
Medizinproduktehersteller sind verpflichtet, die Aufbewahrungsdauer von Dokumenten und Aufzeichnungen zu beachten und dabei gesetzliche Aufbewahrungsfristen einzuhalten.
Dieser Beitrag verschafft Ihnen einen Überblick über die regulatorischen Anforderungen an die Aufbewahrungsfristen für die verschiedenen Dokumentenklassen.
1. Aufbewahrungsfristen für verschiedene Dokumentenklassen
Die gesetzlichen Anforderungen an die Aufbewahrungsdauer unterscheiden meist folgende Typen von Dokumenten:
- Technische Dokumentation (z.B. Produktspezifikationen, klinische Bewertung, Risikomanagementakte, Gebrauchstauglichkeitsakte)
- Konformitätserklärungen, Bescheinigungen der benannten Stellen
- Dokumentation des Qualitätsmanagementsystems, insbesondere Vorgabedokumente (z.B. Prozess- und Verfahrensanweisungen) und Aufzeichnungen
2. Aufbewahrungsdauer: Regulatorische Anforderungen
Die Kategorieseite „Regulatory Affairs“ verschafft einen Überblick über alle regulatorischen Anforderungen an Medizinprodukte und IVD.
a) Medizinprodukterichtlinie MDD
Die MDD schreibt im Anhang VII zur Konformitätserklärung im zweiten Abschnitt:
„Der Hersteller stellt die in Abschnitt 3 beschriebene technische Dokumentation zusammen. Der Hersteller oder sein Bevollmächtigter hält diese Dokumentation zusammen mit der Konformitätserklärung für mindestens fünf Jahre ab der Herstellung des letzten Produkts zur Einsichtnahme durch die nationalen Behörden bereit. Bei implantierten Produkten beträgt dieser Zeitraum mindestens 15 Jahre ab der Herstellung des letzten Produkts.“
Nahezu gleichlautende Anforderungen finden sich in den Anhängen III (Baumusterprüfung) und VIII („Produkte für besondere Zwecke“).
b) Medizinprodukteverordnung MDR
Technische Dokumentation und Konformitätserklärung
Der zentrale Artikel 10 in der MDR („Allgemeine Pflichten der Hersteller“) verdoppelt im Vergleich zur MDD die Aufbewahrungsfrist für nicht-implantierbare Produkte:
„(8) Die Hersteller halten den zuständigen Behörden die technische Dokumentation, die EU-Konformitätserklärung sowie gegebenenfalls eine Kopie von gemäß Artikel 56 ausgestellten einschlägigen Bescheinigungen einschließlich etwaiger Änderungen und Nachträge noch mindestens zehn Jahre, nachdem das letzte von der EU-Konformitätserklärung erfasste Produkt in Verkehr gebracht wurde, zur Verfügung. Bei implantierbaren Produkten beträgt dieser Zeitraum mindestens 15 Jahre ab Inverkehrbringen des letzten Produkts.“
Diese Aufbewahrungsdauer müssen auch die Importeure bezüglich Konformitätserklärungen und Bescheinigungen beachten.
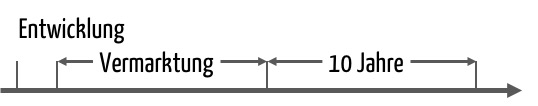
Gelten für die Produkte, die zu einem System oder einer Behandlungseinheit gehören, unterschiedliche Zeiträume, ist der längste Zeitraum ausschlaggebend (Artikel 22).
