beeinflussen das Alter oder die Art der Lagerung der Reagenzien das Ergebnis (Haltbarkeit, Haltbarkeit nach Anbruch, Transportstabilität)?
- Arbeitet das Produkt sicher und ohne Beeinträchtigung seiner Leistung mit anderen Produkten, Reagenzien, Geräten, Software-Systemen zusammen (Kompatibilität)?
Software-Hersteller sollten zudem ausreichend Tests durchführen, um nachzuweisen, dass die Software die Anforderungen an die IT-Security erfüllt.
Schritt 5: Prüfen der Kompatibilität des IVDs mit anderen Produkten
Eine grundlegende Sicherheits- und Leistungsanforderung gemäß Anhang I der IVDR besagt, dass die Kombination eines IVDs mit anderen Produkten oder Systemen die vorgesehene Leistung nicht beeinträchtigen darf (s. Anhang I, Abschnitt 13.1.).
„Wenn ein Produkt zur Verwendung in Kombination mit anderen Produkten oder Ausrüstungen bestimmt ist, muss die Kombination einschließlich der Verbindungen sicher sein und darf die vorgesehene Leistung der Produkte nicht beeinträchtigen. Jede Einschränkung der Anwendung im Zusammenhang mit solchen Kombinationen wird auf der Kennzeichnung und/oder in der Gebrauchsanweisung angegeben“
IVDR, Anhang I, Abschnitt 13.1.
Kombinationen können z. B. sein:
- Verwendung eines IVD-Assay mit einem Laborgerät
- Kompatibilität eines IVD-Analysesystems mit der Methode zur Probenaufarbeitung
- Nutzung von Rohdaten, die durch verschiedene Geräte erzeugt werden, durch eine IVD-Software
Während der Verifizierungsphase müssen IVD-Hersteller die Kompatibilität und Interoperabilität des IVDs mit anderen Produkten prüfen und belegen.
Definitionen
„die Fähigkeit eines Produkts – einschließlich Software -, bei Verwendung zusammen mit einem oder mehreren anderen Produkten gemäß seiner Zweckbestimmung
- seine Leistung zu erbringen, ohne dass seine bestimmungsgemäße Leistungsfähigkeit verloren geht oder beeinträchtigt wird, und/oder
- integriert zu werden und/oder seine Funktion zu erfüllen, ohne dass eine Veränderung oder Anpassung von Teilen der kombinierten Produkte erforderlich ist, und/oder
- konfliktfrei und ohne Interferenzen oder nachteilige Wirkungen in dieser Kombination verwendet zu werden“
Quelle: IVDR, Artikel 2 (18)
„Fähigkeit von zwei oder mehr Produkten – einschließlich Software – desselben Herstellers oder verschiedener Hersteller,
- Informationen auszutauschen und die ausgetauschten Informationen für die korrekte Ausführung einer konkreten Funktion ohne Änderung des Inhalts der Daten zu nutzen und/oder
- miteinander zu kommunizieren und/oder
- bestimmungsgemäß zusammenzuarbeiten“
Quelle: IVDR, Artikel 2 (19)
Anforderungen der ISO 13485:2016
Ebenso wie die IVDR fordert die ISO 13485:2016 in den Kapiteln 7.3.6 und 7.3.7 explizit die Verifizierung und Validierung des Zusammenspiels eines Produkts mit anderen Systemen bzw. Produkten.
4. In 5 Schritten zur Validierung von IVD
Schritt 1: Zielsetzung festlegen
Die Validierung eines IVDs hat zur Aufgabe, den die Zweckbestimmung (Intended Use) zu überprüfen.
Schritt 2: Validierungstätigkeiten auswählen
Üblicherweise erfolgt die Validierung von IVD anhand von klinischen Leistungsstudien. Während dieser „klinischen Studien“ testen die beabsichtigten Nutzer (je nach Zweckbestimmung z. B. Laborpersonal, Patholog:innen, Humangenetiker:innen) das IVD in seiner tatsächlichen Anwendungsumgebung (z. B. in einem medizinischen Labor). Die Validierung des IVDs beruht auf Proben bzw. Daten (bei Software) von Patienten, für die das IVD gemäß seiner Zweckbestimmung vorgesehen ist.
