
Das Medical Device Single Audit Program (MDSAP) wurde ins Leben gerufen, um einen Wunsch vieler Medizinproduktehersteller zu erfüllen: Statt vieler Audits und Inspektionen durch die Behörden verschiedener Länder soll es nur noch eines geben.
Die Teilnahme am MDSAP soll ausreichen, um die Wirksamkeit und Konformität von QM-Systemen (z. B. Konformität mit ISO 13485 oder 21 CFR part 820) nachzuweisen.
Ob dieser Wunsch wirklich in Erfüllung geht und was der Preis dafür ist, erfahren Sie in diesem Artikel.
1. MDSAP: Eine Einführung
a) Ziele des MDSAP-Programms
Das International Medical Device Regulators Forum IMDRF, ein Zusammenschluss internationaler Behörden und Gesetzgeber, hatte erkannt: Die Vielzahl der Audits und Inspektionen von QM-Systemen, die man in den verschiedenen Zielmärkten fordert, sind in mehrfacher Hinsicht suboptimal.
Von einer Vereinheitlichung durch das MDSAP erhofft(e) man sich viele Vorteile.
QM-Overhead bei Herstellern vermeiden, Marktzulassung beschleunigen
Die Hersteller müssen viele, oft redundante Audits und Inspektionen über sich ergehen lassen. Dies führt zu einem QM-Overhead und bedeutet Aufwände, Kosten und Zeitverzug bei der Inverkehrbringung. Das MDSAP hat zum Ziel, diese Aufwände zu minimieren.
Redundante Aufwände; Anforderungen einheitlich interpretieren
Die Aufwände betreffen auch die internationalen „Regulators“: Sie müssen fast identische Anforderungen an QM-Systeme formulieren und kontinuierlich weiterentwickeln, wie das beispielsweise bei der MDR kürzlich geschehen ist. Auch streben die „Regulators“ an, die QM-Anforderungen (wie die der ISO 13485) noch einheitlicher zu interpretieren.
Bewertung vereinheitlichen
Den Auditoren wird ein Bewertungsschema an die Hand gegeben. Damit soll gewährleistet werden, dass verschiedene Auditoren bei der Bewertung eines Sachverhalts z. B. einer Nichtkonformität möglichst zum gleichen Ergebnis gelangen.
Redundante Aufwände für die Überprüfung der QM-Systeme minimieren
Die in hohem Maße redundanten Audits und Inspektionen bedeuten nicht nur für die Hersteller Aufwände, sondern auch für die Behörden selbst, z. B. die FDA. Diese Aufwände für das Auditieren möchte man minimieren. Auch strebten die „Regulators“ an, beim Auditieren einheitlicher vorzugehen.
Auditzeiten verringern
Laut Health Canada soll das MDSAP sogar dazu beitragen, die Dauer der Audits zu verringern: bei Firmen bis 45 Mitarbeitenden um 10 %, bei kleinen Firmen mit bis zu 15 Mitarbeitenden sogar um 20 %.
Schwarze Schafe leichter identifizieren, Produktsicherheit erhöhen
Die Behörden waren (und sind) schlecht abgestimmt waren. Daher gelingt es den schwarzen Schafen unter den Herstellern, Schwächen, die eine Behörde entdeckt hat, vor anderen Behörden zu verstecken. Folglich besteht ein weiteres Ziel des MDSAP darin, Informationen (insbesondere Auditergebnisse) zwischen den teilnehmenden Behörden auszutauschen. Damit erhoffen sich diese, schwarze Schafe und damit unsichere Produkte schneller zu identifizieren und so die Patientensicherheit zu erhöhen.
Die Vereinheitlichung betrifft die Audits und deren Dokumentation. Das MDSAP fordert hingegen keine Vereinheitlichung der Dokumentation der Hersteller.
b) Teilnehmer
In der ersten Phase des Medical Device Single Audit Program haben sich folgende Länder beteiligt:
| Land | Behörde | QM-Vorgaben |
| Australien | Therapeutic Goods Administration (TGA) | Australian Therapeutic Goods (Medical Devices) Regulations (TG(MD)R Sch3) |
| Brasilien | Agência Nacional de Vigilância Sanitária (ANVISA) | Brazilian Good Manufacturing Practices (RDC ANVISA 16/2013) |
| Kanada | Health Canada | ISO 13485:2016 |
| Japan | Ministry of Health, Labour and Welfare (MHLW) and Pharmaceuticals and Medical Devices Agency (PMDA) | Ordinance on Standards for Manufacturing Control and Quality Control of Medical Devices and In Vitro Diagnostic Reagents (MHLW Ministerial Ordinance No. 169) |
| USA | Food and Drug Administration (FDA) | QSR – 21 CFR Part 820 |
Die EU und die WHO beschränken sich auf eine Beobachterrolle.