QM-Dokumentation
Die Anforderungen an das Qualitätsmanagementsystem und dessen Dokumentation beschreibt der Anhang IX. Dort geht das Kapitel III mit den Verwaltungsbestimmungen auf die Aufbewahrungsdauer ein:
„Der Hersteller oder — falls der Hersteller keine eingetragene Niederlassung in einem Mitgliedstaat hat — sein Bevollmächtigter hält während eines Zeitraums, der frühestens zehn Jahre — im Falle von implantierbaren Produkten frühestens 15 Jahre — nach dem Inverkehrbringen des letzten Produkts endet, für die zuständigen Behörden folgende Unterlagen bereit:
- die EU-Konformitätserklärung,
- die in Abschnitt 2.1 fünfter Spiegelstrich genannte Dokumentation und insbesondere die aus den Verfahren gemäß Abschnitt 2.2 Absatz 2 Buchstabe c hervorgehenden Daten und Aufzeichnungen,
- die Informationen über die Änderungen gemäß Abschnitt 2.4,
- die Dokumentation gemäß Abschnitt 4.2 und
- die Entscheidungen und Berichte der Benannten Stelle gemäß diesem Anhang.“
Die im zweiten Punkt referenzierten Dokumente meinen „die Dokumentation über das Qualitätsmanagementsystem des Herstellers,“ einschließlich aller Aufzeichnungen sowie Prozess- bzw. Verfahrensbeschreibungen zu
- Verifizierung, Validierung
- Konformitätsbewertung
- Bestimmung und Nachweis der grundlegenden Sicherheits- und Leistungsanforderungen einschließlich Nachweis entsprechender „Common Specifications“
- Risikomanagement
- Klinische Bewertung, Post-Market Clinical Follow-up
- Produktidentifikation (UDI)
- Spezifikationen auf verschiedenen Herstellungsstufen
- „Handhabung von Änderungen der Auslegung oder des Qualitätsmanagementsystems“
Vereinfachend kann man sagen, dass die Aufbewahrungsdauer (10 Jahre) für die technische Dokumentation sowie für alle wesentlichen Vorgabedokumente (z.B. SOPs) und Aufzeichnungen des QM-Systems einzuhalten sind.
Bei Produkten, die nach Anhang X in den Markt gebracht werden (Baumusterprüfung), müssen die Hersteller bzw. die Bevollmächtigten die Aufbewahrungsdauer zumindest einhalten für:
- Technische Dokumentation gemäß den Anhängen I und II
- Baumusterprüfbescheinigungen, Gutachten
- Änderungsanträge
Für die Konformitätsbewertungsverfahren nach Anhang XI (Produktprüfung) gelten vergleichbare Anforderungen.
Lesen Sie hier mehr zum Thema Medical Device Regulation MDR.
c) ISO 13485:2016
Die ISO 13485:2016 ist nicht so konkret wie die MDD bzw. MDR. Sie fordert, dass die Organisation die Aufbewahrungsdauer festlegen muss. Diese Zeitspanne muss so gewählt sein, dass die Unterlagen „mindestens während der von der Organisation bestimmten Lebensdauer des Medizinprodukts zugänglich sind“. Bei der Wahl der Aufbewahrungsfrist müssen die Hersteller sicherstellen, dass diese
- nicht kürzer ist als „die Aufbewahrungszeitspanne aller sich ergebenden Aufzeichnungen“, die wiederum nicht kürzer sein dürfen als „2 Jahre ab Freigabe des Medizinprodukts“
- nicht kürzer ist als das die regulatorischen Anforderungen verlangen.
Damit sind die Anforderungen der MDR an die Zeitspanne der Aufbewahrung in der Regel höher als die der ISO 13485:2016.
d) EK MED 3.9 A1
Auch die ZLG fühlte sich bemüßigt, die Anforderungen an die Aufbewahrungsfristen zu diskutieren. Sie bezieht sich im EK-MED 3.9 A1 auf die MDD, auf einen Leitfaden der EU aus dem Jahr 1994 und auf die völlig veraltete und längst zurückgezogene DIN EN 46001. Diese wiederum enthält die Anforderung, die inzwischen von der ISO 13485 übernommen wurde:
„Diese Dauer muss sicherstellen, dass Spezifikationen, nach denen Medizinprodukte hergestellt wurden, mindestens für die vom Lieferanten festgelegte Lebensdauer des Medizinproduktes verfügbar sind.“
Eine verlässliche Antwort auf die Fragen nach den Aufbewahrungsfristen gibt die Behörde nicht.
