
Im 21 CFR part 820 formuliert die FDA die Anforderungen an die Qualitätsmanagementsysteme u.a. von Medizinprodukteherstellern. Damit ist bzw. war der 21 CFR part 820 (Quality System Regulation QSR) das Pendant zur ISO 13485.
1. QMSR: Die Änderungen des 21 CFR part 820
Denn die FDA hat am 02.02.2024 beschlossen, den 21 CFR part 820 weitgehend zu „entkernen“ und durch einen Verweis auf die ISO 13485 zu ergänzen.
- Dieser Verweis steht im neuen Abschnitt § 820.7 (“Incorporation by reference”).
- Die Tabelle 1 zeigt, welche Abschnitte dadurch obsolet wurden.
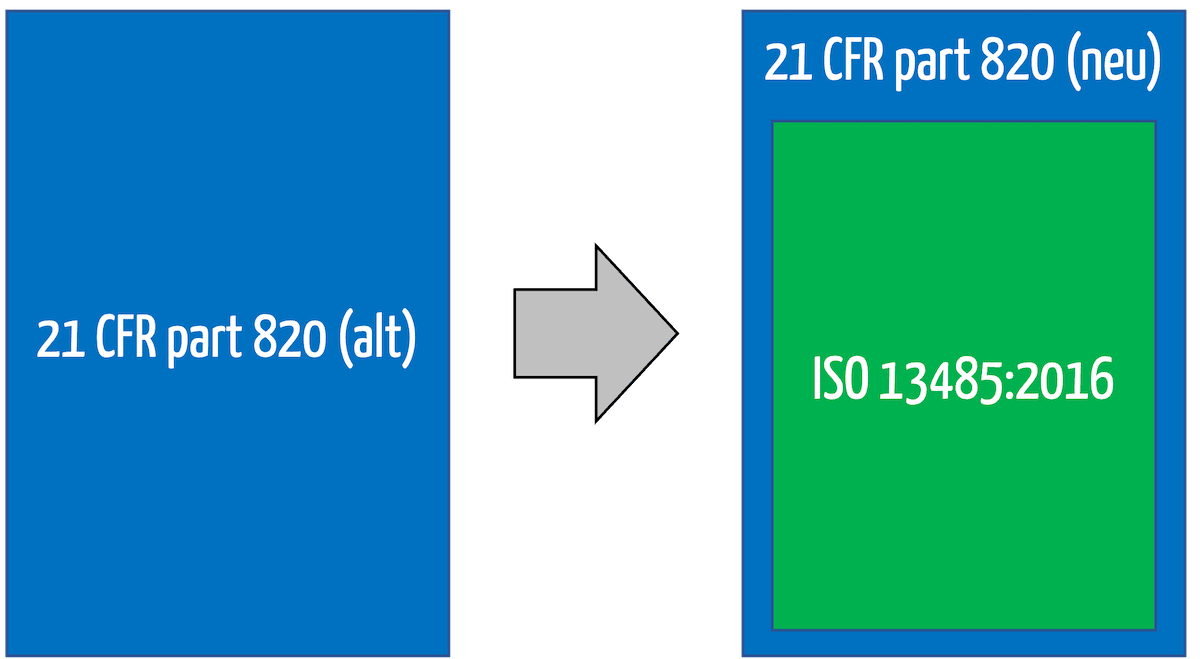
Weil die ISO 13485 von Qualitätsmanagementsystemen spricht, hat sich die FDA entschieden, den Titel des 21 CFR 820 umzubenennen. Dieser heißt nun nicht mehr Quality System Regulation (QSR). Vielmehr spricht die FDA nun von der Quality Management System Regulation (QMSR).
