
Die FDA stellt klare Anforderungen an das Human Factors Engineering (HFE) bzw. Usability Engineering von Medizinprodukten.
- Für welche Produkte gelten diese Anforderungen?
- Welche regulatorischen Dokumente müssen Sie kennen?
- Wie setzen Sie die FDA-Vorgaben an das Human Factors Engineering praktisch um?
Dieser Artikel gibt Ihnen einen vollständigen Überblick über die FDA-Anforderungen zur Gebrauchstauglichkeit und zeigt, wie Sie diese erfolgreich in Ihrer Produktentwicklung umsetzen.
1. Einführung
1.1 Wann müssen Sie die FDA-Anforderungen an Human Factors erfüllen?
Die FDA-Vorgaben zum Human Factors Engineering gelten nicht pauschal für alle Medizinprodukte. Sie hängen von verschiedenen Faktoren ab:
1.1.1 Produktklassifizierung und Software
Grundsätzlich müssen alle Produkte die Anforderungen an die Gebrauchstauglichkeit erfüllen, d. h., es muss immer ein Usability Engineering File (UEF) bzw. Human Factors Engineering Report (HFE-Report) vorhanden sein. Das bedeutet aber nicht, dass auch eine Human Factors Evaluation (also Studien) notwendig ist.
Die Aussage, dass das Usability Engineering File für Class-I-Produkte nicht notwendig ist, ist somit nicht korrekt. Vielmehr würde im UEF mit dem niedrigen Risiko begründet, dass keine Human Factors Validation (summative Evaluation) notwendig ist.
Die FDA leitet die HFE-Anforderungen aus den Quality System Regulations (QSR) ab, insbesondere aus:
- Design Input: „Address the intended use of the device, including the needs of the user and patient“
- Design Validation: „Ensure that devices conform to defined user needs and intended uses“
1.1.2 Risiko durch Use Errors
Die FDA fordert das Human Factors Engineering explizit, wenn die Risikoanalyse zeigt, dass Anwendungsfehler zu schwerwiegenden Schäden (serious harm) führen können.
Serious Harm: Die FDA versteht darunter Tod, schwere Verletzungen oder schwerwiegende unerwünschte Ereignisse, die aus der Nutzung oder Fehlnutzung des Medizinprodukts resultieren können („Includes both serious injury and death“).
Serious injury: An injury or illness that is life-threatening, results in permanent impairment of a body function or permanent damage to a body structure, or necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. Permanent means irreversible impairment or damage to a body structure or function, excluding trivial impairment or damage.
In diesem Fall müssen Human-Factors-Daten in der Premarket Submission (PMA, 510(k)) eingereicht werden.
1.1.3 Spezifische Produktkategorien
Für bestimmte Hochrisiko-Produkte ist das HFE obligatorisch. Die FDA hat eine Liste von Produkten veröffentlicht, bei denen sie Human Factors als besonders kritisch einstuft:
- Beatmungsgeräte
- Dialysegeräte
- Infusionspumpen
- Defibrillatoren
- Blutglukose-Messgeräte
- sowie weitere Produkte
Die FDA hat die „List of highest priority devices“ entfernt, die sich im FDA-Guidance-Dokument„List of Highest Priority Devices for Human Factors Review“ befand.
Dieses Dokument wurde durch die neue Guidance „Content of Human Factors Information in Medical Device Submissions“ ersetzt.
1.1.4 FDA-Ermessen
Die Behörde kann HFE-/UE-Daten auch in folgenden Situationen anfordern:
- bei Produktänderungen
- nach Meldung von Problemen
- bei bestimmten Zulassungsverfahren (insbesondere PMA)
- bei Änderung der Nutzergruppe
Bei Unsicherheiten empfiehlt die FDA eine Presubmission (Q-Submission), um vorab zu klären, welche HFE-Aktivitäten für Ihr spezifisches Produkt erforderlich sind. Dies kann Zeit und Kosten bei der späteren Zulassung sparen.
1.2 Regulatorischer Rahmen: Diese Dokumente müssen Sie kennen
Die FDA-Anforderungen an das Human Factors / Usability Engineering basieren auf einem umfassenden regulatorischen Framework:
1.2.1 Verbindliche Regularien
- 21 CFR part 820.30 (Design Controls): Definiert grundlegende Anforderungen an Design Input und Design Validation
- Quality System Regulations (QSR): Übergeordnete Qualitätssystem-Anforderungen
Der 21 CFR part 820, insbesondere part 820.30, wird im Rahmen der Umstellung auf die Quality System Management Regulations durch die ISO 13485 abgelöst.
1.2.2 Anerkannte Standards
- IEC 62366-1:2015: Internationale Norm zum Usability Engineering Process
- AAMI/ANSI HE75:2009: Human Factors Engineering – Design of Medical Devices
- ISO 14971:2019: Risikomanagement
Die 2026-er Guidance „Content of Human Factors Information in Medical Device Submissions“ verweist auf „ANSI/AAMI/ISO 14971:2019 Medical devices—Application of risk management to medical devices“. Bei der Dokumentation sollten Sie dies berücksichtigen und ggf. Unterschiede zur aktuellen Version dokumentieren.
1.2.3 Guidance Documents der FDA
Die FDA hat zwei zentrale Leitdokumente zum Human Factors Engineering veröffentlicht:
1. Applying Human Factors and Usability Engineering to Medical Devices (Februar 2016)
- Beschreibt den HFE-Prozess
- Definiert Methoden und Aktivitäten
- Legt Validierungsanforderungen fest
2. Content of Human Factors Information in Medical Device Submissions (Mai 2026)
- Spezifiziert einzureichende Dokumente
- Führt risikobasierte Kategorisierung ein
Das Guidance Document soll Herstellern helfen, auf Basis eines risikobasierten Ansatzes zu entscheiden, welche Human-Factors-Informationen sie in ihre Zulassungsanträge aufnehmen sollten. Zudem gibt es eine empfohlene Struktur für den Human-Factors-Bericht vor, einschließlich Beispiele und Vorlagen in den Anhängen.