e) FDA
Die FDA benennt die Anforderungen an die Aufbewahrungsdauer im 21 CFR part 820.180. Dort schreibt die Behörde:
„(b) Record retention period. All records required by this part shall be retained for a period of time equivalent to the design and expected life of the device, but in no case less than 2 years from the date of release for commercial distribution by the manufacturer.“
Zusammenfassung
| Regulatorische Vorgabe | Technische Dokumentation | QM-Unterlagen | Konformitätserklärung, Bescheinigungen |
| MDD | 5 Jahre ab Herstellung des letzten Produkts (Implantate: 15 Jahre) | 5 Jahre ab Herstellung des letzten Produkts (Implantate: 15 Jahre) | |
| MDR | 10 Jahre ab Inverkehrbringung des letzten Produkts (Implantate: 15 Jahre) | 10 Jahre ab Inverkehrbringung des letzten Produkts (Implantate: 15 Jahre) | 10 Jahre ab Herstellung des letzten Produkts (Implantate: 15 Jahre) |
| ISO 13485:2016 | Gemäß regulatorische Anforderungen, aber mindestens Lebensdauer des Medizinprodukts, aber nicht kürzer als 2 Jahre nach (erstmaliger) Freigabe | Gemäß regulatorische Anforderungen, aber mindestens Lebensdauer des Medizinprodukts, aber nicht kürzer als 2 Jahre nach (erstmaliger) Freigabe | |
| FDA 21 CFR part 820-170 | Mindestens Lebensdauer des Produkts, aber nicht kürzer als 2 Jahre nach erstmaliger Inverkehrbringung | Mindestens Lebensdauer des Produkts, aber nicht kürzer als 2 Jahre nach erstmaliger Inverkehrbringung |
3. Herausforderungen beim Einhalten der Aufbewahrungsfristen
a) Bestimmung der Aufbewahrungsfrist
Die Anforderungen der MDR sind ziemlich klar. Allerdings sollten die Hersteller bei sehr langlebigen Produkte sicherstellen, dass deren Lebensdauer die Aufbewahrungsfrist gemäß MDR nicht übersteigt.
Das wäre bei Produkten der Fall, die eine spezifizierte Lebensdauer von mehr als 10 Jahren bzw. von mehr als 15 Jahren bei Implantaten haben. In diesen Fällen würde nämlich die ISO 13485 die längeren Aufbewahrungsfristen fordern.
b) Lesbarkeit
Für die Hersteller bedeuten diese langen Aufbewahrungsfristen einen ziemlichen Aufwand. Ein Produkt, das über 10 Jahre produziert wird und das eine vorgesehene Lebensdauer von 10 Jahren hat, zwingt die Hersteller die Dokumente für mindestens 20 Jahre aufzubewahren.
Papier verlangt nach viel Lagerraum und verpflichtet, die Lesbarkeit fortlaufend zu gewährleisten. Eine geeignete Klimatisierung der Lagerräume ist eine Grundvoraussetzung.
Bei digitalen Aufzeichnungen besteht die Herausforderung darin, die Formate über so lange Zeiträume lesbar zu machen. Können Sie heute noch eine Lotus-1-2-3 Datei öffnen? PDF-A kann dabei nur ein Teil der Lösung sein.
Falls die Unterlagen verschlüsselt vorliegen, weil sie beispielsweise personenbezogene Daten enthalten, müssen Verschlüsselungen regelmäßig aktualisiert werden.
c) Hersteller geht in Konkurs
Die Pflicht zum Einhalten der Aufbewahrungsdauer entkommen die Hersteller nicht durch Konkurs. Die MDR fordert von den EU Mitgliedsstaaten, dass diese wiederum die Hersteller verpflichten, in so einem Fall die Dokumentation den Behörden zu übergeben. Dann müssen die Behörden die Aufbewahrungsfristen einhalten.
d) Betreiber
Wird der Hersteller zum Betreiber, gelten zudem völlig andere regulatorische Vorgaben wie beispielsweise die Röntgenverordnung, die Berufsordnung der Ärzte u.v.m. Dies sollten beispielsweise Firmen beachten, die Medical Apps herstellen und dafür einen Server betreiben.
4. Zusammenfassung und Fazit
So sehr man die MDR verdammen mag, sie stellt nun wenigstens klare Anforderungen an die Aufbewahrungsdauer:
In der Regel sind alle Unterlagen, die im Kontext der Herstellung, Produktion und Überwachung des Medizinprodukts erstellt werden, mindestens für 10 Jahre nach der letzten Inverkehrbringung des Produkts aufzubewahren.
