
Das IMDRF (International Medical Device Regulators Forum) möchte zur Harmonisierung der international unterschiedlichen Vorschriften beitragen und dadurch die Zulassung von Medizinprodukten vereinheitlichen und vereinfachen.
Dazu veröffentlichen die Freiwilligen des IMDRF Leitfäden, die zwar nicht verbindlich sind, aber Hilfestellungen geben. Dieser Artikel stellt Ihnen ausgewählte Leitfäden vor.
Sie finden hier die Liste aller IMDRF-Dokumente.
1. Über das IMDRF
Das IMDRF bezeichnet sich selbst als eine freiwillige Gruppe von Regulierungsbehörden für Medizinprodukte aus der ganzen Welt. Diese haben sich zusammengeschlossen, um auf der soliden Basisarbeit der Global Harmonization Task Force on Medical Devices (GHTF) aufzubauen und die internationale Harmonisierung und Konvergenz der Regulierungsvorschriften für Medizinprodukte zu beschleunigen.
Als Ziel hat sich das IMDRF gesetzt,
- die internationale Regulierung für Medizinprodukte zu harmonisieren, um
- damit die Effizienz und Effektivität bei den Beteiligten (Hersteller, Behörden, Benannte Stellen) zu steigern
- und einen Beitrag zur Sicherheit der Patienten zu leisten.
Das International Medical Device Regulators Forum hat wesentlich zur Entwicklung des Medical Device Single Audit Programs MDSAP beigetragen.
Lesen Sie hier mehr zum Thema Medical Device Single Audit Program.
2. Ausgewählte IMDRF-Dokumente
a) Leitfaden zur Anwendung von QM-Systemen bei Standalone-Software
In seinem neusten Leitfaden Software as a Medical Device (SaMD): Application of Quality Management System geben die Autoren Medizinprodukteherstellern, die Standalone-Software entwickeln, Hinweise, wie sie den Forderungen insbesondere der ISO 13485 gerecht werden.
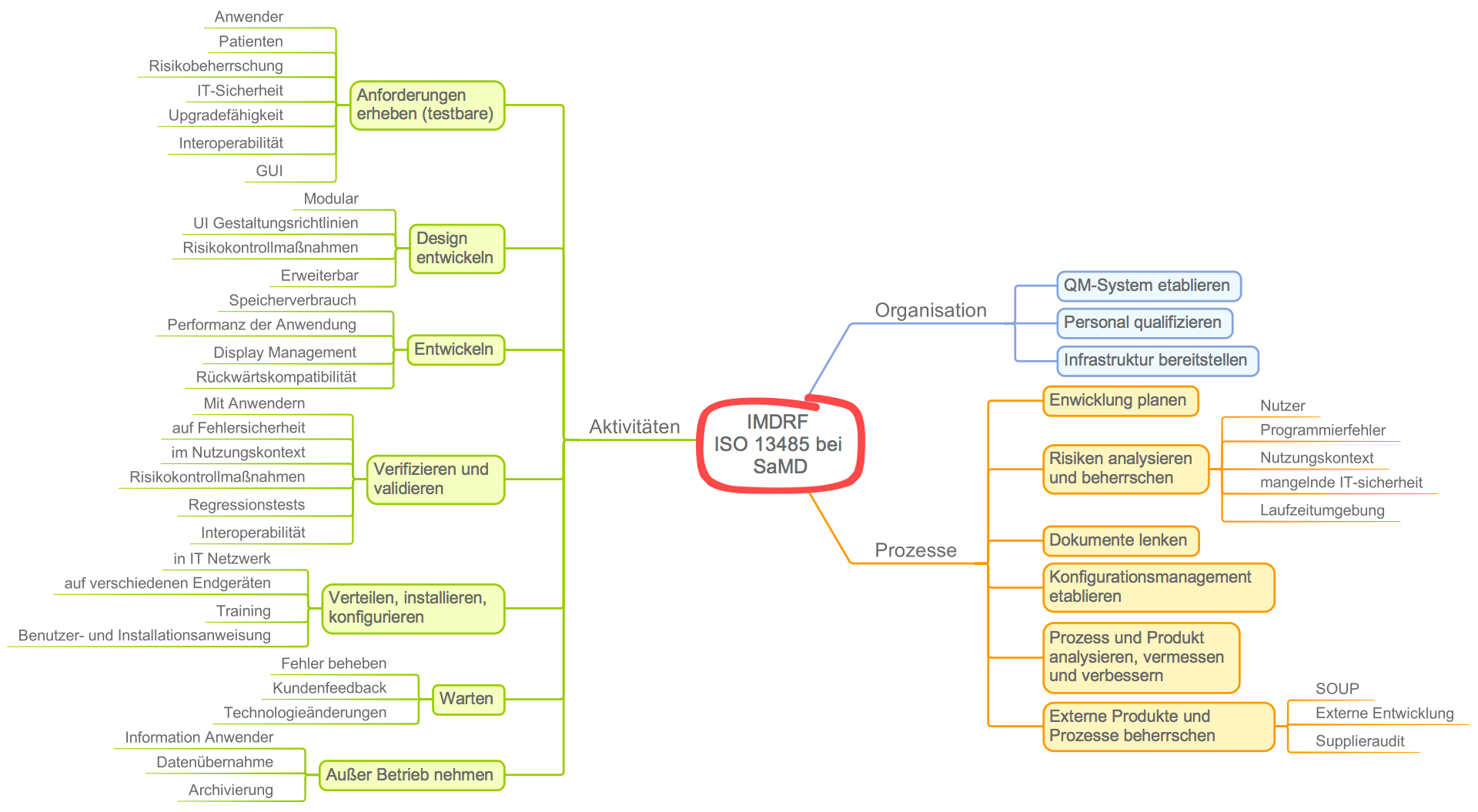
Die Autoren des IMDRF gliedern die Anforderungen in drei Bereiche:
- Anforderungen an die Organisation, z. B. die Verantwortlichkeit, qualifiziertes Personal bereitzustellen
- Anforderung, Prozesse zu etablieren, z. B. einen Entwicklungsprozess, Risikomanagementprozess oder Verbesserungsprozess