Falls für ein IVD wissenschaftliche Peer-review-Literatur verfügbar ist, in der die klinischen Leistungsparameter dieses Produkts belegt werden, können Hersteller neben klinischen Leistungsstudien ggf. auch diese Publikationen als Quelle nutzen (s. IVDR, Anhang XIII, Abschnitt 1.2.3.).
Für CE-markierte IVD, die sich bereits auf dem Markt befinden, können Hersteller unter bestimmten Voraussetzungen (z. B. dieselbe Zweckbestimmung, Einverständnis der Patienten, Studienplan, Rohdaten verfügbar) Daten aus der diagnostischen Routineanwendung nutzen, um die klinische Leistung ihres IVDs nachzuweisen (s. IVDR, Anhang XIII, Abschnitt 1.2.3.).
Schritt 3: Die für die Validierung relevanten Fragen bestimmen
Bei der Validierung von IVD sind beispielsweise folgende Fragen zu beantworten, um die klinische Leistung des Produkts nachzuweisen (s. IVDR, Anhang I, Abschnitt 9.1.b)):
- Wie gut kann das IVD tatsächlich erkrankte Personen identifizieren (diagnostische Sensitivität)?
- Wie spezifisch kann das IVD erkennen, dass ein krankheitsassoziierter Analyt nicht vorhanden ist (diagnostische Spezifität)?
- Wie gut unterscheidet das IVD zwischen richtig-positiven und falsch-positiven Ergebnissen in einer Patientenpopulation mit bestimmter Prävalenz der Krankheit (positiv prädiktiver Wert)?
- Wie gut trennt das IVD zwischen richtig-negativen und falsch-negativen Ergebnissen in einer Patientenpopulation mit bestimmter Prävalenz der Krankheit (negativ prädiktiver Wert)?
- Wie wahrscheinlich ist es, dass ein positives Ergebnis bei einer erkrankten Person eintritt im Vergleich zu der Wahrscheinlichkeit, dass das positive Ergebnis bei einer gesunden Person auftritt (positives Likelihood-Verhältnis, vgl. auch negatives Likelihood-Verhältnis)?
Schritt 4: Klinische Leistungsstudien für IVD durchführen
Um die in Schritt 3 aufgeworfenen Fragen zu beantworten, führen die Hersteller u. a. folgende Aktivitäten durch:
- Sie planen die klinische Leistungsstudie, führen eine Fallzahlberechnung durch und erstellen einen klinischen Leistungsstudienplan (Clinical Performance Study Protocol (CPSP)).
- Sie stellen das Analysesystem (z. B. IVD-Assay, (IVD-)Gerät und IVD-Auswertesoftware) unter Routinebedingungen im medizinischen Labor bereit.
- Sie rekrutieren ein hinreichend großes Kollektiv repräsentativer, gut charakterisierter Patientenproben. Bei Validierung einer Standalone-Software erheben sie repräsentative Daten. Es muss eine Einwilligung der Patienten für die Teilnahme an der Leistungsstudie vorliegen.
- Sie vergleichen die Ergebnisse, die sie mit ihrem neuen IVD erhalten, mit einem Referenzstandard bzw. einer Vergleichsmethode. Dabei ist das in Routine befindliche und CE-gekennzeichnete IVD der Benchmark, an dem sich das neue Produkt messen muss. Die Wahl des Referenzstandards müssen die Hersteller begründen. Dabei berücksichtigen sie ihr Produkt und seine Zweckbestimmung sowie den Stand der Technik in der diagnostischen Praxis.
- Soweit anwendbar, lassen sie mit einem hinreichend großen Probenkollektiv Gesunder die Normalwertbereiche für unterschiedliche Parameter bestätigen und legen den produktspezifischen Cutoff-Wert fest.
- Sie führen die Leistungsstudie unter kontrollierten, dokumentierten und reproduzierbaren Bedingungen durch und erfüllen dabei die Anforderungen der IVDR gemäß Anhang XIII, Abschnitt 2 sowie der ISO 2019:2019.