1.3 Human Factors vs. Usability Engineering: Begriffsklärung
Die FDA definiert HFE als „Application of knowledge about human behavior, abilities, limitations, and other characteristics to the design of medical devices (including software), systems and tasks to achieve adequate usability.“
Die FDA verwendet die Begriffe Human Factors Engineering und Usability Engineering weitgehend synonym:
- Human Factors Engineering (HFE): Traditioneller FDA-Begriff, betont die Sicherheitsaspekte
- Usability Engineering: International gebräuchlicher Begriff (IEC 62366-1)
In Ihrer Dokumentation können Sie beide Begriffe verwenden. Wichtig ist, dass Sie konsistent bleiben und bei der ersten Verwendung klarstellen, dass Sie die Begriffe synonym verwenden.
Die FDA spricht inzwischen selbst durchgängig von HFE/UE, z. B. dem „HFE/UE report“.
Kernpunkt: Bei beiden Ansätzen geht es primär um Risikominimierung durch gebrauchstaugliche Gestaltung, nicht um Marktattraktivität oder Kundenzufriedenheit.
2. Der HFE-/UE-Prozess nach FDA
Die FDA beschreibt einen systematischen Prozess für das Human Factors Engineering, der sich stark am Risikomanagement orientiert. Dieser Prozess stellt sicher, dass Nutzungsrisiken frühzeitig erkannt und minimiert werden.
2.1 Überblick über den HFE-/UE-Prozess
Der Human-Factors-/Usability-Engineering-Prozess der FDA folgt einem iterativen Ansatz, der parallel zur Produktentwicklung läuft. Er orientiert sich am etablierten Risikomanagement nach ISO 14971, fokussiert aber speziell auf nutzungsbedingte Risiken.
Im Gegensatz zur IEC 62366-1 beschreibt die FDA weniger den Prozess selbst, sondern die konkreten Aktivitäten und Methoden.
Die vier Hauptphasen des Prozesses sind:
- User Research (Nutzer, Nutzungsumgebung und User Interface)
- Risikoanalyse und Identifikation kritischer Aufgaben
- Formative Evaluation während der Entwicklung & Risikominimierung
- Validierung der Gebrauchstauglichkeit / Human Factors Validation Testing
2.2 Phase 1: User Research
Die FDA fordert, alle relevanten Merkmale der Nutzer, Nutzungskontexte und der Benutzeroberfläche systematisch zu erfassen und zu dokumentieren.
- Nutzer: Berücksichtigt werden müssen alle intendierten Nutzergruppen wie Fachpersonal (z. B. Ärztinnen und Ärzte, Pflegekräfte), technisches Personal (z. B. für Wartung, Installation, Reinigung) sowie Laien, Patient:innen und deren Angehörige. Entscheidend sind Eigenschaften wie physische und sensorische Fähigkeiten, kognitive Voraussetzungen, Erfahrungsstand, Sprach- und Lesekompetenz sowie Motivation und Schulungsgrad.
- Nutzungskontexte: Die Analyse umfasst alle Einsatzorte des Produkts (z. B. Klinik, OP, häusliche Umgebung, Notfalleinsatz) und deren Bedingungen. Typische Einflussfaktoren sind Licht- und Lärmverhältnisse, räumliche Gegebenheiten, mögliche Ablenkungen oder Notfallsituationen.
- Benutzeroberfläche: Alle Elemente der Interaktion müssen betrachtet und beschrieben werden – vom physischen Gerätedesign und Bedienelementen über Displays, Alarme und Softwareoberflächen bis hin zu Gebrauchsanweisungen und Training. Entscheidend ist, dass die Bedienlogik den Erwartungen und Fähigkeiten der Nutzer entspricht und Fehler aktiv verhindert werden.
Nutzen Sie Methoden wie Interviews, Beobachtungen und Fokusgruppen, um ein realistisches Bild der Nutzer und ihrer Arbeitsumgebung zu erhalten. Nur so können nutzungsbedingte Risiken frühzeitig identifiziert und adressiert werden.
Die Berücksichtigung der Nutzer, der Nutzungskontexte und der Benutzeroberfläche bildet die Basis für ein gebrauchstaugliches und sicheres Medizinproduktdesign. Die Analyse der Nutzer und der Nutzungsumgebungen zieht sich dabei als roter Faden durch die gesamte Risikoanalyse: Sie bildet die Grundlage zur Identifikation von kritischen Aufgaben (Critical Tasks) und muss eng mit der Risikoanalyse verknüpft werden. Nur so ist gewährleistet, dass für jede kritische Aufgabe die jeweils relevanten Nutzergruppen definiert werden können. Dies ist essenziell, um gezielt die richtigen Probanden für die spätere Human Factors Validierung auf Basis der Critical Tasks auszuwählen.
Die sorgfältige Dokumentation des User Research ist nicht nur für die spätere Risikoanalyse und Validierung unerlässlich, sondern wird von der FDA explizit in der Submission gefordert. Dabei müssen die Variabilität und die potenziellen Einschränkungen aller Nutzergruppen sowie die realen Umgebungsbedingungen im gesamten Entwicklungsprozess berücksichtigt werden.
2.3 Phase 2: Risikoanalyse und Identifikation kritischer Aufgaben
2.3.1 Fokus auf sicherheitsrelevante Aufgaben
Die FDA fordert zunächst die Identifikation aller sicherheitsrelevanten Aufgaben (Tasks) am System. Dieser aufgabenorientierte Ansatz deckt sich mit dem Konzept der IEC 62366-1.
Die FDA definiert eine kritische Aufgabe als „A user task which, if performed incorrectly or not performed at all, would or could cause serious harm to the patient or user, where harm is defined to include compromised medical care“, bei der ein Anwendungsfehler zu ernsthaften Schäden führen könnte.