Damit haben wir Klarheit. Dem „Eiern“ deutscher Behörden (siehe EK MED 3.9 A1) wurde zumindest beim Thema Aufbewahrungsdauer von Dokumenten und Aufzeichnungen ein Ende bereitet. Das ist gut.



Guten Tag,
vielen Dank für die gute Zusammenstellung. In der Zusammenfassung ist jedoch ein Fehler. Bei der MDR ist die Aufbewahrungsfrist 10 (15) Jahre nach letztmaliger Inverkehrbringung und nicht nach letzmaliger Herstellung. Je nach Shelflife können zwischen Herstellung und Inverkehrbringung durchaus einige Jahre liegen…
Viele Grüße
Britta Cyron
Sie haben Recht! Wird korrigiert! Danke, liebe Frau Cyron!
Wenn Papierdokumente an ein dafür zertifiziertes Unternehmen zur elektronischen Archivierung gegeben werden und die Rückverfolgbarkeit und Lesbarkeit sichergestellt ist, können diese dann nach dem Scannen entsorgt werden?
Ja, das ist okay. Danke für die spannende Frage!
Vielen Dank. Sehr gute Zusammenfassung für ein oft gefragtes Thema. Gilt sowohl für papierbasierte Dokumente (Daten) als auch elektronische Daten.
Sehr geehrter Herr Johner
ein aufschlussreicher Artikel wie immer.
einen Aspekt möchte ich ergänzen: der Kunde hat möglicherweise eigene Anforderungen, welche über die RA Anforderungen hinausgehen. Einer unserer Kunden verlangt z.B. 15 Jahre Aufbewahrungsdauer. Sicher ist sicher.
Das ist eine wichtige Ergänzung! Danke, lieber Herr Pianegonda!
Um die Sache nicht einfacher zu machen: Auch Dokumente und Aufzeichnungen von Unterlieferanten (beispielsweise Netzteile) unterliegen evtl. den Aufbewahrungspflichten. Der Hersteller hat dann die Wahl, sich diese regelmäßig aushändigen zu lassen, oder aber den Lieferanten zur Aufbewahrung mit allen Aspekten (Dokumententyp, Konkursregelung, u.a.) vertraglich zu verpflichten und die Überprüfung in die Lieferantenaudits einzubauen.
Geht dies über internationale Lieferketten kann das zur Herausforderung werden.
Ich hätte eine Frage.
Es heißt immer „mindestens für 10 Jahre nach der letzten Inverkehrbringung des Produkts“
Bei uns wird damit verstanden, dass bei einem MP, das eine Haltbarkeit von 1 Jahr hat und seit 15 Jahren produziert wird, auch die Dokumente für die hergestellten MP von vor 15 Jahren aufgehoben werden müssen.
Das würde aber bedeuteten,so lange ein Produkt produziert wird egal mit welcher Haltbarkeit, dass alle Dokumente aufgehoben werden müssen bis man die Produktion stoppt und ab dem Inverkehrbringen des letzten Produkts zusätzlich 10 Jahre Lagerungsfrist abgewartet hat. So würden aber teilweise 30 Jahre alte Dokumente gelagert. Das kann doch nicht so gemeint sein oder?
Sehr geehrter Herr Larp,
danke für Ihre Frage!
Ich fürchte, dass es so ist. Es macht auch ein stückweit Sinn. Stellen Sie sich vor, ein Patient erleidet einen Schaden mit einem Produkt, das in einer Klinik bereits seit knapp 10 Jahren im Einsatz ist. Dann wollen Sie den Behörden, ggf. dem Staatsanwalt schon nachweisen können, ob Ihr Produkt den grundlegenden Anforderungen entsprach. Dass das Produkt 20 Jahre unverändert verkauft wurde, sollte daran nichts ändern.
Das Beispiel ist sicher nicht alltäglich, weil Produkte selten so lange in den Verkehr gebracht werden.
Aber es gilt trotzdem zu unterscheiden:
Die Entwickungsunterlagen halten Sie in der Tat solange vor. Die Produktionsunterlagen können Sie hingegen 10 Jahre nach Inverkehrbringung des jeweiligen Produkts vernichten.