Die FDA bewertet die Pilotphase, die im Jahr 2017 endete, als Erfolg:
“Based on its evaluation of the MDSAP Final Pilot Report, the MDSAP Regulatory Authority Council determined that the MDSAP Pilot had satisfactorily demonstrated the viability of the Medical Device Single Audit Program. […] FDA will continue to accept MDSAP audit reports as a substitute for routine Agency inspections.”
Die FDA übernimmt die Anforderungen der ISO 13485 weitgehend, wie Sie im Artikel zum 21 CFR part 820 nachlesen können.
c) Anerkennung
Bisher nehmen die o.g. fünf Länder am Medical Device Single Audit Program MDSAP teil.

Diese Länder verpflichten sich zwar, die Ergebnisse der MDSAP-Audits anzuerkennen, es gibt aber leichte Unterschiede, Einschränkungen und Ausnahmen:
| Land | Anerkennung |
| Australien | Ja, aber nicht für Medizinprodukte, die Arzneimittel oder Materialien menschlichen oder tierischen Ursprungs enthalten |
| Brasilien | Ja, auch initial, aber beispielsweise nicht, wenn vorausgegangenes ANVISA-Audit nennenswerte Abweichungen festgestellt hat |
| Kanada | Ja. Ab 2019 gestattet Health Canada sogar nur MDSAP-Audits. |
| Japan | Ja, aber z. B. nicht für Medizinprodukte, die Materialien menschlichen oder tierischen Ursprungs enthalten |
| USA | Ja, aber nur für „Routine-Inspektionen“ (auch initial), nicht für spezielle Inspektionen (z. B. „for cause inspections“) |
Fazit: Einzig Kanada setzt konsequent auf dieses Vorgehensmodell. Kanada erkennt seit 2019 nur noch QM-Systeme an, die über ein MDSAP auditiert wurden.
d) Auditierende Organisationen
Die Audits führen die sogenannten „Auditing Organizations“ (AO) durch. Zu diesen AOs zählen auch einige benannte Stellen wie der TÜV Süd, der TÜV Rheinland und DQS-med. Benannte Stellen sind aber nicht automatisch als AO autorisiert.
Die FDA führt auf ihrer Webseite eine Liste der Auditing Organizations und eine Liste der von der ANVISA anerkannten AOs.
2. MDSAP Audits
a) Anforderungen und Ausschlüsse
Der Anforderungskatalog basiert stark auf der ISO 13485:2016. Er berücksichtigt zudem die Anforderungen der teilnehmenden Länder, die durch die ISO 13485:2016 nicht abgedeckt sind. Ähnlich wie die ISO 13485 ist auch der MDSAP-Anforderungskatalog prozessorientiert und die Audits erfolgen nach Prozessgruppen.
Es gibt vier Primärprozesse bzw. Prozessgruppen und drei Unterstützungsprozesse:
- Primärprozesse
- Management
- Measurement, Analysis and Improvement
- Design and Development
- Production and Service Controls
- Support Processes
- Purchasing
- Device Marketing Authorization and Facility Registration
- Medical Device Adverse Events and Advisory Notices Reporting
Wenn eine Organisation nicht über alle Prozesse verfügt (z. B. nicht entwickelt), müssen die entsprechenden Prozesse auch nicht auditiert werden. Ein Hersteller, der ein Outsourcing von Prozessen vornimmt, profitiert von dieser Vereinfachung nicht.
Ein Hersteller darf Anforderungen ausschließen, die für einen Zielmarkt spezifisch sind, falls er in diesem Zielmarkt keine Medizinprodukte verkauft.
b) Aufgaben der Auditoren
Ein Audit nach MDSAP berücksichtigt die Abhängigkeiten der Prozesse. Beispielsweise ist der Output des Entwicklungsprozesses der Input des Herstellungsprozesses. Entlang dieser Reihenfolgen müssen die Audits erfolgen. Sie müssen risikobasiert durchgeführt werden und den kompletten Produktlebenszyklus (bis zur Außerbetriebnahme) betrachten.