- Anforderungen an einzelne Aktivitäten (und damit Methoden), z. B. das Requirements Engineering oder die Software-Architektur
Das IMDRF-Dokument verdient sicher das Prädikat „nützlich“. Gleichzeitig führt dieser Leitfaden in die Irre.
- Abdeckung der ISO 13485: Medizinproduktehersteller könnten vermuten, dass sie die Anforderungen der ISO 13485 einhalten, wenn sie konform mit diesem Leitfaden des IMDRF arbeiten. Dieser Leitfaden ist aber nicht vollständig und erhebt auch nicht den Anspruch darauf. Viele Aspekte der Norm berücksichtigt er nicht, beispielsweise das für Software-Entwickler entscheidende Thema der Messwerkzeuge/Messmittel.
- Abdeckung anderer Normen: Der IMDRF-Leitfaden hat nur die ISO 13485 im Blick. Würde man sich also nur an dessen Vorgaben halten, wären die Anforderungen insbesondere der IEC 62304 und der IEC 62366 nicht erfüllt. Es stellt sich schon die Frage, weshalb das Mapping der ISO-13485-Anforderungen, das die IMDRF-Autoren speziell für Software durchführen, gerade die softwarespezifischen Normen ignoriert.
- Konzeptionelle Integrität und Korrektheit: Die Unvollständigkeit des Dokuments ist kein Zufall, sondern wahrscheinlich der Tatsache geschuldet, dass die Autoren „nur“ Erfahrungen zusammengetragen haben. Ein Modell (abgesehen von dem der drei Bereiche) ist nicht erkennbar. Das IMDRF-Dokument lässt kein wirkliches Verständnis eines modernen Usability and Requirements Engineerings erkennen. Verschiedene Anforderungsklassen sind nicht getrennt, Aktivitäten wie das UI-Design den falschen Phasen bzw. Rollen zugedacht.
Fazit: Der IMDRF-Leitfaden Software as a Medical Device (SaMD): Application of Quality Management System enthält viele hilfreiche Gedanken. Dem Anspruch, damit eine normenkonforme Software-Entwicklung anzuleiten, wird er aber nur bedingt gerecht.
b) IMDRF-Leitfaden zu Risiken von „Software as a Medical Device“
Ein weiterer Leitfaden des IMDRF versucht sich an einem Klassifizierungsschema für Risiken durch „Software as a Medical Device“. Possible Framework for Risk Categorization and Corresponding Considerations nennen die Autoren ihr Werk.