Mit den besten Grüßen
Christian Johner
PS: Die Regelungen habe nicht ich geschrieben 🙂
Sehr geehrter Herr Johner,
Es hat allerdings wenig Sinn für ein Produkt, das nur eine Haltbarkeit von einem Jahr aufweist, die Dokumente 30 Jahren nach der Nutzung des Produkts noch irgendwo gelagert zu haben. Es gibt immerhin Verjährungsfristen ( § 1 ProdHaftG Verjährung innerhalb von drei Jahren…). Die Rückverfolgung ist so gut wie unmöglich, da die Daten für ein genutzes MP in einer Klinik ebenfalls nicht so lange aufgehoben werden. Für die meisten Daten einer Klinik wie Laborbefunde usw. sind dies 10 Jahre. Darüberhinaus ist immer fraglich ob die Daten noch entsprechend begutachtet werden können, da Rückstellmuster nicht mehr vorhanden sein können bzw. aufgrund der kurzen Haltbarkeit nicht mehr erneut auswertbar sind.
Ich möchte nochmals betonen es geht hier um ein Produkt welches nur ein Jahr Haltbarkeit aufweist und im Normalfall auch nach einem Jahr verbaucht ist.
Vielen Dank soweit für Ihre Hilfe und natürlich weiß ich, dass Sie die Gesetze nicht machen 😉
Mit besten Grüßen
Die FDA Anforderungen sind in im Kapitel 21CFR820.180 genannt nicht in 21CFR820.170
Sehr geehrter Herr Johner,
gibt es analog zur Dokumentenaufbewahrung eine bekannte Regelung für Aufbewahrungsfristen von Verifizierung- / Validierung-Mustern?
Danke.
Mit besten Grüßen
Sven F.
Muster, die für die Verifizierung und Validierung genutzt werden, zählen zur Testumgebung bzw. sind sogar das Testobjekt. Das kann ich Ihrer Schilderung nicht entnehmen.
Sie haben die Pflicht, die Ergebnisse der Verifizierung und Validierung reproduzieren zu können. D.h. es gelten die gleichen Aufbewahrungsfristen, es sei denn, Sie können darlegen, dass mit anderen „Mustern“ das Ergebnis ebenfalls reproduziert werden kann.
Hallo,
Ich habe eine inhaltliche Frage zu dem Gesetz:
VERORDNUNG (EU) 2017/745 DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 5. April 2017 über Medizinprodukte, zur Änderung der Richtlinie 2001/83/EG, der Verordnung (EG) Nr. 178/2002 und der Verordnung (EG) Nr. 1223/2009 und zur Aufhebung der Richtlinien 90/385/EWG und 93/42/EWG des Rates
Können Sie mir Ausskunft geben, ob in dem Bereich der Zahnmedizin/Zahntechnik vorbereitende Produkte (keine Sonderanfertigungen), welche
kurzzeitig in den Körper (Mundraum) eingebracht werden, genauso dokumentiert werden müssen wie die entgültigen Sonderanfertigungen?
Bei den Zwischenprodukten handelt es sich z.B. um individuelle Abformlöffel aus Kunststoff, Kunstoffbasen mit einem Wachsüberzug in den Patienten einbeißen.
Hier könnte es unter Umständen zu einer Materialabgabe im Nanobereich kommen.
Müssen die Inhaltsstoffe dieser Produkte auch auf der Konfomitätserklärung dokumentiert werden?
Vielen Dank für Ihre Antwort.
Sehr geehrter Herr Sanders,
danke für Ihre spannende und nicht ganz einfach zu beantwortende Frage!
Mir ist nicht ganz klar, was Sie mit „vorbereitenden Produkten“ meinen. Wenn das die Zwischenprodukte sind, von denen Sie ebenfalls sprechen, dann scheinen das entweder Medizinprodukte oder Zubehör zu sein. Beide müssen die Anforderungen der MDR erfüllen.
Das Thema „Materialabgabe im Nanobereich“ gilt es im Kontext der Biokompatibilitätsprüfungen zu adressieren. Diese Materialien müssen nicht in der Konformitätserklärung genannt werden, aber ggf. im „Labeling“ und natürlich in der technischen Dokumentation.