2.3.2 Methoden zur Risikoanalyse
Die FDA nennt konkrete Methoden zur Identifikation und Analyse von Nutzungsrisiken:
- FMEA (Failure Mode and Effects Analysis): Systematische Analyse möglicher Fehler und deren Auswirkungen
- FTA (Fault Tree Analysis): Top-Down-Analyse von unerwünschten Ereignissen
- Aufgabenanalyse (Task Analysis): Zerlegung von Arbeitsabläufen in Einzelschritte
- Experten-Review: Bewertung durch Human-Factors-Spezialisten
- Kontextanalyse: Untersuchung der realen Nutzungsumgebung
- Interviews und Fokusgruppen: Direkte Befragung von Anwendern
Kombinieren Sie mehrere Methoden für eine umfassende Risikoanalyse. Die FDA empfiehlt besonders die Aufgabenanalyse als Basis, ergänzt durch FMEA für die systematische Fehlerbetrachtung.
2.3.3 Zu berücksichtigende Informationsquellen
Die FDA fordert explizit, folgende Quellen in die Risikoanalyse einzubeziehen:
- Rückmeldungen von Kunden und Anwendern
- Berichte vom Service, vom Verkaufspersonal und von Trainern
- Ergebnisse vorausgegangener HFE-Studien (auch von Vorgängerprodukten)
- Wissenschaftliche Publikationen und Fachartikel
- Behördenmeldungen (FDA MAUDE-Datenbank, BfArM-Datenbank etc.)
Die MAUDE-Datenbank der FDA (Manufacturer and User Facility Device Experience) enthält Berichte über unerwünschte Ereignisse mit Medizinprodukten und ist öffentlich zugänglich. Sie ist eine wertvolle Quelle für die Identifikation bekannter Nutzungsprobleme.
2.4 Phase 3: Formative Evaluation & Risikominimierung
2.4.1 Ziele der formativen Evaluation
Die FDA fordert formative Evaluationen während der Entwicklung mit folgenden Zielen:
- Ideen für Testszenarien der späteren Validierung generieren
- Gefährliche Situationen frühzeitig identifizieren
- Input für Verbesserungen der Benutzeroberfläche sammeln
- Besseres Verständnis möglicher Anwendungsfehler entwickeln
Die explizite Forderung nach formativen Evaluationen während des Designprozesses geht über die Anforderungen der IEC 62366-1 hinaus. Die FDA erwartet, dass Sie diese Aktivitäten dokumentieren und in Ihrer Submission zusammenfassen.
2.4.2 Methoden der formativen Evaluation
Die FDA nennt zwei Hauptmethoden:
- Durchführung mit repräsentativen Nutzern (nicht nur Experten)
- Systematisches Durchgehen von Aufgaben
- Identifikation von Verständnisproblemen und Fehlerquellen
Simulated Use Testing
- Tests mit Prototypen oder Mockups
- Simulation realistischer Nutzungsszenarien
- Beobachtung und Befragung der Testpersonen
Beginnen Sie mit einfachen Papier-Prototypen oder Mock-ups. Die FDA erlaubt ausdrücklich Tests an nichtfinalen Versionen. So können Sie früh und kostengünstig Erkenntnisse gewinnen.
2.4.3 Risikokontrollmaßnahmen
Während der formativen Evaluation identifizierte (unvertretbare) Risiken müssen die Hersteller minimieren. Die FDA folgt der etablierten Hierarchie der Risikokontrolle:
- Inhärent sicheres Design: Eliminierung oder Reduktion von Risiken durch das Design selbst
- Schutzmaßnahmen: Alarme, Warnungen, Sicherheitsmechanismen bei Produkt oder in Produktion
- Hinweise für die Anwendung: Gebrauchsanweisung, Labels, Training
Die FDA akzeptiert Risikominimierung durch Hinweise in der Gebrauchsanweisung nur, wenn Sie deren Wirksamkeit nachweisen können. Gleiches gilt für das „Additional Training“.
Konkrete Beispiele für Designmaßnahmen:
- Vereinfachung von Arbeitsabläufen
- Eindeutige Kennzeichnung und Beschriftung
- Fehlertolerante Gestaltung (z. B. Rückgängig-Funktionen)
- Physische Barrieren gegen Fehlbedienung
- Konsistente Benutzerführung
2.5 Phase 4: Human Factors Validation Testing
2.5.1 Anforderungen an die Validierung
Die FDA stellt konkrete Anforderungen an die Human Factors Validation:
Praktische Definition: Die summative Bewertung der Gebrauchstauglichkeit mit dem finalen Produkt unter realistischen Bedingungen. Die IEC 62366-1 nennt dies Summative Evaluation.
Offizielle Definition der FDA: Testing conducted at the end of the device development process to assess user interactions with a device user interface to identify use errors that would or could result in serious harm to the patient or user. Human factors validation testing is also used to assess the effectiveness of risk management measures. Human factors validation testing represents one portion of design validation.
Kernforderungen:
- Alle kritischen Aufgaben müssen validiert werden.
- Test mit vorher definierten Erfolgskriterien
- Nachweis, dass keine ernsten Nutzungsprobleme auftreten
- Erhebung objektiver Daten (Beobachtung) und subjektiver Daten (Befragung)
- Überprüfung der Effektivität („For the device to be considered to be optimized with respect to use safety and effectiveness“).
2.5.2 Testbedingungen und Teilnehmende
Produkt
- Test mit dem endgültigen Produkt
- einschließlich finalem Labeling (Gebrauchsanweisung, Beschriftungen)
- in der finalen Konfiguration
Teilnehmende
- Repräsentative Nutzer aus allen relevanten Nutzergruppen
- Keine Mitarbeitenden des Herstellers
- Mindestens 15 Teilnehmende pro Nutzergruppe
- Nutzer müssen aus den USA stammen: Professionelle Nutzer müssen im US-amerikanischen Gesundheitssystem arbeiten, daher muss die Evaluation meist in den USA erfolgen. Es reicht nicht, US-Bürger in Deutschland zu rekrutieren, die nicht (mehr) im US-Gesundheitssystem tätig sind. Auch bei Laien sollte der kulturelle Hintergrund berücksichtigt werden.