Für jede der oben genannten Prozessgruppen beschreibt das MDSAP-Audit-Modell Folgendes:
| Element | Beispiel (Ausschnitte) |
| Name des Prozesses | Management-Prozess |
| Ziel des Audits dieses Prozesses | Sicherstellen, dass adäquate Ressourcen für die Entwicklung, Herstellung […] zur Verfügung stehen […] |
| Erwartete Ergebnisse (= Anforderungen) | Verpflichtung, dass in angemessener Weise Personal und Ressourcen für die Infrastruktur des QM-Systems bereitstehen […] |
| Verweis auf Prozesse, die von diesem Prozess abhängig sind | Messung, Analyse und Verbesserung, Entwicklung, Einkauf, Produktion, Inverkehrbringung, […] |
| Aufgaben des Auditors | Überprüfen Sie die Organisationsstruktur und entsprechende Dokumente, um sicherzustellen, dass Sie Vorgaben enthalten zu Verantwortlichkeiten, Personal, Ressourcen […] |
| Für jede Aufgabe Verweise auf die entsprechenden Teilkapitel, Artikel und Absätze der ISO 13485:2016 und der anderen Anforderungen wie 21 CFR part 820 und RDC ANVISA 16/2013 | SO 13485:2016: 5.1, 5.5.1, 5.5.2, 6.1, 6.2; TG(MD)R Sch3 P1 1.4(5)(b); […] |
| Hinweise für jede Aufgabe, ggf. länderspezifische Anforderungen | keine |
| Hinweise für die Auditoren (diese finden sich im Companion Document) | Eine Methode, um zu bestätigen, dass ausreichende Ressourcen zur Verfügung gestellt werden, besteht darin, den QMB um Beispiele zu kürzlich erfolgten Anfragen nach Ressourcen zu bitten und ihn beschreiben zu lassen, wie mit diesen Anfragen verfahren wurde. |
c) Häufigkeit von Audits
Die MDSAP-Audits erfolgen nach der gleichen dreijährigen Frequenz wie die Audits zu 93/42 EWG bzw. ISO 13485.
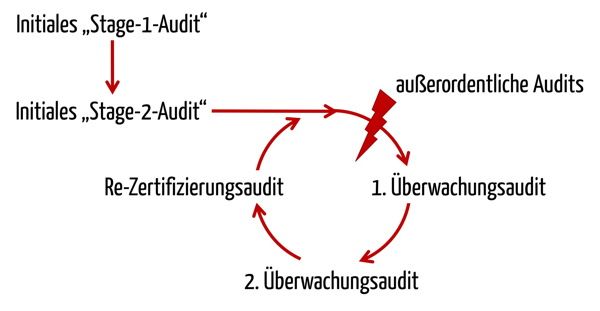
Bei dieser Abfolge orientiert sich das MDSAP stark an der ISO 17021:2015, die auch die Grundlage für die Audits nach ISO 13485 darstellt. Wie stark sich das MDSAP an der ISO 13485 orientiert, findet sich auch in der Beschreibung des Audit-Modells:
“The purpose of a Stage 2 audit is to determine if all applicable requirements of ISO 13485:2016 and the relevant regulatory requirements from participating regulatory authorities have been implemented.”
d) Dauer von Audits
Die Dauer der MDSAP-Audits berechnet sich nach der Anzahl der Aufgaben. Diese Anzahl wiederum hängt von den Ausschlüssen ab, z. B. wenn ein Hersteller keine Anforderungen an sterile Produkte erfüllen muss.
Im ungünstigsten Fall müssen die Auditoren 90 Aufgaben erledigen, für die sie je nach Aufgabentyp zwischen 12 und 35 Minuten benötigen dürfen. Für die Aufgaben in der Prozessgruppe „Management“ sind je 28,8 Minuten vorgesehen.
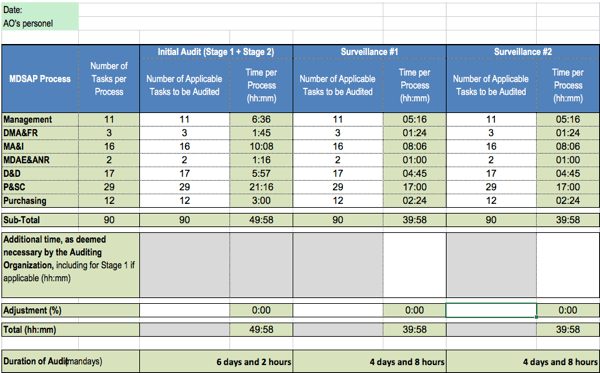
Für die Auditierung der länderspezifischen Anforderungen erhöhen sich die Aufwände.
e) Auditergebnisse
Für die Dokumentation der Planung, Durchführung und Bewertung von Audits stehen Templates bereit. Damit werden Audit- und Abweichungsberichte einheitlich gestaltet und automatisiert auswertbar.
