
Die FMEA, die Failure Mode and Effect Analysis (auf Deutsch: Fehlermöglichkeits- und -einflussanalyse), ist ein Verfahren, um zu bekannten Ursachen unbekannte Auswirkungen zu untersuchen.
Bei Medizinprodukten nutzt man die FMEA beispielsweise bei der Risikoanalyse, um die Folgen einer fehlerhaften Komponente zu analysieren, insbesondere die sich daraus ergebenden Gefährdungen.
1. Wie Sie die FMEA nutzen sollten
a) FMEA bei der Entwicklung
Nutzen Sie die Failure Mode and Effect Analysis, um bisher unbekannte Gefährdungen zu identifizieren, die sich aus hypothetischen Fehlern ergeben:
- Beginnen Sie bei dieser Analyse mit den Inputs und den Komponenten des Produkts.
- Notieren Sie mögliche Fehlerzustände bei diesen Inputs und Komponenten.
- Finden Sie heraus, welche bislang unbekannten Auswirkungen (z. B. Produktfehlverhalten bzw. Gefährdungen) diese Fehlerzustände haben können.
Das Beispiel sei ein Beatmungsgerät:
- Komponente: Software der zentralen Steuereinheit
- Fehlerzustand der Komponente: Software stürzt ab, wenn Sensoren unerwartete Werte liefern
- Auswirkungen / Gefährdungen / Risiko: Die abgestützte Software bringt das Beatmungsgerät zum Stillstand, d. h. die Beatmung setzt aus (Gefährdung und Gefährdungssituation). Mit einer gewissen Wahrscheinlichkeit verstirbt (Schweregrad des Schadens) der Patient. Das ist das Risiko.
Lesen Sie hier mehr zu den Begriffen Gefährdung und Gefährdungssituation und Schweregrad von Schäden.
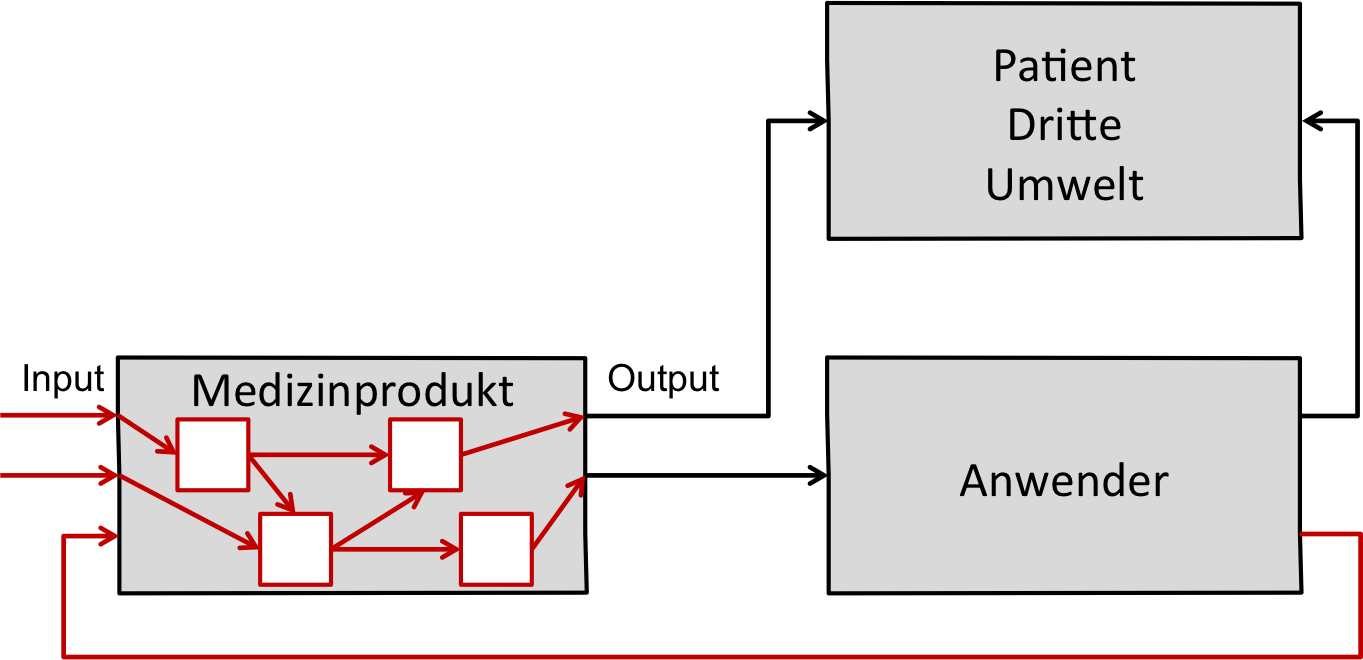
Man bezeichnet die Methode wegen des Weges von der Ursache zur Wirkung als ein Bottom-Up-Verfahren.