Planen Sie frühzeitig die Rekrutierung von US-Nutzern ein. Das Johner Institut kann Sie bei der Rekrutierung der Probandinnen und Probanden sowie bei der Organisation und Durchführung von Usability-Tests mit US-amerikanischen Anwendenden unterstützen.
Testumgebung:
- Repräsentative Nutzungsumgebung
- Realistische Bedingungen (Licht, Lärm, Ablenkungen)
- Training der Teilnehmenden entsprechend dem realen Szenario
2.5.3 Dokumentation der Validierung
Die Validierung muss umfassend dokumentiert werden:
- Testprotokoll mit allen Aufgaben
- Erfolgskriterien und deren Begründung
- Beobachtungsprotokolle
- Auswertung aller Nutzungsprobleme
- Bewertung von Restrisiken
- Begründung, warum verbleibende Probleme akzeptabel sind
Die FDA erwartet eine detaillierte Diskussion aller beobachteten Nutzungsprobleme, auch wenn diese nicht zu Fehlern führten. Dies hilft der Behörde, die Gründlichkeit Ihrer Evaluation zu beurteilen.
3. FDA Human Factors Submission Requirements
Die FDA macht im Guidance Content of Human Factors Information in Medical Device Submissions, das im Mai 2026 veröffentlicht wurde, klare Vorgaben, welche Human-Factors-Dokumente bei der Zulassung einzureichen sind. Der Umfang hängt dabei vom Risiko des Produkts ab.
Während die Entwurfsversion dieses Guidance-Dokuments auf einen Draft Guidance zu „Combination Products“ verwies, sind die Kombinationsprodukte in der finalen Version explizit aus dem Geltungsbereich ausgeschlossen.
3.1 Die drei Dokumentationskategorien
3.1.1 Übersicht / Entscheidungsbaum
Die FDA unterscheidet drei Kategorien der Dokumentation von Human Factors bzw. Usability Engineering, basierend auf dem Risiko des Produkts bzw. der Produktänderung:
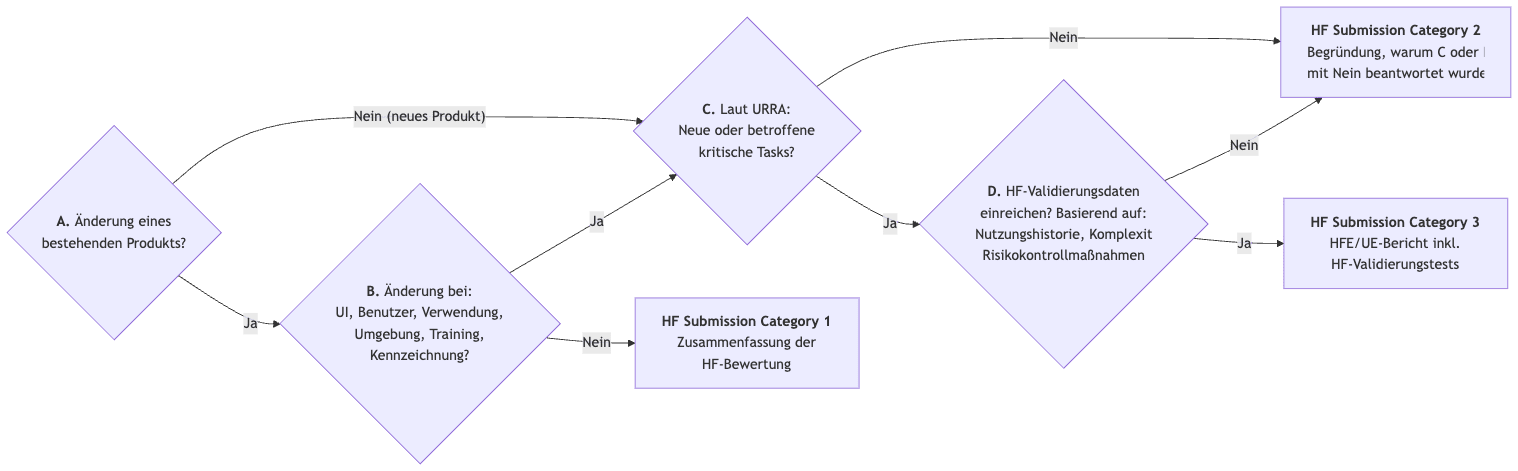
3.1.2 Neuer Entscheidungspunkt D
Neu und zu einer Vereinfachung beitragend ist der „Decision Point D“. Bei ausreichend verfügbaren Daten kann auch bei neuen Produkten oder Produkten mit Änderungen, die zu neuen oder geänderten kritischen „Tasks“ führen, eine Einreichung gemäß Kategorie 2 ausreichen.
Die FDA gibt dazu Beispiele. So verzichtet die Behörde selbst bei neuen Produkten mit kritischen „Tasks“ auf die Einreichung von Validierungsdaten:
- Example C.1: Ein „balloon catheter deliver system“ würde keine neuartigen technischen Features enthalten und die Nutzer seien im Umgang mit solchen Produkten geübt.
- Example C.4: Bei einer Blutlanzette sieht die FDA das User Interface „of low complexity“ und ähnlich mit anderen Produkten mit gleicher Zweckbestimmung. Auch würde sich die Zweckbestimmung nicht von diesen anderen Produkten unterscheiden.