Mit dieser Änderung sind die bisherigen Anforderungen an ein Design History File, einen Device Master Record und einen Device History Record zwar nicht mehr explizit erwähnt. Dennoch verlangt auch die ISO 13485 ähnliche Aufzeichnungen.
a) Vergleich des alten und neuen 21 CFR part 820
Subparts A-D
| 21 CFR part 820 alt (QSR) | 21 CFR 820 neu (QMSR) |
| Subpart A – General Provisions
§ 820.1 – Scope. § 820.3 – Definitions. § 820.5 – Quality system. | Subpart A – General Provisions § 820.1 – Scope. § 820.3 – Definitions. § 820.7 – Incorporation by reference. § 820.10 – Requirements for a quality management system. |
| Subpart B – Supplemental Provisions | |
| Subpart B – Quality System Requirements
§ 820.20 – Management responsibility. § 820.22 – Quality audit. § 820.25 – Personnel. | Gelöscht
(reserviert für künftige Ergänzungen) |
| § 820.35 Control of records | |
| Subpart C – Design Controls
§ 820.30 – Design controls. | Gelöscht
(reserviert für künftige Ergänzungen) |
| Subpart D – Document Controls
§ 820.40 – Document controls. | Gelöscht
(reserviert für künftige Ergänzungen) |
Subparts E-O
| 21 CFR part 820 alt | Geplante Änderungen |
| Subpart E – Purchasing Controls
§ 820.50 – Purchasing controls. | Gelöscht |
| Subpart F – Identification and Traceability
§ 820.60 – Identification. § 820.65 – Traceability. | Gelöscht |
| Subpart G – Production and Process Controls
§ 820.70 – Production and process controls. § 820.72 – Inspection, measuring, and test equipment. § 820.75 – Process validation. | Gelöscht |
| Subpart H – Acceptance Activities
§ 820.80 – Receiving, in-process, and finished device acceptance. § 820.86 – Acceptance status. | Gelöscht |
| Subpart I – Nonconforming Product
§ 820.90 – Nonconforming product. | Gelöscht |
| Subpart J – Corrective and Preventive Action
§ 820.100 – Corrective and preventive action. | Gelöscht |
| Subpart K – Labeling and Packaging Control
§ 820.120 – Device labeling. § 820.130 – Device packaging. | 820.45 Device labeling and packaging controls. |
| Subpart L – Handling, Storage, Distribution, and Installation
§ 820.140 – Handling. § 820.150 – Storage. § 820.160 – Distribution. § 820.170 – Installation. | Gelöscht |
| Subpart M – Records
§ 820.180 – General requirements. § 820.181 – Device master record. § 820.184 – Device history record. § 820.186 – Quality system record. § 820.198 – Complaint files. | Gelöscht Gelöscht § 820.35 Control of records (nur bezüglich UDI) Gelöscht § 820.35 Control of records |
| Subpart N – Servicing
§ 820.200 – Servicing. | § 820.35 Control of records (nur bezüglich Servicing Records) |
| Subpart O – Statistical Techniques
§ 820.250 – Statistical techniques. | Gelöscht |
b) Unterschiede im Vergleich zur ISO 13485
Die Quality Management System Regulations im part 820 und die ISO 13485 sind nicht ganz deckungsgleich:
| Aspekt | Unterschiede |
| Anwendungsbereich | Der Anwendungsbereich (§ 820.1) unterscheidet sich insgesamt. Bei den Produkten der Klasse I (ausgenommen z.B. Produkte, die Software enthalten) verzichtet die FDA auf die Anforderungen des Kapitel 7.3 der ISO 13485 („Design and Development“). |
| Begriffsdefinitionen | Die FDA fügt in § 820.3 der ISO 13485 eigene Definitionen hinzu wie beispielsweise „Component“, „Finished Device“, „Remanufacturer“. Die Begriffe „Implantable medical device“, „Manufacturer“, „Organization“, „Rework“ und „Safety and Performance“ definiert sie anders. D.h. sie überschreibt die entsprechenden Begriffe der ISO 13485. |
| Produktidentifizierung, Nachvollziehbarkeit und Labeling | Hier ist die FDA spezifischer und fordert die UDI wie im § 830 beschrieben sowie die „Traceability“ gemäß § 821. Die Labeling-Anforderungen sind genau wie bei der MDR auch spezifischer als die der ISO 13485. Sie finden sich im neuen § 820.45. |
| Vigilanz und Behördenkommunikation | Für die spezifischeren Anforderungen verweist die FDA auf die Parts § 803 und § 806. |
| Dokumentation | Die FDA legt genauer den Inhalt der Aufzeichnungen von Kundenbeschwerden und Service-Tätigkeiten fest. |
Sie finden im Federal Register die Begründung und die genaue Beschreibung der Änderungen. Sie müssen dazu sehr weit nach unten scrollen oder auf der Webseite nach dem Begriff suchen „4. Revise part 820 to read as follows:“
Eine Übersicht über alle Anforderungen der FDA finden Sie hier unter dem Schlagwort FDA.
c) Zeitpunkt und Übergangsfristen
Die „final rule“ zur QMSR wurde am 02. Februar 2024 veröffentlicht. Gültig und damit anwendbar wird diese mit einer Frist von 2 Jahren d.h. zum 02. Februar 2026. Bis dahin ist noch die alte QSR zu befolgen.