Wenn Sie Unterstützung benötigen, dann geben Sie gerne Bescheid z.B. mit einer kurzen Mail an [email protected]. Dann können sich das unsere Experten für die Biokompatibilität und die klinische Bewertung ansehen. Wir haben Erfahrungen auch im Bereich Dentalprodukte.
Mit den besten Grüßen, Christian Johner
Guten Tag Herr Johner,
zu welcher Kategorie werden Wartungsdokumente/Serviceberichte/Sicherheitskontrollen von bereits bei Kunden installierten Medizinprodukten gezählt, wenn man diese als Hersteller durchführt?
Zählen sie zur „Technischen Dokumentation“ oder zur „QM-Dokumentation“?
Oder ist es nochmal ein anderer Bereich? Und gelten dafür dann andere Fristen?
Viele Dank für Ihre Hilfe.
Viele Grüße
Lew Mazlis
Sehr geehrter Herr Mazlis,
im Englischen wären das Dokumente des „Device Master Records“. Ansonsten wäre zu prüfen, ob diese Dokumente im QM-System gefordert sind, dann könnten Sie diese bei den Aufzeichnungen einsortieren.
Zur technischen Dokumentation gehören sie eigentlich nicht, weil man darunter meist die Unterlagen versteht, die für die Einreichung (also „pre-market“) notwendig sind.
Es spricht nichts dagegen, die Dokumente bei den Service-Unterlagen (> QMS) oder den produktspezifischen Unterlagen (Einzelprodukt) aufzubewahren.
Viele Grüße, Christian Johner
Hallo Herr Prof. Dr. Johner,
welche Aufbewahrungsfristen gelten für Distributoren für ihre QM-Unterlagen (Auditpläne/Dokumentenmatrix/Qualifikationsmatrix/Schulungspläne etc.) nach ISO 13485?
Vielen Dank.
Herzliche Grüße nach Konstanz
Markus Wand
Sehr geehrter Herr Wand,
danke für die spannende Frage! Die ISO 13485 unterscheidet keine Hersteller und Distributoren (Händler). Sie kennt nur Organisationen.
Die Anforderungen an die Aufbewahrungsfrist hängen vom Dokumententyp ab. Wenn das Dokument etwas mit der Prüfung des Medizinprodukts zu tun hat (Händler haben auch solche Fristen), dann müssen diese Dokumente während der Lebensdauer der Produkte verfügbar bleiben.
Bei den von Ihnen genannten Dokumenten können Sie die Zeitspanne festlegen. Das sollte aber mindestens bis nach der nächsten Rezertifzierung sein, also nicht nur bis zum nächsten Audit. Eine Ausnahme könnten die Qualifikationsmatrizen sein, weil diese wieder etwas mit der Prüfung der Produkte zu tun haben können.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
Sehr interessante Diskussion zum Thema Aufbewahrungsfristen!
Vielleicht können Sie mir weiterhelfen?
Wo kann ich etwas über die Aufbewahrungsfristen von Klinischen Studien sowie des dabei verwendeten Implantate finden?
Vielen Dank im Voraus
Mit freundlichem Gruß
Ulrike Weinbrenner
Guten Tag Herr Johner,
wir beziehen eine (unkritische) Komponente von einem Lieferanten aus Indien, welche wir in ein Medizinprodukt verbauen. Muss der Lieferant dieser Komponente, welche für sich alleine kein Medizinprodukt ist, die Herstellungsdokumente ebenfalls 10 Jahre aufbewahren? Oder ist es ausreichend, wenn wir als Hersteller des Medizinprodukts die Spezifikation der Komponente 10 Jahre aufbewahren?
Vielen Dank im Voraus für Ihre Hilfe!
Beste Grüße,
Karl-Heinz
Sehr geehrter Herr Suddera,
danke für die spannende Frage! Darauf gibt es keine globale Antwort. Bei einer unkritischen Komponente, insbesondere bei einer „Katalogware“ sehe ich die Aufbewahrungspflicht tendenziell eher bei Ihnen. D.h. ich würde Ihre letzte Frage mit „ja“ beantworten, wobei die Dokumentation ggf. auch die Wareneingangskontrolle umfassen sollte.