Schritt 5: Gebrauchstauglichkeit prüfen
Die Validierung von IVD muss auch die Gebrauchstauglichkeit umfassen. Die Hersteller müssen sicherzustellen, dass die spezifizierten Nutzer im spezifizierten Nutzungskontext (z. B. medizinisches Labor) mit dem IVD tatsächlich den spezifizierten Zweck – die Analyse von Proben oder die Auswertung von Daten – erreichen können.
Dazu müssen die Hersteller die Gebrauchstauglichkeiit aller sicherheitsrelevanten Nutzungsszenarien im Usablity-Test konform mit der IEC 62366 oder dem FDA Human Factors Engineering prüfen. Diese Nutzungsszenarien umfassen bei einem IVD-Gerät typischerweise die Probenvorbereitung, die Beladung des Geräts mit Proben und Reagenzien, das Erstellen der Arbeitslisten, deren Abarbeitung und Auswertung sowie das Verstehen der Gebrauchsanweisung. Bei einer IVD-Software stehen während der Usability-Validierung die Interaktionen der Nutzer mit dem User Interface im Vordergrund.
5. Typische Fehler bei der Verifizierung und Validierung von IVD vermeiden
a) Unterschätzen des Aufwands
Die Hersteller unterschätzen den Zeitaufwand. Die Verifizierung und Validierung von IVD beanspruchen oft 50 % der Zeit von Entwicklungsbeginn bis Inverkehrbringung. Bei der Rekrutierung geeigneter Proben für eine klinische Leistungsstudie können oft Monate vergehen.
b) Den Gesamtprozess außer Acht lassen
Es ist sowohl bei der Verifizierung als auch bei der Validierung von IVD essenziell, dass Hersteller stets den Kontext der Anwendung ihres IVDs betrachten. Dies umfasst den gesamten Prozess: von der Probenahme über die Probenaufbereitung und Analyse bis hin zur Auswertung und Berichterstellung. Das heißt, die Hersteller müssen stets jede relevante Hardware, Software, Reagenzien und Proben sowie Nachbarsysteme wie Laborinformationssysteme (LIS) bei ihren Experimenten und Tests zum Sicherheits- und Leistungsnachweis berücksichtigen.
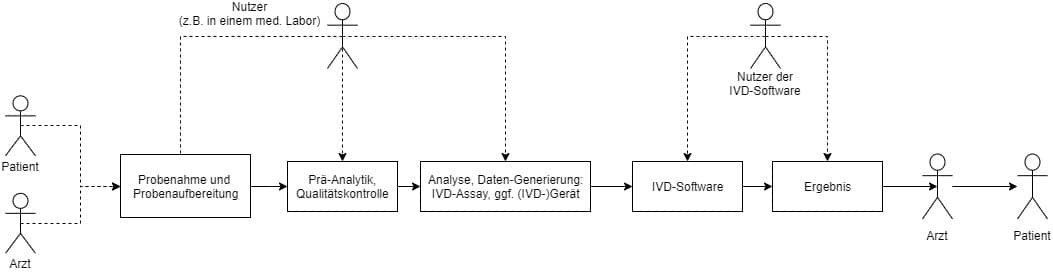
c) Unnötige V&V-Aktivitäten
Die Produktanforderungen (Design Input Requirements, DIR) müssen die Hersteller verifizieren. Viele „Anforderungen“ sind jedoch eher den Business Requirements (BR) zuzurechnen, z. B.:
- Reagenzienverbrauch
- Abfallmengen
- Gewicht
Für die Zulassung müssen die Hersteller „nur“ die Umsetzung der DIR nachweisen. Eine Trennung von DIR und BR hilft, den Verifizierungsaufwand zu minimieren.
Zudem passiert es häufig, dass Hersteller Verifizierung und Validierung nicht präzise trennen und daher Prüfungen doppelt durchführen.
d) Fehlende Verifizierungs- und Validierungsaktivitäten
Die Hersteller berücksichtigen bei der Verifizierung und Validierung der IVD nicht die Gebrauchstauglichkeit und die Kompatibilität mit anderer Hardware, Software und Reagenzien oder die Integration und Interoperabilität mit anderen Systemen.