Die Gedanken finde ich prinzipiell gut. Nicht ganz klar sind mir aber die Konsequenzen, die es zu ziehen gilt, wenn man die Risiken klassifiziert hat. Für mich gibt es nämlich nur zwei Kategorien von Risiken: Inakzeptable Risiken sowie Risiken, die man akzeptiert, nachdem man alle möglichen Maßnahmen zur Reduzierung des Risikos angewendet hat.
Diesen Leitfaden hat die MDCG genutzt, um die Konsequenzen der MDR Regel 11 bei der Klassifizierung von Software abzumildern.
c) Weitere IMDRF-Dokumente
Cybersecurity
Die Leitlinie Principles and Practices for Medical Device Cybersecurity beschreibt auf 46 Seiten ziemlich genau das, was man auch findet in:
- FDA Guidances zur Cybersecurity
- AAMI TIR57
- UL 2900
Weshalb es dieses weiteren Dokuments bedarf, erschließt sich mir auch nach mehrmaligem Lesen nicht.
Weitere Leitlinien sind:
- Principles and Practices for the Cybersecurity of Legacy Medical Devices: Dieses Dokument wendet sich sowohl an die Hersteller als auch an die Betreiber. Die Anforderungen sind vergleichsweise anspruchsvoll.
- Principles and Practices for Software Bill of Materials for Medical Device Cybersecurity: Dieses Dokument wendet sich an die Hersteller. Es enthält nicht nur die Verpflichtung, eine SBOM zu erstellen, sondern auch, diese zu verteilen und aktuell zu halten. Sie verbindet diese Forderungen mit dem „Vulnerability Management“.
Personalized Medical Devices
Die Leitlinie Personalized Medical Devices – Regulatory Pathways hat nicht primär etwas mit personalisierter Medizin zu tun. Vielmehr beschreibt sie, wann Produkte als Sonderanfertigungen gezählt werden dürfen und welche Anforderungen Hersteller in diesem Fall einhalten müssen. Beispiele sind 3D-gedruckte Medizinprodukte.
In der ersten Ausgabe hat die Qualitätssicherung beim IMDRF selbst nicht gegriffen: Nicht einmal die Seitenorientierung der Bilder stimmt:
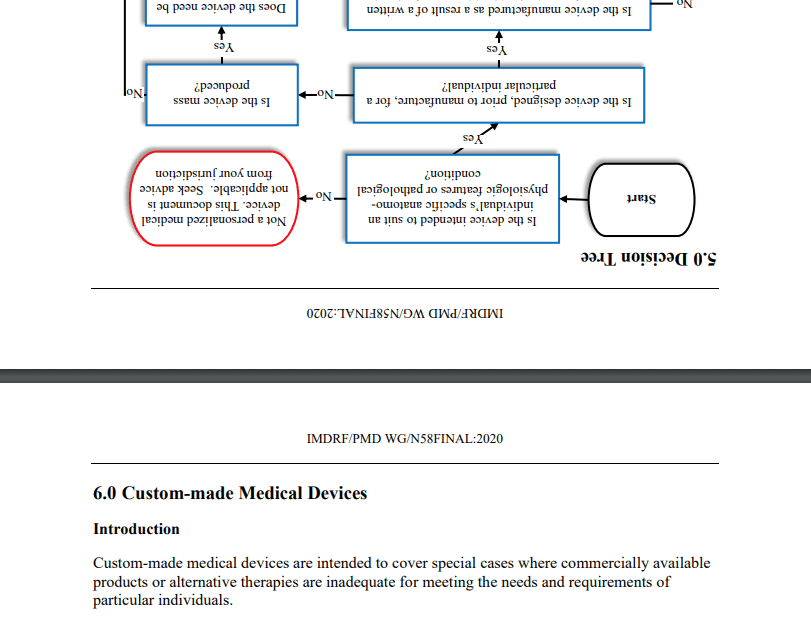
Das IMDRF überlässt es den Lesern, die Anforderungen mit denen z. B. der MDR abzugleichen. Das ist besonders deshalb herausfordernd, weil das IMDRF mit „Pathways“ argumentiert, die die MDR so nicht kennt.
