
Diese FAQ geben Antworten auf die häufigsten Fragen, die Firmen wie Medizinproduktehersteller zu Qualitätsmanagementsystemen (QM-Systemen) und zur ISO 13485 haben.
Beachten Sie auch den Übersichtsartikel zu QM-Systemen und der ISO 13485. Er verlinkt auf viele weitere Artikel zu den einzelnen Aktivitäten im Rahmen des Qualitätsmanagements.
Fehlt Ihnen die Antwort auf eine Frage? Dann nutzen Sie gerne unser kostenloses Micro-Consulting.
1. Frage: Wer benötigt ein QM-System?
Sowohl die EU-Medizinprodukteverordnungen MDR und IVDR als auch die FDA stellen Anforderungen an die QM-Systeme. Diese Anforderungen betreffen u. a.:
- Medizinproduktehersteller
- Händler und Importeure, die laut Artikel 16 MDR/IVDR Herstellerpflichten haben
- Organisationen, die Medizinprodukte als neu aufbereiten oder sterilisieren

Diese Anforderungen formulieren die MDR bzw. IVDR in den Artikeln 10 (Abschnitt 9) bzw. in den Anhängen IX.
Bisher spezifizierte die FDA ihre Anforderungen an QM-Systeme im 21 CFR part 820.
Mit der neuen Quality Management System Regulation (QMSR) verfolgt die FDA das Ziel der Harmonisierung mit den ISO 13485:2016 Anforderungen.
Die QMSR ist nun final und gilt ab dem 02.02.2026. Das heißt, die Hersteller haben zwei Jahre Zeit, um die Vorgaben umzusetzen.
Die Übergangsfrist für die Umsetzung der Anforderungen der neuen QMSR endet am 02.02.2026. Unser Expertenteam hilft Ihnen, sich auf die QMSR vorzubereiten.
2. Frage: Muss das QM-System die Anforderungen der ISO 13485 erfüllen?
Ja, denn die EN ISO 13485 ist die einzige Norm, die für die MDR und IVDR harmonisiert ist. Mit der FDA-QMSR muss das QM-System nun auch für den amerikanischen Markt der ISO 13485:2016 sowie einigen zusätzlichen FDA-Anforderungen entsprechen.
Beachten Sie auch unseren Artikel zu den geplanten Änderungen der FDA.
3. Frage: Sind auch andere Normen wie die ISO 9001 erlaubt?
Andere Normen sind erlaubt, sie eignen sich aber nicht allein für den Nachweis, dass die gesetzlichen Anforderungen an das QM-System erfüllt sind. Daher ist die ISO 9001 eine Norm, die Medizinproduktehersteller eher zusätzlich erfüllen.
Weitere Managementnormen wie die ISO 27001 haben nicht die Qualität im Fokus, sondern beispielsweise die IT-Sicherheit. Diese Normen können oder müssen zusätzlich erfüllt werden.
Achten Sie in solchen Fällen darauf, kein paralleles, sondern ein(!) integriertes Managementsystem zu etablieren. Nutzen Sie unsere Hilfe.
4. Frage: Muss das QM-System nach ISO 13485 zertifiziert sein?
Hersteller müssen eine Benannte Stelle in die Konformitätsbewertung einbeziehen, es sei denn, die Produkte fallen in die Klasse I.
Das übliche Konformitätsbewertungsverfahren ist das nach Anhang IX der MDR bzw. IVDR. Dazu benötigen die Hersteller ein Anhangszertifikat.
Streng genommen ist kein ISO 13485-Zertifikat vorgeschrieben. Aber weil die ISO 13485 die Anforderungen des Artikels 10 und des Anhangs IX der MDR bzw. IVDR fast vollständig abdeckt, ist es nicht sinnvoll, auf das ISO-13485-Zertifikat zu verzichten.
Damit ist die kurze Aussage, dass ein ISO-13485-Zertifikat für alle Hersteller (außer für Hersteller von Klasse-I-Produkten) für den europäischen Markt verpflichtend ist, ausreichend korrekt.
Für die FDA zählt ab 2026 zwar ebenso die Konformität mit der ISO 13485:2016, jedoch ist ein Zertifikat nicht zwingend erforderlich.
Üblicherweise nutzen die Hersteller von Produkten der Klassen IIa und höher das Konformitätsbewertungsverfahren nach Anhang IX. Dieses bedingt ein zertifiziertes QM-System. Bei anderen Konformitätsbewertungsverfahren ist das nicht zwingend der Fall. Allerdings eigenen sich diese nur in speziellen Konstellationen.
Melden Sie sich, um schnelle Hilfe dabei zu bekommen, das für Sie passende Konformitätsbewertungsverfahren auszuwählen.