Das MDSAP legt die Fristen fest, innerhalb derer die auditierenden Organisationen (AOs) die Berichte erstellen und verteilen müssen. Diese Fristen sind ambitionierter, als wir das von vielen Benannten Stellen gewohnt sind.
3. Updates
- März 2018: Die FDA erwägt, als QM-Standard die ISO 13485 anstatt des 21 CFR part 820 anzuerkennen bzw. einzufordern. Der entsprechende Hinweis ist auf der offiziellen Seite zwar verschwunden; andere Quellen wie das AAMI erwähnen dies aber weiterhin. Die Notwendigkeit eines MDSAP würde dadurch stark relativiert.
- 13. April 2018: Health Canada kündigt auf seiner Webseite an, die Anforderungen von MDSAP nicht sofort und in aller Härte einfordern zu wollen.
- 21. Februar 2021: Die FDA hat die Liste der Verfahren und Formulare auf ihrer Webseite überarbeitet.
- 1. April 2023: Das MDSAP AU P0002.008 ist veröffentlicht und auch auf der Webseite der FDA zu finden.
- 6. August 2024: Das MDSAP AU P0002.009 ist veröffentlicht und auch auf der Webseite der FDA zu finden.
4. Fazit
Für die Hersteller kann es einen großen Vorteil bedeuten, wenn mit dem MDSAP die Anzahl der Audits und Inspektionen durch die Behörden der verschiedenen Zielmärkte verringert wird. Dennoch waren die Hersteller in der MDSAP-Pilotphase zurückhaltend. Dies mag mit folgenden Nachteilen und Herausforderungen zusammenhängen:
- Die Transparenz bedeutet, dass negative Auditergebnisse allen teilnehmenden Ländern zur Verfügung stehen und diese dann zusätzliche Inspektionen veranlassen können. Die FDA beispielsweise behält sich dieses Recht explizit vor.
- Die Liste der Anforderungen wird tendenziell eher breiter, weil sie die Vereinigungsmenge der Anforderungen verschiedener Länder darstellt.
- Das interne Qualitätsmanagementteam muss sich stärker mit internationalen Anforderungen beschäftigen.
- Einer der wichtigsten Zielmärkte ist außen vor: Europa.
Wer Medizinprodukte in Kanada in den Markt bringen will, hat keine Wahl: Das Medical Device Single Audit Program wird der einzige Weg sein. Für alle anderen Hersteller bedeutet die Teilnahme eine Abwägung von Vor- und Nachteilen, zumindest, solange Europa sich nicht von seiner Beobachterrolle verabschiedet und das MDSAP anerkennt.
Der Umstieg auf MDSAP ist zeitintensiv. Starten Sie daher rechtzeitig.
Die relativ transparenten und einheitlichen Regelungen zu den Tätigkeiten, zur Dokumentation und zur Bewertung dürften dazu beitragen, dass Audits weltweit einheitlicher ablaufen.
Die versprochene Anpassung der Aufwände an das Risiko der Medizinprodukte eines Herstellers und an dessen Größe lässt aber zu wünschen übrig. Damit wird das MDSAP im Vergleich zu ISO 13485-Audits gerade für kleine Hersteller mit weniger kritischen Produkten zu einer eher höheren Hürde.
Das Johner Institut unterstützt Medizinproduktehersteller und deren Dienstleister dabei, schlanke QM-Systeme zu etablieren und auf Audits und Inspektionen vorzubereiten.
Änderungshistorie
- 2024-10-24: Hinweis auf Aktualisierung des MDSAP AU P0002.009 ergänzt
- 2024-05-17: Hinweis auf Aktualisierung des MDSAP AU P0002.008 ergänzt
- 2022-03: Im Kapitel 1c inhaltlichen Fehler korrigiert (FDA akzeptiert MDSAP auch für initiale Inspektion, siehe Kommentare)
- 2021-03: Kapitel „Updates“ eingeführt



Vielen Dank, sehr gute Zusammenfassung.
Sehr informativer Artikel, vielen Dank.
Spannend dürfte es werden, wenn es um die Details des Bewertungsschemas geht…spontan fallen mir da mehrere Varianten ein:
– SPICE / ISO/IEC 15504 Part 7 – „Assessment of organizational maturity“ nach MedicalSpice/MDevSpice
– oder CMMI
In meinen Augen könnten beide gut für diesen Zweck dienen
Die 15504 schätze ich ebenfalls. Die Breite der Tätigkeiten in den Firmen ist leider so breit, dass so spezifische Kriterien wie in der Norm kaum definiert werden können. Dazu bedarf es weiterer Normen. Beispiele sind Tätigkeiten wie Schweißen, Transportieren, Service, Sterilisieren, Platinenbestücken, Risikoanalyse usw. usw.. Würde man das alles mit aufnehmen, gäbe es für viele Hersteller viele nicht anwendbare Anforderungen.