b) FMEA beim Austausch oder bei Änderungen von Komponenten
Als Medizinproduktehersteller sollten Sie die FMEA immer als bevorzugtes Verfahren zur Risikoanalyse (präziser: Gefährdungsanalyse) anwenden, um die Auswirkungen zu analysieren, die der Austausch oder die Änderung einer Komponente nach sich ziehen kann.
Die FMEA sollten Sie auch dann nutzen, wenn Sie die Release Notes und Bug-Reports Ihrer SOUPs lesen und mögliche Folgen dieser Fehler bewerten müssen. Das verlangt übrigens die IEC 62304.
Lesen Sie hier mehr zur Software-FMEA.
Es ist möglich, die FMEA auch auf einzelne Komponenten anzuwenden, die aus Subkomponenten bestehen.
Komponenten-FMEA ist keine Risikoanalyse!
Allerdings dient diese Form der FMEA nicht der Risiko- bzw. Gefährdungsanalyse, weil die „Komponenten-FMEA“ nicht Gefährdungen als Auswirkungen diskutiert, sondern das Fehlverhalten der Komponente an deren „Außenkante“. Dieses Fehlverhalten kann wie beim Medizinprodukt selbst durch einen Fehler innerhalb der Komponente oder einen fehlerhaften Input an dieser Komponente verursacht sein.
In anderen Worten: Die Komponenten-FMEA ist keine Risikoanalyse, weil sie nicht Folgen im Sinne von Gefährdungen bzw. Schäden untersucht, sondern im Sinne einer fehlerhaften Komponente. Sie kann also nur die äußere Fehlverhalten und dessen Wahrscheinlichkeiten analysieren – d. h. die (unerwünschten) Effekte. Daher der Name Failure Mode Effect Analysis.
c) FMEA bei der Produktion
Das Gleiche gilt für die Failure and Effect Analysis bei der Produktion: Auch diese ist kein Risikoanalyseverfahren, weil ebenfalls keine Schäden bzw. Wahrscheinlichkeiten als Folge von Produktionsfehlern untersucht werden, sondern Fehler im produzierten Produkt bzw. in einer Komponente des Produkts (als Folge eines Produktionsfehlers).
Bei der Prozess- bzw. Produktions-FMEA untersucht der Hersteller Prozess- bzw. Produktionsschritt für Prozess- bzw. Produktionsschritt. Für jeden dieser Schritte werden die Auswirkungen möglicher Fehler analysiert. Mögliche, „nach außen sichtbare“ Fehler sind:
- Falsche physikalische, chemische oder biologische Beschaffenheit der Komponente
- Komponente gibt in nicht spezifizierter Weise Informationen bzw. Daten weiter (zu wenig, zu viel, zu früh, zu spät, falsche Reihenfolge usw.)
- Komponente gibt in nicht spezifizierter Weise Energien oder Materialien ab (zu wenig, zu viel, zu früh, zu spät, falscher Ort usw.)