2. Der 21 CFR part 820 vor Referenzierung der ISO 13485
a) Übersicht
Die Quality System Regulations bestehen (vor der Referenzierung) aus den Subparts A bis O, welche die Abschnitte 1 bis 250 umfassen (siehe Tabelle 1 und Abbildung 2).
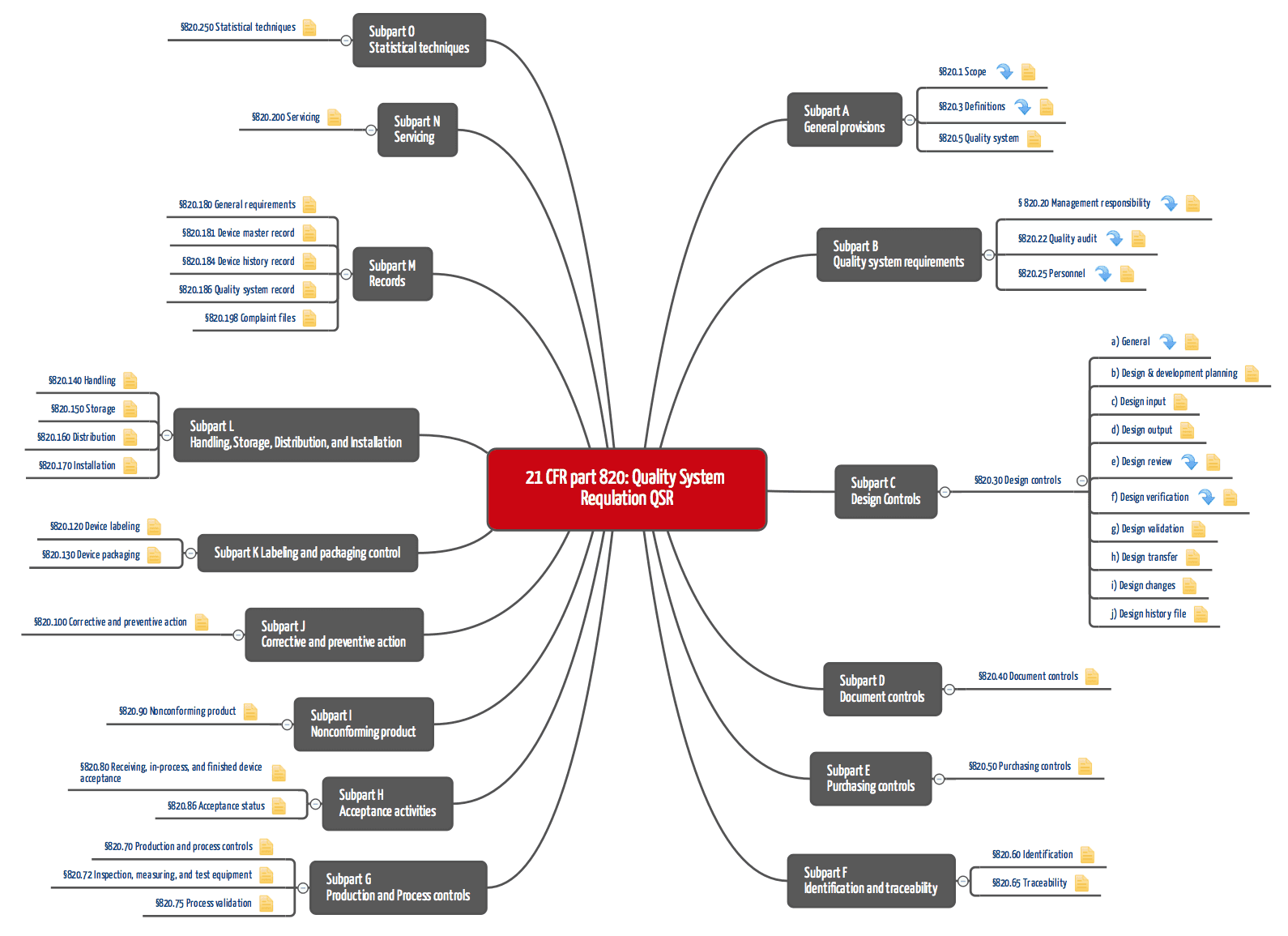
Der „part 820“ fordert ein vollständiges Qualitätsmanagementsystem, das voraussetzt, dass die „üblichen“ Verfahrensanweisungen dokumentiert und umgesetzt werden. Dazu gehören:
- Dokumentenlenkung
- Beschaffung
- Entwicklung
- Produktion
b) Anwendbarkeit des 21 CFR part 820
Je nach Klasse des Medizinprodukts müssen die Hersteller die im „Food, Drug & Cosmetic Act“ festgelegten General Controls (§ 501 ff) und ab Klasse II zusätzlich auch die „Special Controls“ erfüllen. Bereits die „General Controls“ beinhalten „Good manufacturing practice requirements“, die sich auf die Entwicklung, Produktion, Verpackung, Lagerung und Installation der Geräte beziehen.
Genau diese Anforderungen an „Current good manufacturing practice (CGMP)“ sind Gegenstand der Quality System Regulation QSR. Damit sind diese Regelungen von allen Medizinprodukteherstellern und auch weiteren Akteuren wie den Contract Manufacturers einzuhalten. Ausgenommen bzw. GMP-exempt sind nur einige wenige Klasse I-Produkte. Die Einhaltung der CGMP überprüft die FDA per Inspektion.
c) Anforderungen an die Entwicklung
Für Entwicklungsabteilung relevant ist v. a. der Bereich 820.30 mit den Design Controls. Das gilt sogar für Entwicklung von Medizinprodukten der Klasse I, wenn diese Software enthalten oder Software sind.