5. Frage: Was sind die Anforderungen der ISO 13485?
Die ISO 13485 verpflichtet die Hersteller u. a. zu:
- QM-Handbuch erstellen inklusive Qualitätsziele und Qualitätspolitik
- Qualitätsmanagementbeauftragten etablieren
- Beschreibung von Vorgabedokumenten wie Verfahrensanweisungen, u. a. zu/zur/zum
- Dokumentenlenkung, einschließlich Dokumentenfreigabe und Aufbewahrungsfristen
- Managementbewertung
- Durchführung interner Audits
- Korrekturmaßnahmen und Vorbeugungsmaßnahmen
- Umgang mit Messmitteln
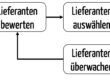
- Lenkung von Lieferanten, z. B. mit Qualitätssicherungsvereinbarungen (QSV)
- Arbeiten konform mit diesen Verfahrensanweisungen
Die Vorgabedokumente lassen sich beispielsweise in Verfahrens- und Arbeitsanweisungen, Templates, Formblätter und Checklisten unterteilen. Die ISO 13485 fordert die Dokumentation von 25 Verfahren.
6. Frage: Wie gelangt man zur Zertifizierung?
Schritt 1: Zertifizierer (Benannte Stelle) auswählen
Die Deutsche Akkreditierungsstelle DaKKs publiziert eine Liste der Benannten Stellen bzw. Zertifizierer, die für eine ISO-13485-Zertifizierung akkreditiert sind. Beachten Sie, dass die zertifizierten Stellen nur für einen gewissen Typ von Medizinprodukten eine ISO 13485-Zertifizierung durchführen dürfen (z. B. für In-vitro-Diagnostika, nicht aktive Medizinprodukte, extra-korporale Kreisläufe).
Schritt 2: Qualitätsmanagementsystem etablieren
Dazu zählt (wie unter Frage 5 beschrieben):
- QM-Handbuch erstellen
- Verfahrensanweisungen beschreiben
- QM-Beauftragten etablieren
Wie Sie mit unserer Hilfe in kürzester Zeit ein schlankes QM-System etablieren können, finden Sie auf dieser Seite beschrieben (zu den „7 Schritten“ scrollen).
Schritt 3: Qualitätsmanagementsystem leben
Um ein Audit beantragen zu können, müssen Sie nicht nur ein QM-System definiert haben, sondern auch schon nach den Regeln dieses (neuen) QM-Systems arbeiten. Es sollte beispielsweise bereits eine Technische Dokumentation für ein Medizinprodukt (weitestgehend) bestehen. Typischerweise sollten Sie auch ein internes Audit sowie eine Managementbewertung durchgeführt haben.
Schritt 4: Benannte Stelle zum Audit einladen
Anschließend laden Sie die ausgewählte Benannte Stelle zu einem Audit ein. Wenn Sie dieses Audit bestehen, wird Ihnen die Benannte Stelle (mit hoher Wahrscheinlichkeit) ein ISO-13485-Zertifikat und ggf. ein Anhang-XI-Zertifikat ausstellen. U. a. damit sind Sie befugt, Medizinprodukte zu vermarkten.
Wir haben für Sie beschrieben, wie der Weg von der ersten Kontaktaufnahme bis zum ISO-13485-zertifizierten System verlaufen kann.
7. Frage: Wie lange dauert es bis zum Zertifikat?
In typischerweise neun bis zwölf Monaten führen wir Firmen zum ISO-13485-Zertifikat (mehr zum Ablauf). Derzeit kann die Überlastung der Benannten Stellen diesen Zeitrahmen etwas verzögern. Entscheidend ist v. a. der Willen der Organisation, das QM-System zu etablieren und zu leben.
8. Frage: Was kostet es und welche Aufwände fallen an?
Der Aufwand hängt stark von der Größe der Organisation und den zu zertifizierenden Bereichen ab. Kleine und mittlere Firmen wenden 30 bis 90 Tage für den Aufbau ihres QM-Systems auf.
Mit der Unterstützung des Johner Instituts (Auditgarant & Templates und/oder Beratung) gelingt es den Firmen, diese Dauer signifikant zu verringern.
Das QM-System dauerhaft zu pflegen, bedeutet weitere Aufwände; diese werden aber durch niedrigere Fehlerfolgekosten (Rückrufe, Nachbesserungen, unnötige Iterationen bei der Entwicklung) kompensiert.
Mehr über die Auditdauer erfahren Sie in unserem Fachartikel Auditdauer: Wie lange Ihr Auditor bleiben darf.
Für die Zertifizierung Ihres Qualitätsmanagementsystems fallen in jedem Fall externe Zertifizierungskosten an. Für die Vorbereitung, das Vor-Ort-Audit und die Nachbereitung des Audits stellt die Zertifizierstelle, abhängig von der Größe des Unternehmens, Kosten zwischen 20.000 und 60.000 EUR in Rechnung. Die Prüfung der Technischen Dokumentation ist inbegriffen.
Falls Sie unsere Unterstützung beanspruchen möchten, fallen weitere Kosten an, die abhängig von der Größe Ihres Unternehmens, dem aktuellen Stand Ihres Qualitätsmanagementsystems und dem Umfang Ihrer Eigenleistung sind. Gerne erstellen wir Ihnen ein passgenaues Angebot.