Die Kenntnis, mit welcher Wahrscheinlichkeit zu welchen Fehlern im Produkt eine fehlerhafte Produktion führt, hilft im Rahmen der Entwicklungs-FMEA, die Wahrscheinlichkeiten des Fehlers in einer Komponente genauer abzuschätzen.
Die Produktion besteht aus einem oder mehreren Prozessen. Lesen Sie hier mehr zur Prozess-FMEA (pFMEA).
d) FMEA im Rahmen des QM-Systems
Neben der Produktion legt das QM-System weitere Prozesse und Verfahren fest, mit denen die Organisationen die Produkte und Dienstleistungen entwickeln und den Kunden zur Verfügung stellen. Entsprechend lässt sich die Failure Mode and Effect Analysis auf andere Prozesse übertragen.
Sie beantwortet die Frage „Was wäre, wenn …?“, wenn also ein Prozessschritt oder eine Aktivität nicht gemäß den Vorgaben erfolgt oder aus einem anderen Grund nicht das gewünschte Ergebnis liefert.
Die Methode verfolgt die Ursachenkette weiter bis zum Prozessergebnis, ggf. sogar weiter bis zu Risiken für die Patienten, Anwender und Dritten oder Risiken für die Konformität des Unternehmens.
2. Tipps zur Anwendung der FMEA
a) Wesentliche Voraussetzung erfüllen: Eine dokumentierte System- bzw. Software-Architektur
Die FMEA setzt also im Gegensatz zur PHA eine Systemarchitektur bzw. Software-Architektur voraus. Erst wenn Sie diese Architekturen dokumentiert haben, kennen Sie die einzelnen Komponenten. Nur mit einer Architektur können Sie nachvollziehen, wie sich ein Fehler in einer dieser Komponenten auf das Gesamtsystem (Medizinprodukt) auswirkt.
Die Architekturen sollten aber nicht nur erkennen lassen, mit welchen anderen Komponenten eine Komponente interagiert, sondern auch, wie diese Interaktion erfolgt. Um eine FMEA anwenden zu können, müssen Sie also wissen, welche
- Daten/Informationen,
- Materialien oder Stoffe bzw.
- Energien
eine Komponente zur nächsten weitergeben kann. Bei „Datenschnittstellen“ sollten Sie diese Information der Komponentenspezifikation entnehmen können.
b) Ergebnisse tabellarisch dokumentieren
Medizinproduktehersteller dokumentieren die Risiken meist tabellarisch und nennen diese Tabelle nicht ganz zutreffend die „FMEA-Tabelle“. Dieser Begriff ist irreführend, weil die Tabelle geeignet ist, die Ergebnisse aller Risikoanalyseverfahren (z. B. auch die der Fault-Tree-Analysis, FTA) zu dokumentieren. Regelmäßig hat diese Tabelle folgende Spalten:
- ID
- Komponente/Input
- Fehler in Komponente/Input
- Innere Ursachenkette
- Äußeres Fehlverhalten
- Gefährdung
- Wahrscheinlichkeit des Schadens
- Schweregrad des Schadens
- Risiko vor Maßnahmen
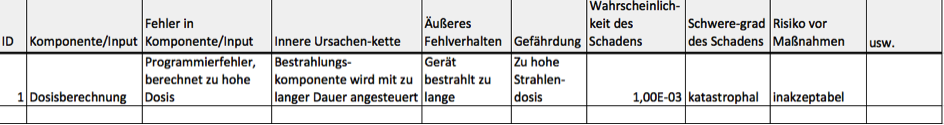
Ebenfalls in dieser Tabelle dokumentieren Hersteller üblicherweise die Maßnahmen zur Risikobeherrschung.
c) FMEA mit HAZOP kombinieren
Um mögliche Fehlverhalten von Inputs oder Komponenten systematisch zu identifizieren, lässt sich die FMEA mit der HAZOP-Analyse kombinieren. Dieses Verfahren beschreibt die IEC 61882. Es arbeitet u. a. mit Leitfragen wie „zu früh?“ „falsche Reihenfolge?“, „zu schnell?“ usw.