Hingegen würde die FDA beim „Decision Point D“ bei anderen Konstellationen zur Entscheidung kommen, dass umfangreiche Validierungsdaten eingereicht werden müssen:
- Example C.3: Bei einem Injektionsgerät, bestimmt zur Aspiration und Injektion, für das es kein Predicate Device gibt und das „unique design features“ hat, geht die FDA von einem anderen Risikoprofil aus.
- Example C.6: Auch bei einem neuartigen geführten Ultraschall-Gerät für medizinisches Fachpersonal und „non-expert users“ geht die FDA nicht davon aus, dass die Anwender über die notwendigen Erfahrungen verfügen, was ebenfalls die Einreichung von Usability-Validierungsdaten bedingt.
Die FDA schreibt explizit, dass Hersteller das Gerätedesign verifizieren und validieren sowie Human-Factors-Informationen im QMS dokumentieren sollen (21 CFR 820, ISO 13485), unabhängig davon, ob diese Informationen eingereicht werden müssen.
3.2 Umfang der einzureichenden Dokumente
3.2.1 Übersicht der Anforderungen nach Kategorie
| Dokumente | Kategorie 1 | Kategorie 2 | Kategorie 3 |
|---|---|---|---|
| HF-Zusammenfassung | ✓ | ✓ | ✓ |
| Beschreibung der Zweckbestimmung *) | – | ✓ | ✓ |
| Beschreibung des User Interface | – | ✓ | ✓ |
| Bekannte Nutzungsprobleme | – | ✓ | ✓ |
| Formative Evaluationen | – | – | ✓ |
| Risikoanalyse (HF-Fokus) | – | – | ✓ |
| Validierungsdetails | – | – | ✓ |
*) inkl. „intended device users“, „uses“, „use environments“ und „training“
3.2.2 Kategorie 1: HFE/UE-Zusammenfassung
Für Kategorie 1 genügen eine kurze Zusammenfassung und eine Erklärung, dass es keine Änderungen am UI, der Zweckbestimmung, dem Training oder Labeling gab bzw. dass es ein Software-Bug-Fix im Backend war.
3.2.3 Kategorie 2: Moderate Dokumentation
Zusätzlich zur Zusammenfassung sind einzureichen:
Beschreibung der Zweckbestimmung
- Intended User (Nutzergruppen und deren Charakteristika)
- Intended Use Environment (Nutzungsumgebung)
- Training-Anforderungen
Beschreibung des User Interface
- Überblick über die Benutzerschnittstelle
- Hauptfunktionen und Bedienkonzept
- Screenshots oder Fotos der wichtigsten Screens/Bedienelemente
Zusammenfassung bekannter Nutzungsprobleme
Diese Informationen können auch stammen aus:
- Post-Market-Daten von Vorgängerprodukten
- Literaturrecherche zu ähnlichen Produkten
- Erkenntnisse aus Kundenbeschwerden
Wichtig ist, dass die Risikoanalyse zeigt, dass die Änderungen keine kritischen Aufgaben betreffen oder neue entstehen. Diese Stellungnahme sollte bei der Kategorisierung angegeben werden.
3.2.4 Kategorie 3: Vollständige HF-Dokumentation
Für Hochrisikoprodukte fordert die FDA das komplette HF-Portfolio, also zusätzlich zu den bereits genannten Dokumenten:
Zusammenfassung der formativen Evaluationen
- Durchgeführte formative Tests
- Wichtigste Erkenntnisse
- Resultierende Designänderungen
Risikoanalyse mit HF-Fokus:
- Use-related Risk Analysis (URRA)
- Identifikation und Beschreibung der Critical Tasks
- Risikokontrollmaßnahmen
„Systematic use of available information to identify use related hazards and to estimate the use-related risk“
Der Abnormal Use (absichtliche Fehlanwendung) ist out of scope, aber ggf. Teil des vorhersehbaren Missbrauchs.
Details der HF-Validierung
- vollständiges Testprotokoll
- Teilnehmenden-Demografie
- Testergebnisse für alle kritischen Aufgaben
- Analyse und Bewertung aller Nutzungsprobleme
- Diskussion von Restrisiken
3.3 Struktur des HFE/UE Reports
3.3.1 Empfohlener Aufbau nach FDA
Die FDA empfiehlt für den Human Factors Engineering Report die folgende Struktur :
- Conclusion and high-level summary
- Aussage, dass Produkt sicher ist
- Zusammenfassung der Ergebnisse, insbesondere der Risiken
- Duchgeführte Aktivitäten
- Beschreibung der Nutzer, Nutzungsumgebung und Training
- Zusammenfassung der Zweckbestimmung
- Nutzerprofile
- Nutzungsumgebung mit kritischen Faktoren
- Trainingskonzept und -materialien
- Beschreibung der Nutzungsschnittstelle
- Grafische Darstellung, z. B. Fotos oder Zeichnungen des Produkts
- Beschreibung der Interaktion von Nutzern mit dem Produkt (z. B. Abfolgen von Tätigkeiten)
- Begleitinformationen
- Ggf. Beschreibung der Änderungen
- Zusammenfassung bekannter Nutzungsprobleme
- Scope: Vorgängerprodukt, ähnliches Produkt
- Relevanz für das aktuelle Produkt
- Falls möglich: Aussage, dass keine Nutzungsprobleme bekannt sind
- Beschreibung der Analyse, z. B. formative Bewertung
- Methodenbeschreibung
- Wichtige Ergebnisse
- Folgende Designentscheidungen
- Risikoanalyse
- Mögliche Use Errors
- Gefährdungen
- Risiken und mögliche Schäden
- Beschreibung der kritischen Aufgaben
- Detaillierte Task-Beschreibungen
- Zugehörige Use Scenarios
- Änderungen am User Interface
- Details der HFE/UE-Validierung
- Studiendesign und Methodik
- Durchführung
- Ergebnisse
- Diskussion und Schlussfolgerungen
Nutzen Sie diese Struktur als Vorlage für Ihren Report. Die FDA-Reviewer erwarten diese Gliederung und können so effizient die benötigten Informationen finden.