Vielen Dank für diese Zusammenfassung!
Verstehe ich es richtig, dass, wenn man Produkte in Kanada und mindestens einem weiteren Teilnehmerland vertreibt, bis 2019 das MDSAP-Audit für den Scope Kanada+x absolviert haben muss? Oder würde zum jetzigen Zeitpunkt ein Audit mit Einbezug der kanadaspezifischen Anforderungen genügen?
Es genügt Kanada, weil Kanada bereits die erste Erweiterung (zur ISO 13485) ist. Sie müssen kein weiteres Land im Audit berücksichtigen.
Die FDA-homepage ist zu MDSAP nur mäßig übersichtlich, aber es gibt dort ein Link zu einer FAQ von 2017-08-23. Dort steht unter Frage 95,
„Q: Can the manufacturer exclude a jurisdiction from the scope of an MDSAP audit?
A: A manufacturer may exclude the requirements of a jurisdiction where the organization does not in tend to supply medical devices. In other words, audit criteria under the MDSAP include at a minimum ISO 13485 and the medical device regulations that are applicable in any of the participating regulatory authority’s jurisdiction where the organization supplies medical devices.“
Dies würde ich so verstehen, dass „Kanada+x“ Auditkriterium wäre.
Eine praktische Herausforderung für den Hersteller ist es, für die jeweiligen Teilnehmerländer herauszufiltern, welche Anforderungen im einzelnen anwendbar sind (also „x“, abhängig von der Natur des Produktes). Dies wird die AO in der Regel schon im Vorfeld abfragen, um ein seriöses Angebot abgeben zu können.
Kanada verlangt ab 2019 MDSAP Berichte/Zertifikate fuer all die Produktklassen, fuer die bisher ein CMDCAS Zertifikat notwendig war. Aus diesem Grund sind alle Hersteller, die auf dem kanadischen Markt aktiv sind bzw. es werden wollen, dazu angehalten, zumindest ihr MDSAP Audit bis Ende 2018 durch eine MDSAP Auditorganisation erfolgreich durchgefuehrt zu haben. Tueckisch ist dabei, dass eine MDSAP Auditorganisation aufgrund der gegenwaertigen Anforderungen/Regeln dazu angehalten wird, im MDSAP Audit alle regulatorischen Anforderungen all der MDSAP Laender abzudecken, in denen der Hersteller bereits Produkte vertreibt oder vertreiben moechte. Das waeren maximal Anforderungen fuer 5 MDSAP Laender, die in einem solchen Audit abgedeckt werden muessen, obwohl offiziell fuer den Hersteller bisher nur fuer ein MDSAP Land (Kanada) der Nachweis verpflichtend ist (fuer neue Lizenzen bzw. jaehrliche Lizenzerneuerungen).
Fuer die anderen MDSAP Laender ist das Einreichen eines MDSAP Berichtes/Zertifikates aber (noch) nicht unbedingt verpflichtend. Der Hersteller hat immer noch die Wahl, je nach den landesspezifischen Zulassungsbedingungen zu agieren. Wenn der Hersteller diese „Kuer“ fuer sich selbst nicht umgehend auch zur „Pflicht“ macht bzw. die anderen MDSAP Laender nicht bald dem Aktionismus von Kanada folgen, ist das erste MDSAP Audit fuer so einen Hersteller eine relativ hohe Investition.
Hallo Herr Johner,
bekommt man durch das MDSAP Audit automatisch das 13485:2016 Zertifikat?
Viele Grüße,
Klaus Behrenberg
Danke für die Frage, Herr Behrenberg!
Die Frage müsste Ihnen der Zertifizierer beantworten. Besonders wichtig ist zudem, ob Sie die Anhangs-Zertifikate bekommen – die sind für die Konformitätsbewertung entscheidend. Diese Anhangs-Zertifikate bekommt man üblicherweise nicht automatisch.
Beste Grüße, Christian Johner
Üblicherweise, bekommt man nur die Zertifikate, die man beantragt hat – unter der Voraussetzung, dass das Audit durchgeführt und bestanden wurde sowie das Projekt. On der Zertifizierungsstelle freigegeben wurde.