3. Vor- und Nachteile der FMEA
a) Vorteile
Die FMEA hat den Vorteil, ein sehr systematisches Verfahren zu sein. Man kann bei gegebener System- bzw. Software-Architektur jede Komponente untersuchen und „abhaken“.
Dieses Verfahren ist einfach zu verstehen und entspricht der Denkweise vieler Entwickler, die mit „ihrer“ Komponente beginnen können.
b) Nachteile
Die FMEA ist ungeeignet, um logische Verknüpfungen vieler Fehler zu untersuchen und zu beschreiben. Dazu eignet sich die Fault-Tree-Analysis (FTA).
Auf Basis einer FMEA lässt sich auch nicht beurteilen, wie granular Komponenten analysiert werden müssen. Viele Medizinproduktehersteller ersticken in „Risikotabellen“ mit hunderten und tausenden Zeilen.
Weil Entwickler diese Methode gut anwenden können, überträgt man ihnen oft die vollständige Risikoanalyse. Dafür sind aber Entwickler – in ihrer Rolle als Entwickler – weder qualifiziert noch geeignet.
Viele Hersteller wenden die FMEA unter Verwendung der Risikoprioritätszahl RPZ an. Diese Definition des Begriffs Risiko stimmt nicht mit der Definition in der ISO 14971 überein. Lesen Sie hier mehr über dieses Problem.
4. Fazit und Zusammenfassung
Die Failure-Mode and Effect Analysis (FMEA) ist eine Methode, die beim Risikomanagement zum Einsatz kommen sollte. Die ISO 14971 stellt diese Methode als eine der wichtigsten vor.
In verschiedenen Industrien wird die FMEA unterschiedlich angewendet und bezeichnet. In der Automobilbranche dienen die identifizierten „Failure Modes“ als Input für die eigentliche Risikoanalyse. Hingegen wird in der Welt der Medizinprodukte die FMEA häufig genutzt, um Risiken und nicht nur „Failure Modes“ zu identifizieren.
Unabhängig davon, welchen Ansatz Sie als Hersteller verfolgen: Sie müssen die Ursachenketten bis zum eigentlichen Schaden nachverfolgen und deren potenzielle Schweregerade und Wahrscheinlichkeiten abschätzen. Genau das sind definitionsgemäß die Risiken.
In den Videotrainings des Auditgarant lernen Sie, eine Risikoanalyse (auch FMEA) systematisch zu erstellen und zu dokumentieren. Die Premium-Version enthält fertige Templates.
Änderungshistorie
- 2025-05-14: Kleine redaktionelle Änderungen wie fehlende Verlinkungen ergänzt
- 2023-10-02: Beispiel in Kapitel 1, Kapitel 1.d) sowie Zusammenfassung (Kapitel 4) ergänzt. Redaktionelle Änderungen.
- 2020-10-20: Erste Version erstellt

Hallo Herr Prof. Johner,
ich darf Sie auf den fast neuen Band FMEA des VDA aufmerksam machen.
https://webshop.vda.de/QMC/de/aiag-vda-fmea-handbuch
Dieser wurde gründlich überarbeitet und hat nun nicht mehr die RPZ, sondern die Aufgabenpriorität (AP).
Diese ist unterteilt in hoch, mittel und niedrig. Die AP ergibt sich immer noch aus B(edeutung) * A(uftreten) * E(ntdeckung). Wobei jetzt die Bedeutung wesentlich stärker gewichtet ist.
Mit vielen Grüßen
Oliver Graus
Einmal mehr, herzlichen Dank Herr Graus!
Ich werde demnächst den Beitrag überarbeiten. Danke für den Hinweis!