„Seek advice from your jurisdiction“ ist ebenso wenig hilfreich. Genau deswegen liest man doch das Dokument.
Die Ausgabe 2023 hat viele dieser Schwächen behoben.
3. IMDRF Codes
Der IMDRF hat sich die Arbeit gemacht, die Begrifflichkeiten im Kontext von Zwischenfällen zu definieren und in ein kontrolliertes Vokabular zu überführen, dem sogar Codes zugeordnet werden. Konkret geht es um Begriffe in diesen Domänen:
- Zwischenfälle
- Ursachen dafür
- Resultierende Patientenprobleme
- Involvierte Komponenten
Die Begriffe sind hierarchisch gruppiert, stellen also eine Taxonomie dar:
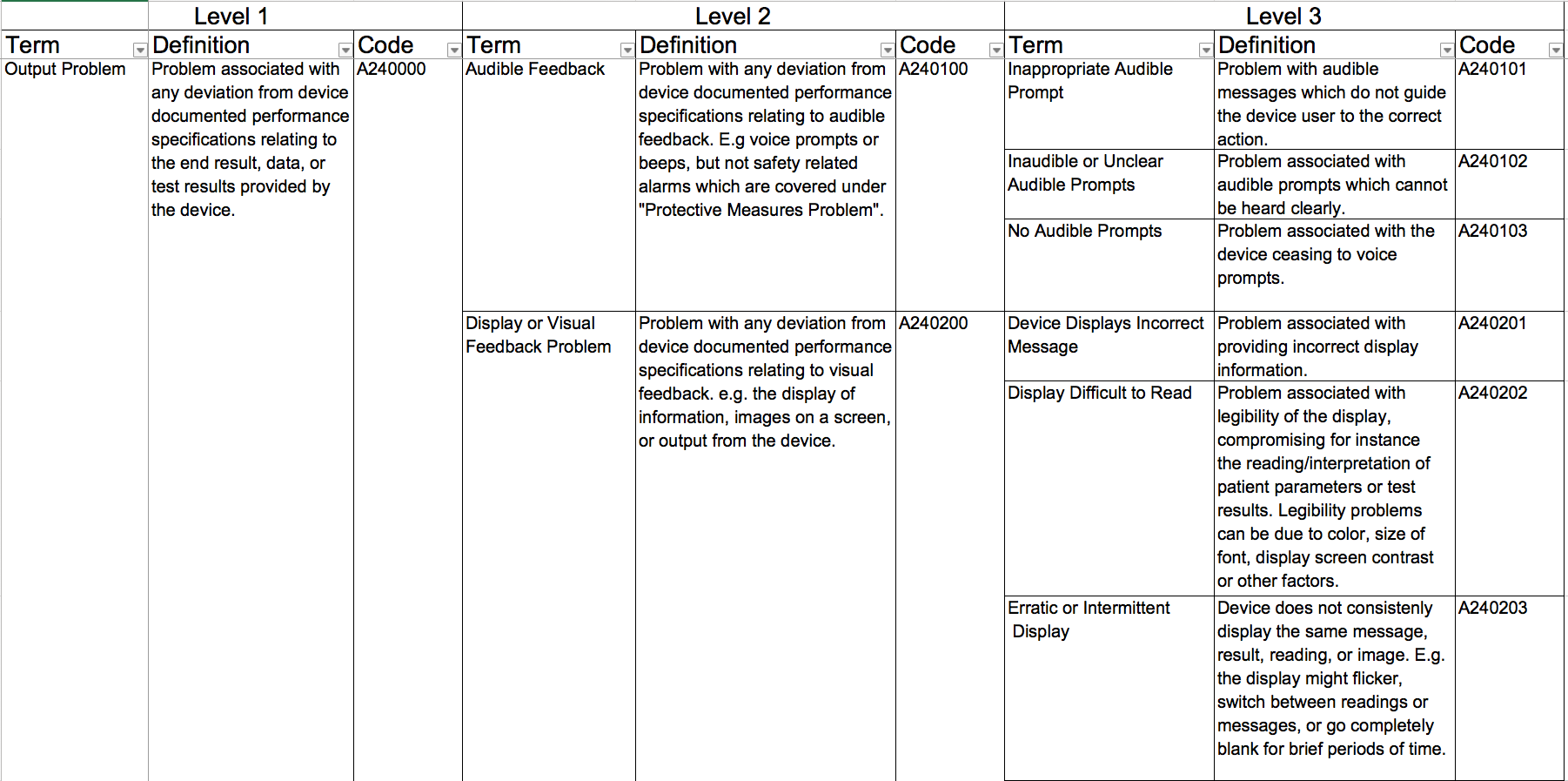
Das IMDRF hat diese Codes publiziert, die beispielsweise für „Incident Reports“ gemäß MDR bzw. MEDDEV 2.12-1 genutzt werden müssen.