Ein MDSAP Zertifikat muss auch nicht zwangsläufig zu einem ISO 13485:2016 Zertifikat führen, da die ersten MDSAP-Projekte auf der Basis von ISO 13485:2003 durchgeführt wurden. MDSAP (ISO13485:2003) läuft auch erst am 31.03.2019 offiziell aus. Manche Stellen bieten es deshalb gar nicht mehr auf der 2003er Version an, bzw. informieren Ihre Kunden über die notwendige zeitnahe Umstellung. Geschenkt bekommt man ein ISO 13485:2016 Zertifikat jedenfalls nicht 🙂
Ist es richtig, dass ich für alle MDSAP-Länder, in die ich verkaufe, auch ein MDSAP-Audit durchführen muss?
Kanada ist klar, aber sind die restlichen Länder auch Pflicht?
Sehr geehrte Frau Bochtler,
für die anderen Länder besteht keine Pflicht. Es ist eher eine Möglichkeit, sich Redundanzen zu ersparen. Wenn man nur in einem der anderen Länder in Verkehr bringen will, kann man auch den „lokalen Zulassungsweg“ beschreiten.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
wir haben gerade ein MDSAP-Audit absolviert und unsere benannte Stelle bestand darauf, dass für alle MDSAP-Länder, in die unsere Firma Medizinprodukte der Klasse 2a verkauft, auch ein MDSAP-Audit durchgeführt werden muss!
Können Sie bitte einen Hinweis geben, aus dem hervorgeht, dass diese Pflicht nicht besteht?
Gruß
Liebe(r) Frau/Herr CHE,
ich bin mir nicht ganz sicher, ob ich Ihre Frage richtig verstehe, ich versuche es aber mal:
Während eines MDSAP-Audits werden alle Anforderungen der MDSAP-Länder überprüft, in denen Ihre Firma Medizinprodukte auf den Markt bringt. Es dürfen keine Jurisdiktionen ausgeschlossen werde.
Dies lässt sich aus Chapter 2, Task 1 des MDSAP Audit Approach schließen, des Weiteren wird auf diese Frage im FAQ Papier (Version 018 2022-08-22, Frage 87) eingegangen.
Ich hoffe ich konnte helfen, lassen Sie mich bitte wissen, falls Sie noch Fragen haben.
Schöne Grüße,
Andreas Kalchschmid-Lehmann
Hallo Herr Prof. Dr. Johner,
reicht es aus nur noch ein MDSAP ISO 13485 Zertifikat zu haben oder benötige ich dennoch weiterhin auch ein EN ISO13485:2016 Zertifikat, um Produkte in Europa zu verkaufen ?
Besten Dank und viele Grüße
Sehr geehrter Robert,
Sie benötigen in Europa genau genommen ein Anhangs-Zertifikat (Anhang II MDD bzw. Anhang IX MDR), wenn Sie die entsprechenden Konformitätsbewertungsverfahren durchlaufen. Allerdings macht man alles (Anhangs-Zertifikat, 13485-Zertifikat, MDSAP-Zertifikat) i.d.R. in einem Aufwasch. Wenn Sie allerdings nur ein MDSAP-Zertifikat hätten, würde das nicht genügen.
Viele Grüße, Christian Johner
Hallo Herr Prof. Johner,
die FDA hat vor einiger Zeit einen neuen MDSAP Audit Approach veröffentlicht (MDSAP AU P0002):
https://www.fda.gov/medical-devices/medical-device-single-audit-program-mdsap/mdsap-audit-procedures-and-forms
Dieses Dokument kombiniert die Dokumente „Audit Model“ und „Companion Document“.
Das könnten Sie hier in dem Artikel noch erwähnen.
Beste Grüße
Das ist ein sehr wichtiger Hinweis, Herr von Rüden!
Vielen herzlichen Dank dafür!
Ich aktualisere den Artikel gleich nächste Woche.
Mit nochmaligem Dank und mit vielen Grüßen, Chrisitan Johner
Lieber Prof. Johner,
bezüglich MDSAP habe ich eine kurze Frage. Da ja die Zertifizierung nach MDSAP relativ langwierig ist, kann man die Produktzulassung in den einzelnen Ländern parallel beantragen, oder muss das zwingend seriell erfolgen, erst MDSAP, dann Produktzulassung? (Endgültige Produktzulassung dann natürlich erst mit MDSAP Zertifikat.)
Liebe Grüße
Wolf Kürschner
Sehr geehrter Kürschner,
danke für Ihre wichtige Frage.