Beste Grüße, Christian Johner
Hallo Herr Johner,
ich würde den damaligen Hinweis gerne aufgreifen und auf das Thema eingehen. Mittlerweile 3 Jahre später lese ich zwar viel über den AP und man solle diesen nun unbedingt der gewohnten RPZ vorziehen, allerdings macht das niemand. So kommt es mir zumindest vor. So ist es auch bei uns das tägliche Geschäft die RPZ zu verwenden. Es wird quasi erwartet.
Können Sie bei dieser Problematik von RPZ und AP ein wenig Klarheit schaffen? Für die Automobilbranche geschaffen, für die Medizin so nutzbar und nicht vielleicht sogar undbedingt notwendig?
Ich danke Ihnen vielmals.
Herzliche Grüße
Lieber Herr Großman, vielleicht ist das Thema mit dieser Erklärung leichter nachzuvollziehen:
Die ISO 14971 erwartet die Darstellung der Risiken und deren Abschätzung und Bewertung in einer vorgegebenen Weise, bei der eben nur die Wahrscheinlichkeit des Auftretens eines Schadens am Menschen (!) und dessen Schweregrad betrachtet wird und diese beiden Parameter NICHT multiplikatorisch verknüpft werden.
Wenn Sie Methoden verwenden, um die möglichen Ursachenketten (z.B. vom auslösenden Fehler bis hoch zur Spezifikationsverletzung am Produkt) zu identifizieren, dann können Sie dort beliebige Methoden zur Analyse und zur Bewertung der Risiken verwenden. Z.B. wird bei der reinen Produktions-Prozess-Risikoanalyse (die nicht bis zum Schaden am Menschen betrachtet!) gerne weiterhin die RPZ oder andere Bewertungsmechanismen angewendet. Wichtig ist, dass Sie beim Übertrag der Risiken in die ISO 14971 Risikoanalyse nur inhaltlich die Ursachenketten übernehmen, die Bewertungen (z.B. RPZ) dabei einfach ignorieren. Sie führen dann die Ereignisketten in ihrem Effekt weiter bis zum Schaden am Menschen aus und bewerten dann mit den Metriken der ISO 14971 neu.
Somit haben Sie immer eine saubere Trennung zwischen den „untergeordneten“ Analysen und der eigentlichen ISO 14971 Analyse.
Die Diskussion, ob AP oder RPZ spielt also im Medizinproduktebereich absolut keine Rolle. Wir benötigen nur die identifizierten Ereignisketten, die auf Fehler folgen und machen jegliche Bewertung neu, mit den Maßstäben und Forderungen der ISO 14971.
Kürzlich hat ein Teilnehmer in meinem Seminar gesagt, er hat es erst dort im Seminar richtig verstanden, als ich es im Detail erklärt und mit Präsentations-Folien veranschaulicht habe. Kommen Sie also gerne im Seminar vorbei, wenn es weiterhin Schwierigkeiten in der Verständlichkeit der getrennten Konzepte gibt.
Liebe Grüße
Christian Rosenzweig
Hallo Herr Rosenzweig,
vielen Dank für Ihre umfangreiche Erläuterung.
Tatsächlich klärt das dieses AP / RPZ Mythos bei uns endlich mal, dafür vielen Dank. Das Seminarangebot werde ich intern mal besprechen und vielleicht sehen wir uns ja bald.
Herzliche Grüße
Peter Großmann
lieber Herr Johner,
bei der Überarbeitung bitte noch den Druckfehlerteufel im Titel verbessern „Defin*in*ition“.
kann passieren, es hat keinen Einfluss auf den immer wieder ausgezeichneten Inhalt.
Herzliche Grüsse
Peter Pianegonda
Sie haben ja so Recht, Herr Pianegonda!
Vielen Dank für Ihren Hinweis, denn ich sofort umgesetzt habe. Damit ist das „inini“ beseitigt. 🙂
Nochmals vielen Dank!
Herzliche Grüße, Christian Johner