3.3.2 Besondere Anforderungen an die Dokumentation
Klarheit über Critical Tasks: Die FDA wünscht eine eindeutige Aussage zum Vorhandensein oder Fehlen von Critical Tasks. Falls keine identifiziert wurden, muss dies klar begründet werden.
Restrisiko-Bewertung: Jedes verbleibende Nutzungsrisiko muss diskutiert und bewertet werden:
- Warum kann das Risiko nicht weiter reduziert werden?
- Warum ist das Risiko akzeptabel?
- Welche Post-Market-Maßnahmen sind geplant?
Nachvollziehbarkeit: Die Dokumentation sollte so gestaltet sein, dass ein FDA-Reviewer ohne Rückfragen verstehen kann,
- warum bestimmte Designentscheidungen getroffen wurden,
- wie Risiken identifiziert und kontrolliert wurden,
- warum das finale Design sicher und effektiv ist.
Inzwischen besteht die FDA nicht mehr auf einem konsolidierten HF-Report, der alle relevanten Informationen enthält. Sie erlaubt explizit Verweise auf andere Dokumente.
4. Praktische Umsetzung
Die erfolgreiche Umsetzung der FDA-Anforderungen erfordert einen strukturierten Prozess, der sowohl FDA- als auch IEC-62366-1-konform ist. Dieses Kapitel zeigt Ihnen, wie Sie die Anforderungen praktisch umsetzen.
4.1 Ein integrierter HFE-/UE-Prozess
4.1.1 Prozessvorschlag für FDA- und IEC-konforme Entwicklung
Der folgende Prozess erfüllt sowohl die FDA-Anforderungen als auch die der IEC 62366-1.

Dieser Prozess integriert die FDA-Phasen (User Research, Risikoanalyse, Formative Evaluation, Validierung) mit dem Usability Engineering Process der IEC 62366-1. So vermeiden Sie doppelte Arbeit und erfüllen beide Regularien gleichzeitig.
4.1.2 Schlüsselaktivitäten im Entwicklungsprozess
Frühe Entwicklungsphase
- User Research und Kontextanalyse
- Erste Risikoanalyse (Use-Related Risk Analysis)
- Definition von Nutzergruppen und Nutzungsumgebung
Konzeptphase
- Identifikation kritischer Aufgaben
- Erste formative Evaluationen (z. B. mit Papierprototypen)
- Iterative Designverbesserungen
Detailentwicklung
- Weitere formative Tests mit funktionsfähigen Prototypen
- Verfeinerung der Risikoanalyse
- Finalisierung der Risikokontrollmaßnahmen
Verifizierung und Validierung
- Summative Evaluation / HF Validation Testing
- Bewertung von Restrisiken
- Finalisierung der HF-Dokumentation
Beginnen Sie die HFE/UE-Aktivitäten nicht erst kurz vor der Zulassung. Die FDA erwartet Nachweise über formative Evaluationen während der gesamten Entwicklung. Späte Änderungen sind teuer und verzögern die Markteinführung.
4.2 Integration der Anforderungen von FDA und IEC 62366-1
4.2.1 Terminologie-Mapping
Die unterschiedliche Terminologie kann verwirrend sein. In Tabelle 1 sind die wichtigsten Entsprechungen gegenübergestellt:
| Begriff der FDA | Begriff in der IEC 62366-1 | Erläuterung |
|---|---|---|
| Human Factors Engineering | Usability Engineering | Gesamtprozess |
| Critical Task | Hazard-Related Use Scenario | Die sicherheitskritische Aufgabe entspricht nicht ganz dem sicherheitsbezogenen Use Scenario. Vielmehr beinhaltet dieses den „critical task“. |
| HF Validation Testing | Summative Evaluation | Abschließende Validierung |
| Use Error | Use Error | Anwendungsfehler (Hinweis beachten) |
| Formative Evaluation | Formative Evaluation | Bewertung während der Entwicklung |
Die FDA definiert Use Error als „User action or lack of action that was different from that expcted by the manufacturer and caused a result that (1) was different from the result exptected by the user and (2) was not caused solely by device failure and (3) did or could result in harm“.
Ohne einen möglichen Schaden ist ein Nutzungsfehler also kein Use Error aus Sicht der FDA.
4.2.2 Dokumentations-Synergie
Ein cleverer Ansatz zur Dokumentation:
Ein Dokument – zwei Regularien
- Erstellen Sie ein HF/Usability Engineering File.
- Strukturieren Sie nach den Anforderungen der IEC 62366-1.
- Ergänzen Sie FDA-spezifische Elemente.
- Erstellen Sie daraus den FDA HF Report.
Die IEC 62366-1 fordert eine Usability-Engineering-Akte mit spezifischen Inhalten. Diese überschneiden sich größtenteils mit den FDA-Anforderungen. Eine geschickte Strukturierung spart erheblichen Dokumentationsaufwand.
4.3 Besonderheiten und praktische Tipps
1. US-Nutzer frühzeitig einplanen
- FDA fordert Tests mit US-amerikanischen Nutzern
- Rekrutierung kann zeitaufwendig sein
- Kulturelle Unterschiede beachten
2. Presubmission nutzen
- Bei bei Unsicherheiten Q-Submission einreichen
- FDA gibt verbindliche Auskunft zu HF-Anforderungen
- Vermeidet teure Überraschungen
3. Realistische Testbedingungen
- Nutzungsumgebung authentisch nachbilden
- Typische Störfaktoren einbeziehen
- Realistisches Training der Testnutzer
Dokumentieren Sie auch „close calls“ (also Beinahe-Fehler) und kleinere Probleme in der Validierung. Die FDA schätzt Transparenz und sieht darin einen Beleg für gründliche Tests.