Kanada ist das einzige Land, das MDSAP zwingend vorschreibt. Dort ist es so, dass man bei der Anmeldung der Produktprüfung bereits ein MDSAP-Zertifikat mit einreichen muss. Es ist demnach sequentiell.
In Japan, Brasilien und Australien muss, abhängig vom Zulassungsverfahren, das QMS-Zertifikat bei der Beantragung mit eingereicht werden. Für die USA reicht man kein MDSAP-Zertifikat ein, zumindest nicht für 510ks und de Novos.
Viele Grüße, Luca Salvatore, Christian Rosenzweig und Margarita Rozhdestvenskaya
PS:
Die Bibel von MDSAP Auditoren: https://www.fda.gov/media/147457/download
Die FAQ (insbesondere Frage 31): https://www.fda.gov/media/90179/download
Upsi, da hab ich glatt das „Herr“ vergessen, und dabei hab ich doch die Blogseite, wie Herr Prof. Johner angeredet werden möchte, gelesen…
Sehr geehrter Herr Prof. Johner,
unter 1c. „Anerkennung“ schreiben sie, dass die USA MDSAP für „Routine-Inspektionen“, nicht aber für initiale Inspektionen anerkennt. Ist damit das initiale MDSAP Audit gemeint und wo ist das festgelegt?
Mit besten Grüssen
Thomas Farner
Lieber Herr Farner,
die FDA hat eine Webseite mit allen gesammelten Dokumenten und Informationen zu MDSAP: FDA – MDSAP
Dort finden Sie ein Dokument, das von der FDA selbst stammt: FDA
Und darin heißt es auf Seite 2:
FDA QS Inspections
U.S. Food and Drug Administration’s (FDA) Center for Devices and Radiological Health, will accept the MDSAP audit reports as a substitute for FDA routine inspections.
However, all other situations listed under the FDA’s Compliance Program Guidance Manual (CPGM) 7382.845, Inspection of Medical Device Manufacturers, still apply.
Auf Seite 3 heißt es dann, dass zu den Routine-Inspektionen auch die „initialen“ gehören. Sie haben also völlig recht mit Ihrer kritischen Frage. Ich passe unseren Blogbeitrag entsprechend an!
Vielen Dank für Ihre wichtige Rückmeldung.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Prof. Johner,
wir haben zwei Benannte Stellen und nur bei einer Benannten Stelle führen wir MDSAP durch.
Gibt es dann evtl. Probleme, da die eine Benannte Stelle im MDSAP Audit ja gar nicht weiß, welche Produkte über die andere Benannte Stelle in den MDSAP Ländern in Verkehr gebracht werden?
Bräuchten wir dann nicht auch ein MDSAP Zertifikat der anderen Benannten Stelle über diese Produkte, die in die MDSAP Länder vertrieben werden?
Ich hoffe, ich konnte mein Problem verständlich darstellen.
Vielen Dank und viele Grüße Steffi
Liebe Steffi,
die Tätigkeit der Benannten Stellen (BS) ist formal unabhängig von der Tätigkeit der MDSAP-Auditing Organization (AO).
D.h. eine Firma kann als BS fungieren, und kann gleichzeitig als AO fungieren. Es wäre auch möglich, dass Sie eine AO haben, die keine BS ist.
Die BS führt Konformitätsbewertungen im Rahmen der MDR/IVDR (EU) durch, die AO prüft, ob das Qualitätsmanagementsystem eines Medizinprodukteherstellers die Anforderungen an das MDSAP-Programm erfüllt.
Bzgl. der Produkte: Die Auditoren der jeweiligen Organisationen prüfen jeweils nach deren Anforderungen, welche Produkte wo in Verkehr gebracht werden.
Die konkrete Antwort auf Ihre Frage lautet also: Nein, hier sind keine Probleme zu erwarten, und Sie benötigen kein MDSAP-Zertifikat von mehreren verschiedenen Stellen.
Hilft Ihnen diese Antwort weiter? Lassen Sie mich gerne wissen, wenn hier noch Fragen offen sind!
Schöne Grüße,
Andreas Kalchschmid-Lehmann
Sehr geehrter Herr Kalchschmid-Lehmann,
wenn ich nun eine Organisation habe die sowohl als BS als auch als AO fungiert, ich also an einer CE Zertifizierung und einer MDSAP Zertifizierung arbeite, wie groß sind dann die Redundanzen bei den Audits als BS und AO ?
Kann man Zeit (und evtl. Geld) sparen, wenn man beide Prozesse gleichzeitig und mit einer einzigen Organisation durchführt ?