Die Erfahrung zeigt, dass auch diese Unterstützung sich rechnet, weil dadurch die Kosten für Nacharbeiten und die Interaktionen mit der Benannten Stelle stark reduziert werden.
9. Frage: Wo erhalte ich Unterstützung?
Die Expertinnen und Experten des Johner Instituts unterstützen Medizinproduktehersteller und deren Dienstleister beim Aufbau und bei der kontinuierlichen Verbesserung von QM-Systemen.
a) Aus- und Weiterbildung
Das Johner Institut bietet Seminare für Einsteiger und Fortgeschrittene an:
- Grundlagenseminar zur ISO 13485
- Seminar zum Internen Auditor
- Seminar zu MDSAP
- Zertifizierter 5-tägiger Auditorenkurs
Die Johner Academy vermittelt das regulatorische Grundlagenwissen (nicht nur zum Qualitätsmanagement) in Hunderten von Videos und mit Selbsttrainings.
b) Hilfe zur Selbsthilfe
Der Auditgarant ist ein Programm, das Hersteller beim Aufbau eines QM-Systems unterstützt. Es kombiniert dazu:
- Ein Onboarding, das einen Überblick über den Fahrplan verschafft
- Videotrainings
- Vollständiger Satz an Mustervorlagen, die nur noch an die eigene Situation angepasst werden müssen
- Wöchentliche Sessions mit Experten, die Ihre Fragen beantworten
c) Beratung und Übernahme von Rollen
Die Beraterinnen und Berater des Johner Instituts unterstützen auch direkt. Sie …
- erstellen QM-Systeme für Sie und/oder mit Ihnen.
- prüfen diese Systeme und bereiten auf Audits vor (z. B. mit Mock-Audits).
- betreiben ein QM-System für Hersteller und treten als Legal-Hersteller auf.
- übernehmen die Rolle des oder der QM-Beauftragten.
Interesse? Dann nehmen Sie gleich Kontakt mit uns auf.
Änderungshistorie
- Februar 2024: Hinweise auf die QMSR der FDA ergänzt.
- August 2023: Antworten zu den Fragen 1-3 etwas ausführlicher formuliert. Redaktionelle Änderungen.
- Mai 2023: Erste Version erstellt


Liebes Johner Team,
vielleicht ist es nur ein Tippfehler, aber müsste es nicht in ihrem Punkt 4 zur Anforderung der Zertifizierung eines QM Systems der Konformitätsbewertung nach Anhang I heißen? Sie schreiben Anhang IX, das wiederum ’nur‘ die Anforderungen an die EU-Konformitätserklärung beschreibt. Freundlichen Gruß
Sehr geehrter Herr Ender,
danke für Ihre Rückmeldung. Ich hoffe ich verstehe Ihre Frage richtig. Falls nicht haken Sie bitte nach.
Der Anhang I der MDR beschreibt die Anforderungen an die Produkte. Der Anhang IX heißt „KONFORMITÄTSBEWERTUNG AUF DER GRUNDLAGE EINES QUALITÄTSMANAGEMENTSYSTEMS UND EINER BEWERTUNG DER TECHNISCHEN DOKUMENTATION“. Die Anforderungen an die EU-Konformitätserklärung beschreibt der Anhang IV.
Daher bin ich der Meinung, dass der Text stimmt. Aber vielleicht habe ich Ihren Gedanken noch nicht nachvollzogen.
In jedem Fall besten Dank für Ihr Lesen und Ihre Rückmeldung!
Beste Grüße, Christian Johner
Liebes Johner Team,
laut Frage 4 ist ein ISO-13465-Zertifikat nicht zwingend für reine Klasse 1 Produkte. Wie führt man dann den Nachweis, die Anforderungen der MDR hinsichtlich QMS zu erfüllen? Wäre ein Audit durch einen Dienstleister z.B. ausreichend?
MfG
Sehr geehrter Herr Hüttel,
ich stimme Ihnen absolut zu, dass ein ISO-13485-Zertifikat nicht notwendig ist. Einen Beweis, dass Sie alle QM-Anforderungen erfüllen, können Sie nur schwer erbringen. Denn auch ein Audit durch einen Dienstleister ist wie jede Prüfung eine Stichproben-Prüfung. Allerdings liefert ein solches externes Audit von einem kompetenten Dienstleister nützliche Evidenz, dass diese Anforderungen erfüllt sind. Manche Behörden geben sich damit zufrieden.
Allerdings ist der Anlass einer Überprüfung durch eine Behörde oft ein Zwischenfall. Viele Zwischenfälle sind dadurch verursacht, dass der Hersteller nicht konform den QM-Vorgaben gearbeitet hat.
Geben Sie gerne Bescheid, wenn wir Ihr QM-System auf Konformität prüfen können.
Viele Grüße
Christian Johner