Die Forderungen im 820.30 sind sehr allgemein gehalten. Daher orientieren sich beispielsweise Hersteller von Medizinprodukten, die Software enthalten, auch an den Guidance-Dokumenten der FDA. Diese beschreiben detailliert, wie etwa der Design Input dokumentiert werden muss.
Die Bedeutung des Design History File DHF (820.30 j) unterschätzen viele Hersteller. Denn dieser Satz an Dokumenten muss den Nachweis ermöglichen, dass die im 21 CFR part 820 beschriebenen Verfahren tatsächlich gelebt wurden und die Dokumentation nicht etwa nachträglich erstellt wurde.
d) Unterschied zwischen 21 CFR 820 und ISO 13485
Die AnsonGroup hat eine Gegenüberstellung der Forderungen der ISO 13485 und der FDA QSR veröffentlicht. Eine weitere Gegenüberstellung finden Sie hier. Die weitgehende Übereinstimmung der beiden Standards ist offensichtlich. Dennoch gibt es Unterschiede zwischen der ISO 13485 und dem part 820 zu beachten:
- Die Anforderungen an die Dokumentation sind höher, auch die logische Gruppierung der Dokumente in das Design History File, den Device Master Record und den Device History Record kennt die ISO 13485 nicht.
- Umgekehrt geht der Anspruch der ISO 13485 bzw. ISO 9001 auf Kundenzufriedenheit und die kontinuierliche Verbesserung des QM-Systems über die Forderungen QSRs hinaus.
- Weiter unterscheiden sich der Umgang mit Beschwerden und das Meldewesen deutlich.
Eine ISO 13485 Zertifizierung erkennt die FDA nicht als Nachweis der Konformität mit den Forderungen des 21 CFR part 820 an. Im Gegensatz zur ISO 13485 gibt es auch keine Zertifizierung nach 21 CFR Part 820.
Bereits vor der Harmonisierung der QSRs mit der ISO 13485 können Hersteller im Rahmen des Medical Device Single Audit Programs MDSAP gleichzeitig die Konformität mit den Anforderungen des 21 CFR part 820 und der ISO 13485:2016 nachweisen.