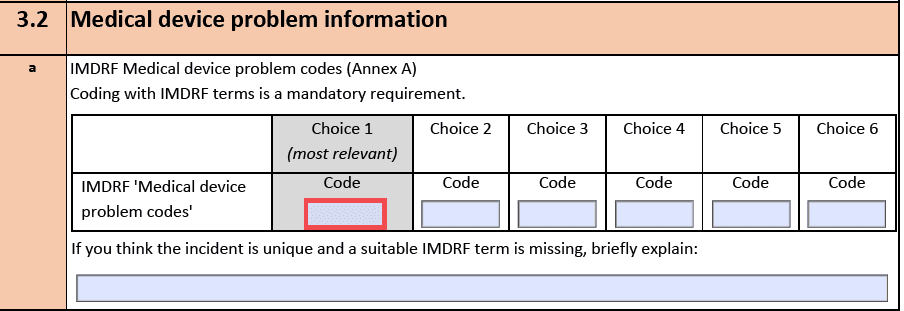
Diese Codes hat das IMDRF auf seiner Webseite publiziert:
- Annex A: Medical Device Problem
- Annex B: Type of Investigation
- Annex C: Investigation Findings
- Annex D: Investigation Conclusion
- Annex E: Health Effects – Clinical Signs and Symptoms or Conditions
- Annex F: Health Effects – Health Impact
- Annex G: Medical Device Component
Beachten Sie, dass die IMDRF diese Codes regelmäßig aktualisiert.
Die FDA nutzt diese Codes auch für ihren Medical Device Report (ebenfalls mit MDR abgekürzt).
- 2023-04-24: Artikel aktualisiert, insbesondere aktuellste IMDRF-Dokumente erwähnt
- 2023-10-25: Kapitel 3 aktualisiert, Verlinkung auf die neueste Ausgabe der Codes



Guten Morgen,
ich stimme zu, der Leitfaden klingt zunächst spannend, denn er scheint „risk-based“ an eine categorization der Software gehen zu wollen. Er bleibt aber an mehreren Stellen hinter den Erwartungen zurück.
– Es werden keine klaren Konsequenzen genannt, für keine der angesprochenen Gruppen. Es werden aber sowohl Hersteller als auch Betreiber angesprochen. Einerseits ist das klar, denn das Dokument soll (darf) keine neuen Requirements ausgeben. Was man aber mindestens sagen kann: Die Inhalte aus Kapitel 9 sollten ein Input in das Risikomanagement der Hersteller werden.
– Referenzen zu ISO 14971 und IEC TR 80002-1 fehlen. Schade, so kann und darf man doch nicht an Risiken mit Software-Medizinprodukten herangehen, dass die konkretesten Normen außer Acht gelassen werden.
– Die ISO 27001 ist zumindest referenziert, die deutlich besser passende 27799 fehlt.
– Die FDA-Guidances für Software sind nicht zitiert.
– Es werden vier Risikoklassen angegeben, die natürlich nicht mit denen nach IEC 62304 oder FDA (level of concern) übereinstimmen können. Schade, sie werden auch nicht in klare Beziehung zueinander gestellt.
Wir machen uns jetzt einmal den „Spaß“ und zitieren bei den nächsten Zulassungen in DE und USA dieses Papier, wenn wir über die Safety Class (oder LoC) sprechen und argumentieren. Mal sehen, was an Kommentaren zurückkommt. Wenn es öffentlichkeitstauglich ist, berichte ich einmal.
Beste Grüße
Peter Knipp