Oder andersherum: Wenn ich eine MSDAP Zertifizierung habe, hilft mir das in irgendeiner Weise beim CE Prozess ?
Vielen Dank
Andreas Schulte
Lieber Herr Schulte,
ja, sofern das Auditteam die entsprechenden Kompetenzen und Autorisierungen aufweist, kann ein sogenanntes kombiniertes Audit durchgeführt werden, d.h. es findet nur ein Audit statt, aber die Auditkriterien umfassen die MDR/IVDR (nötig für die CE-Zertifizierung) und MDSAP.
Gegenüber zwei separaten Audits können so ca. 50% – 80% der Auditzeit (und damit auch Kosten) eingespart werden.
Ich hoffe ich konnte Ihnen hiermit weiterhelfen!
Schöne Grüße,
Andreas Kalchschmid-Lehmann
Sehr geehrter Herr Johner,
im Artikel oben ist zu finden, das Kanada seit 2019 nur noch MDSAP Audits gestattet, nichts desto trotz ist es wohl nicht allen Akteuren klar, das erst ab der Risikoklasse II für Produkte ein MDSAP Programm für das Inverkehrbringen von Produkten in Kanada notwendig ist. Ich schreibe diesen Kommentar, weil die Aussagen sich meiner Meinung wiedersprechen würden, wenn für Kanada nur noch das MDSAP Programm angewendet werden kann, aber Produkte der Risikoklasse I in Kanada trotzdem in Verkehr gebracht werden können.
Danke im Voraus für Ihre Rückmeldung
Ralph Hoffmann
Lieber Herr Hoffmann,
vielen Dank für Ihre Anmerkung!
Die Aussage unter 1c) oben bezieht sich auf die Anerkennung von QMS-Audits. D.h. wenn ein QMS-Audit von Health Canada anerkannt werden soll, dann muss es ein MDSAP-Audit sein, andere Audits (wie z.B. ein Audit nach ISO 13485) wird von Health Canada nicht anerkannt.
Ich hoffe das macht so Sinn, lassen Sie mich gerne wissen, wenn hier noch etwas unklar ist.
Schöne Grüße,
Andreas Kalchschmid-Lehmann
Sehr geehrter Herr Kalchschmid-Lehmann,
vielen Dank für Ihre Rückmeldung, ja mit der Trennung nach Anerkennung des QMS oder Zulassung/Verkaufserlaubnis auf dem Kanadischen Markt, macht Ihre Antwort Sinn.
Viele Grüße
Ralph Hoffmann
Hallo Herr Prof. Johner,
ich habe folgende Aussage gefunden:
Hersteller, die weiterhin ihre Medizinprodukte der Klassen II, III und IV in Kanada vermarkten möchten, müssen daher an MDSAP teilnehmen.
Daraus könnte ich ja ableiten das für Klasse I Produkte keine MDSAP erforderlich ist. Allerdings kann ich in der CMDR aber keinen Nachweis dazu finden. Können Sie mir hier weiterhelfen?
S. Urban
freundliche Grüße
Lieber Herr Urban,
vielen Dank für die Frage!
In der CMDR steht tatsächlich nicht explizit, dass keine QMS nach MDSAP vorhanden sein muss, man kann es sich aber aus Part 1, Section 32 erschließen:
Für Class II Produkte wird in 32 (2) ein „quality management system certificate“ gefordert. genauso für Class III Produkte in 32 (3) und für Class IV Produkte in 32 (4).
Der Absatz 32 (1) dreht sich um Class I Produkte, hier findet man keine Anforderung bzgl. eines QMS-Zertifikates.
Implizit kann man also daraus schließen, dass für Class 1 Produkte kein MDSAP-Zertifikat erforderlich ist.
Ich hoffe die Antwort hilft, liebe Grüße,
Andreas Kalchschmid-Lehmann
Hallo,
der verlinkte Audit Approach ist veraltet, es gibt jetzt MDSAP AU P0002.008 (2023-04-01).
Mit freundlichen Grüßen
Herzlichen Dank „Metzler“!
Sie haben absolut Recht, hier war eine Aktualisierung überfällig. Ich habe sofort das von Ihnen angemerkte Dokument im Artikel erwähnt.
Nochmals vielen Dank!
Beste Grüße, Christian Johner
Dieser Artikel sollte in Hinblick auf den neuen Audit Approach angepasst werden
Lieber Patrick,
vielen lieben Dank für den Hinweis, ich habe die neue Revision im Artikel ergänzt.
Schöne Grüße,
Andreas Kalchschmid-Lehmann