4.4 Verfügbare Ressourcen und Unterstützung
4.4.1 FDA-Ressourcen
Guidance Documents
- Applying Human Factors and Usability Engineering to Medical Devices
- Content of Human Factors Information in Medical Device Submissions
Datenbanken
- MAUDE-Datenbank für gemeldete Nutzungsprobleme
- FDA-Website mit device-spezifischen Guidances
FDA-Kontakt
- Q-Submission Program für Presubmissions
- Division of Human Factors im CDRH
4.4.2 Praktische Arbeitshilfen
Templates und Vorlagen: Das Johner Institut bietet verschiedene Arbeitshilfen:
- Templates für den HF-Report
- Protokollvorlagen für Validierungstests
Im Auditgarant finden Sie vollständige Dokumentenvorlagen: von den Use Scenarios über den Usability-Validierungs-Plan bis zum HF-Report. Diese sind bereits auf FDA-Konformität optimiert.
Fachbücher
- „Usability als Erfolgsfaktor“ von Geis und Johner – speziell auf FDA und IEC 62366-1 ausgerichtet
- AAMI HE75 als umfassendes Nachschlagewerk
4.4.3 Externe Unterstützung
Wann externe Hilfe sinnvoll ist:
- bei fehlender Erfahrung mit FDA-Submissions
- bei Bedarf an US-Nutzern für Tests
- bei knappen internen Ressourcen
- bei komplexen oder neuartigen Produkten
Das Johner Institut bietet umfassende Usability-Services an, einschließlich:
- Durchführung von Usability-Tests mit US-Nutzern
- Review der HF-Dokumentation
- Unterstützung bei Q-Submissions
- Training zu FDA Human Factors
Die Investition in professionelle Unterstützung zahlt sich oft aus. Eine verzögerte oder abgelehnte FDA-Zulassung kostet meist mehr als externe Beratung.
5. Zusammenfassung und Fazit
Die FDA-Anforderungen an Human Factors Engineering sind umfassend, aber gut strukturiert und nachvollziehbar. Mit dem richtigen Verständnis und einem systematischen Vorgehen lassen sich diese Anforderungen effizient erfüllen.
5.1 Die wichtigsten Erkenntnisse im Überblick
5.1.1 Regulatorische Klarheit
Die FDA hat mit ihren beiden Guidance-Dokumenten klare Vorgaben geschaffen:
- Der HFE-Prozess ist im Guidance „Applying Human Factors and Usability Engineering to Medical Devices“ definiert.
- Die einzureichenden Dokumente sind im Guidance „Content of Human Factors Information in Medical Device Submissions“ spezifiziert.
- Der risikobasierte Ansatz ermöglicht angemessenen Dokumentationsaufwand.
5.1.2 Fokus auf Patientensicherheit
Die FDA-Philosophie ist eindeutig:
- Primäres Ziel: Minimierung von Risiken durch Anwendungsfehler
- Sekundär: Benutzerfreundlichkeit und Markterfolg
- Kernprinzip: Risikobasiertes Vorgehen bei allen Entscheidungen
Diese Priorisierung unterscheidet Medical Human Factors von kommerziellem UX-Design.
5.2 Erfolgsfaktoren für die FDA-Zulassung
5.2.1 Strategische Erfolgsfaktoren
1. Frühzeitiger Start: Human Factors Engineering ist kein „Add-on“ am Ende der Entwicklung, sondern integraler Bestandteil des Designprozesses.
2. Risikobasiertes Denken: Konzentrieren Sie Ihre Ressourcen auf die kritischen Aufgaben. Die FDA honoriert fokussierte Arbeit mehr als umfangreiche, aber oberflächliche Dokumentation.
3. Transparenz und Vollständigkeit: Dokumentieren Sie auch Probleme und deren Lösung. Die FDA schätzt einen offenen Umgang mit Herausforderungen.
Eine gut strukturierte und vollständige HF-Dokumentation kann den Review-Prozess der FDA um Wochen verkürzen. Investieren Sie in Qualität statt in Quantität.
5.2.2 Operative Erfolgsfaktoren
Nutzen Sie die FDA-Ressourcen:
- Q-Submission für verbindliche Vorabklärungen
- MAUDE-Datenbank für bekannte Probleme
- Guidance-Dokumente als konkrete Anleitung
Planen Sie US-spezifische Anforderungen ein:
- US-Nutzer für Validierung
- US-Nutzungsumgebung berücksichtigen
- Kulturelle Unterschiede beachten
Dokumentieren Sie prozessbegleitend:
- Nicht erst am Ende zusammenschreiben
- Formative Evaluationen zeitnah dokumentieren
- Designentscheidungen begründet festhalten
5.3 Häufige Fehler und wie Sie diese vermeiden
5.3.1 Die Top 5 Fehler bei FDA HF-Submissions
- Zu spätes Human Factors Engineering
- Fehler: HF erst kurz vor Zulassung
- Lösung: Integration ab Projektstart
- Fehlende US-Nutzer bei Validierung
- Fehler: Tests nur mit europäischen Nutzern
- Lösung: frühzeitige Planung der US-Tests
- Unvollständige Risikoanalyse
- Fehler: Fokus nur auf offensichtliche Risiken
- Lösung: systematische Analyse aller Tasks
- Training als einzige Risikokontrolle
- Fehler: „Wir schulen das weg.“
- Lösung: Design-basierte Maßnahmen priorisieren
- Fehlende formative Evaluationen
- Fehler: nur finale Validierung
- Lösung: iterative Tests während der Entwicklung
Diese Fehler führen regelmäßig zu erheblichen Verzögerungen oder sogar zur Ablehnung von FDA-Submissions. Die Korrektur im Nachhinein ist immer aufwendiger als die richtige Durchführung von Anfang an.
5.4 Ausblick und Empfehlungen
Für Hersteller, die FDA-konforme Human Factors umsetzen wollen:
1. Bestandsaufnahme
- Welche HF-Aktivitäten führen Sie bereits durch?