Es passiert leider auch, dass aufgrund unvollständiger Prüfungen mangelhafte Leistung, Sicherheitslücken und Fehler eines IVDs unentdeckt bleiben und fehlerhafte Produkte in Verkehr gebracht werden. Wie dieser Bericht in der ZEIT zeigt, birgt das enorme Risiken für Patienten und gefährdet Menschenleben.
e) Fehlende oder unvollständige Spezifikationen sowie fehlende Traceability
Die Spezifikationen und die Zweckbestimmung sollten Toleranzgrenzen festlegen, die ggf. von anderen Parametern abhängig sind. Andernfalls ist der Hersteller bei Abweichungen vom festgelegten Wert in Erklärungsnot.
Hersteller sollten nicht vergessen, den Beweis zu führen, dass alle Anforderungen erfüllt wurden. Dies geschieht üblicherweise mit Hilfe einer Traceability-Matrix, die die Ergebnisse der Tests mit den damit nachgewiesenen Anforderungen verknüpft.
f) Falscher Zeitpunkt der „offiziellen“ Verifizierung und Validierung
Die Hersteller müssen die „endgültigen“ Produkte verifizieren und validieren. Eine Änderung des Designs kann bereits durchgeführte Verifizierungs- und Validierungsergebnisse obsolet machen. Das erhöht den Aufwand unnötig.
Ein zu später Beginn der V&V-Aktivitäten birgt das Risiko, dass Probleme beim Design zu lange unentdeckt bleiben. Daher sind frühe „Vortestungen“ und eine iterative Vorgehensweise während der Entwicklung hilfreich.
6. Fazit
Gut spezifizierte Produktanforderungen bzw. Software-Requirements stellen die Grundlage für die nötigen Verifizierungs- und Validierungstätigkeiten für ein IVD dar. Hersteller sollten die Rückverfolgbarkeit zwischen Anforderung und dazugehörigem Verifizierungstest bzw. Validierungsstudie sicherstellen, um nachzuweisen, dass alle Produktanforderungen erfüllt sind.
Wenn Hersteller ihre Strategie zur Verifizierung und Validierung frühzeitig planen, können sie Abhängigkeiten zwischen den Tests, aber auch Synergien identifizieren. Sie sparen Zeit, Geld und Ressourcen, weil sie doppelte Arbeit und unnötige Wiederholungen vermeiden.
Durch eine systematische Vorgehensweise zur Verifizierung und Validierung von IVD sorgen Hersteller dafür, ihren Kunden stets sichere, leistungsstarke und qualitativ hochwertige Produkte zur Verfügung zu stellen. Dies erspart den Anwendern viel Ärger und rettet Menschenleben.
Nemen Sie mit uns Kontakt auf, wenn Sie zielgerichtet und ohne unnötigen Aufwand die Sicherheit und Leistung Ihres IVDs nachweisen wollen.
Gern unterstützen wir – das IVD-Team des Johner Instituts – Sie bei der Planung und normenkonformen Dokumentation Ihrer Verifizierungsaktivitäten und bei der Validierung Ihrer IVD. Wir helfen Ihnen auch bei der Spezifizierung der für Ihre Produkte relevanten Sicherheits- und Leistungsanforderungen.
Sollten Sie Fragen zur Leistungsbewertung von IVD oder zur Durchführung von Usability-Studien haben, wenden Sie sich an unsere Fachexperten – sie helfen Ihnen gerne weiter.
Änderungshistorie
- 2021-04: Artikel vollständig überarbeitet.


Benötige ich zur Produktion eines qPCR Schnelltests eines ISO 13485?
mfg
Ulli Cremers
Lieber Herr Cremers,
Wenn der qPCR-Schnelltest eine in vitro diagnostische Zweckbestimmung hat, sollten Sie die Vorgaben der ISO 13485 für die Entwicklung und Herstellung des Tests einhalten. Die ISO 13485 ist der harmonisierte Standard für die Einhaltung der grundlegenden Anforderungen der EU-IVD-Richtlinie 98/79/EG. Wenn Ihre Frage zudem impliziert, ob Sie eine ISO 13485-Zertifizierung benötigen, müsste ich noch wissen, was die genaue Zweckbestimmung des Tests ist. nutzen Sie dafür gerne unser Microconsulting: https://www.johner-institut.de/micro-consulting/
Beste Grüße, Sebastian Grömminger