Der schnellste und kostengünstigste Weg Ihr ISO 13485-konformes QMS aufzubauen
Mit unseren Videotrainings und dutzenden Vorlagen im Auditgarant erhalten Sie eine umfassende Anleitung zum Aufbau eines schlanken und konformen Qualitätsmanagementsystems.
3. Fazit und Zusammenfassung
Es ist ein großer Fortschritt für die Harmonisierung regulatorischer Anforderungen, dass die FDA ihre Vorgaben an ein QM-System im 21 CFR part 820 im Wesentlichen durch einen Verweis auf die ISO 13485 ersetzt. Sie erklärt im „neuen“ 21 CFR part 820 wie bestimmte Anforderungen z.B. an die Produktidentifizierung und Vigilanz konkret zu erfüllen sind.
Es wäre den Medizinprodukteherstellern dienlich gewesen, wenn die MDR und IVDR ebenfalls diesen Weg gegangen wären.
Benötigen Sie Unterstützung dabei, ein ISO 13485- oder/und FDA-konformes Qualitätsmanagementsystem zu etablieren oder zu verbessern? Oder benötigen Sie Hilfe bei der Umstellung von der QSR auf die QMSR? Wir unterstützen Sie gerne!
Bei Bedarf übernehmen wir auch gerne die Rolle des US Agents.
Melden Sie sich z.B. hier über das Kontaktformular.
Änderungshistorie
- 2025-12-08: In 2.d) weitere Gegenüberstellung verlinkt
- 2024-02-21: Überarbeitung basierend auf der verabschiedeten Version der QMSR vom 02.02.2024


Sehr geehrter Herr Johner,
gibt es zu der Änderung bereits eine Konkretisierung der terminlichen Umsetzung? Bis jetzt konnte ich noch nicht mehr als den besagten Dezember 2023 finden. Auch stellt sich dann noch die Frage der Übergangsfristen.
Sehr geehrter Herr Hoffmann,
leider liegen uns noch keine aktuelleren Informationen vor. Wir gehen weiterhin von einer Verabschiedung des Gesetzes zum Ende diesen Jahres aus. Bis dahin sollten dann auch Regelungen zu den Übergangsfristen definiert worden sein.
Sobald wir aktuelle Informationen dazu haben, werden wir in unserem Newsletter davon berichten.
Beste Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
da ja jetzt das Ende des Jahres in Sicht rückt, wollte ich mich nochmals erkundigen ob Sie hier bereits aktualisierte Informationen vorliegen haben? Wir planen aktuell einen Workshop für die kommenden Wochen um uns mit den Änderungen zu beschäftigen.
Beste Grüße
Ralph Hoffmann
Sehr geehrter Herr Hoffmann,
uns liegen bisher leider noch keine neuen Informationen vor bezüglich der „final rule“. Somit gehen wir aktuell weiterhin vom Ende diesen Jahres aus.
Sobald es Neuigkeiten gibt, werden wir diese direkt im Newsletter veröffentlichen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
jetzt ist es aber so weit, wie im heutigen Journal beschrieben. Haben Sie Zugriff auf eine eventuelle GAP Analyse?
Sehr geehrter Herr Hoffmann,
in der Tat wurde die proposed rule inzwischen akzeptiert. Laut Information des OIRA (https://www.reginfo.gov/public/do/eoDetails?rrid=352211) wurden keine Änderungen vorgenommen. D.h. es sollten keine Unterschiede zwischen proposed und final rule bestehen. Somit sollten auch die Informationen in diesem Artikel bezüglich der Unterschiede zur aktuellen QSR weiterhin korrekt sein.
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
im Artikel wird erwähnt, dass die AnsonGroup eine Gegenüberstellung der Forderungen der ISO 13485 und der FDA QSR veröffentlicht hat. Hätten sie hierfür einen Link, wo ich das Dokument einsehen kann?
Viele Grüße
Martin Kares
Sehr geehrter Herr Kares,
wir sind selbst noch auf der Suche nach dem Dokument. Wir ergänzen den Link, sobald wir ihn haben.
Es gibt aber eine weitere Gegenüberstellung, die Ihre Frage beantwortet.
Mit herzlichen Grüßen, Christian Johner