- Wo bestehen Lücken zu FDA-Anforderungen?
- Welche Ressourcen stehen zur Verfügung?
2. Prozessetablierung
- HF in den Entwicklungsprozess integrieren
- Verantwortlichkeiten festlegen
- Templates und Checklisten erstellen
3. Kompetenzaufbau
- Mitarbeiter schulen
- Externe Expertise einbinden
- Von Best Practices lernen
Das Johner Institut bietet spezielle FDA Human Factors Trainings an, die alle Aspekte von der Planung über die Durchführung bis zur Dokumentation abdecken.
5.5 Abschließende Gedanken
Die FDA-Anforderungen an Human Factors Engineering mögen auf den ersten Blick komplex erscheinen. Mit dem richtigen Verständnis und einem systematischen Vorgehen sind sie jedoch handhabbar. Die Investition in professionelles Human Factors Engineering zahlt sich mehrfach aus:
- Schnellere FDA-Zulassung durch vollständige Dokumentation
- Sichere Produkte mit minimierten Anwendungsrisiken
- Zufriedene Nutzer aufgrund durchdachter Gestaltung
- Reduzierte Haftungsrisiken durch nachgewiesene Sicherheit
Die gute Nachricht: Die Harmonisierung zwischen den FDA-Guidances und der IEC 62366-1 macht es heute einfacher denn je, global konforme Medizinprodukte zu entwickeln.
Sehen Sie Human Factors Engineering nicht als regulatorische Pflicht, sondern als Chance für bessere und sicherere Produkte. Die besten Medizinprodukte entstehen, wenn Sicherheit und Benutzerfreundlichkeit Hand in Hand gehen.
Fazit in einem Satz: Mit einem strukturierten Prozess, frühzeitiger Planung und fokussierter Umsetzung meistern Sie die FDA-Anforderungen an Human Factors Engineering sicher und effizient.
Das Johner Institut unterstützt Sie gerne bei der Umsetzung FDA-konformer Human Factors: von der Planung über die Durchführung von Tests mit US-Nutzern bis zur Erstellung der Zulassungsdokumentation. Kontaktieren Sie uns für eine unverbindliche Beratung.
Änderungshistorie
- 2026-07-07
- Kasten am Ende von 1.1.3 ersetzt
- Kapitel 1.2.3 überarbeitet
- Tipp in 1.3 ergänzt. Konsequenter von HFE/UE statt nur von HFE gesprochen, da die FDA nun erst den Begriff durchgängig verwendet
- Kapitel 3.1. komplett überarbeitet (auch Flussdiagramm)
- In Kapitel 3.3.2 den Kasten mit weiterführenden Informationen gelöscht und Box mit Tipp umformuliert.
- 2025-09-02: Umfangreiche Aktualisierungen und Änderungen

Liebes Johner Institut Team,
den Satz „der neue risikobasierte Ansatz ermöglicht eine differenziertere Betrachtung und kann den Dokumentationsaufwand bei geringem Risiko reduzieren“ verstehe ich nicht ganz.
Die jetzt veröffentlichte Guidance von 2026 (HF Content for Submissions) sagt doch ganz klar in Fußnote 28 „FDA recommends that human factors information be maintained by the manufacturer regardless of whether it is submitted to FDA.“ D.h. selbst wenn ich in HF Kategorie 2 meine HF Tests mit 15 US Usern nicht einreiche, müssten mir diese Tests als Hersteller aber vorliegen?
Oder würden dann meine HF Tests mit EU Usern der FDA ausreichen?
Was ist da Ihre Erfahrung auch vielleicht in Bezug auf FDA Audits?
Ich warte noch auf das Webinar der FDA zur finalen Guidance am 22.07., vielleicht wird man daraus schlauer.
Viele Grüße und danke für den Beitrag,
Anne Schmidt
Liebe Frau Schmidt,
vielen Dank für Ihren aufmerksamen Kommentar. Sie haben vollkommen Recht: Die Kategorien der neuen finalen Guidance (Mai 2026) beziehen sich auf den Umfang dessen, was bei der FDA eingereicht werden muss, nicht auf das, was erstellt werden muss. Fußnote 28 ist da eindeutig.
Allerdings kann der Dokumentationsaufwand aus zwei Gründen dennoch sinken:
1. Risikobasierte Argumentation: Je nach Ergebnis der Risikoanalyse haben Sie die Möglichkeit zu argumentieren, dass für Ihr Produkt keine HF-Validierungsdaten notwendig sind – etwa wenn keine Critical Tasks identifiziert wurden. In diesem Fall entfällt nicht nur die Einreichung, sondern auch die Erstellung dieser Daten.
2. Aufbereitung für die FDA: Auch wenn Ihnen HF-Daten intern vorliegen, bedeutet das Zusammenstellen, Strukturieren und Aufbereiten für eine FDA-Submission einen erheblichen Zusatzaufwand. Wenn Sie in Kategorie 2 fallen, entfällt dieser Aufwand für die Validierungsdetails.
Zur Frage EU- vs. US-Nutzer: Die FDA erwartet grundsätzlich Tests mit Nutzern, die im US-amerikanischen Kontext arbeiten bzw. leben. EU-Daten allein werden in der Regel nicht akzeptiert, da sich Nutzungsgewohnheiten, Ausbildung und Arbeitsumgebungen unterscheiden können. Wenn Sie allerdings risikobasiert argumentieren können, dass keine HF-Validierung notwendig ist, stellt sich diese Frage gar nicht erst.
Zum Thema FDA-Audits: Auch wenn Daten nicht eingereicht werden, kann die FDA im Rahmen einer Inspektion Einsicht in das vollständige Design History File verlangen – inklusive aller HF-Unterlagen. Fußnote 28 ist daher als klare Empfehlung zu verstehen, die Dokumentation unabhängig von der Einreichungskategorie vollständig vorzuhalten.
Herzliche Grüße
Philipp Schleer