Viele Grüße, Christian Johner
Vielen Dank für Ihre Hilfe. Unser Lieferant weigert sich verständlicherweise seine Dokumente 10 Jahre aufzubewahren. Wir werden eine Wareneingangsprüfung durchführen und diese inkl. der Spezifikation 10 Jahre aufbewaren.
Beste Grüße,
Karl-Heinz
Alle im Artikel genannten Literaturstellen handeln von den Vorgabedokumenten – also von Arbeits- und Verfahrensanweisungen usw.
Wie sieht das aber mit der Chargendokumentation aus?
Wir haben das so geregelt, dass wir die Archivierungsfrist für die Chargenaufzeichnungen auf 1 Jahr nach Ablauf der Haltbarkeit der Charge festgelegt haben.
Das ist bisher auch immer von der Benannten Stelle akzeptiert worden.
Ein Kunde von uns ist aber der Meinung, dass die 10 Jahre nach letztem Inverkehrbringen auch für die Chargendokumentaiton zutrifft.
Gibt es für die Chargendokumentation auch irgendwo eine Literaturstelle, die wir angeben können?
Im Voraus schon vielen Dank für die Hilfe!
Sehr geehrte Frau Theiling,
Ihr Kunden bezieht sich möglicherweise auf die Anforderung der MDR bzw. IVDR, die ich im Artikel nenne. Wenn die Chargendokumentation dem entspricht, was z.B. im Anhang II in „4. GENERAL SAFETY AND PERFORMANCE REQUIREMENTS“ oder „6. PRODUCT VERIFICATION AND VALIDATION“, dann wäre das Teil der technischen Dokumentation und unterläge den entsprechenden Aufbewahrungsfristen.
Ich weiß allerdings nicht, was Ihre „Chargenaufzeichnungen“ genau sind. Wenn Sie das gegen die beiden Absätze abgleichen, hätten Sie möglicherweise die Antwort. Andernfalls helfe ich gern weiter.
Viele Grüße, Christian Johner
Hallo Herr Johner,
vielen Dank für die schnelle Antwort.
Bei der Chargendokumentation handelt es sich um die Herstellprotokolle für jede einzelne hergestellt Charge.
Nach meinem Verständnis fällt das eben nicht unter die oben genannten Paragraphen.
Die Vorlagedokumente – also die Herstellanweisungen mit den Blanko Herstellprotokollen bewahren wir selbstverständlich entsprechend der IVDR Angaben auf.
Da die Einzelchargen aber eine definierte Haltbarkeit haben, wir gleichzeitig genau wissen, zu welchem Zeitpunkt welche Versionen der Vorlagedokument gültig waren (und dich auch im Archiv haben), halten ich es eigentlich nicht für notwendig, die Chargenprotokolle der Einzelchargen ebenfalls über diesen langen Zeitraum aufzubewahren. Die Chargen sind ja längest verbraucht und man hat keinerlei Informationsnotwendigkeit mehr. Aus Sicherhietsgründen haben wir hier eine Aufbewahrungsfrist definiert, die ein Jahr länger als das Haltbarkeitsdatum der Einzelcharge ist.
Viele Grüße
Kirsten Theiling
Moin!
Ich habe eine Frage zu Rückstellmustern von hergestellten Medizinprodukten. Da gibt es in einer Firma eine Diskussion über die Anzahl von Rückstellmustern, die aufbewahrt werden müssen, wie lange die aufbewahrt werden müssen, ob das abhängig ist von der Medizinprodukteklasse, und ob man überhaupt welche aufbewahren muss. Ich habe in MDD, MDR, MPG, 13485 dazu nichts gefunden – gibt es da überhaupt (ausserhalb von GMP von Arzneimitteln) irgendwo Vorschiften? Außer man erlegt die sich selbst auf?
Liebe Grüße
Wolf
Sehr geehrter Wolf,
mir sind auch keine Pflichten zu Rückstellmustern bekannt.
Man muss im Rahmen von Audits und Inspektionen auch mal Tests reproduzieren. Das kann dazu führen, dass man dazu Produkte oder Komponenten benötigt. Den Begriff Rückstellmuster habe ich in diesem Kontext noch nicht gehört.
Beste Grüße, Christian Johner
Wie ist die Aufbewahrung von Vertriebsaufzeichnungen auszulegen, insbesondere für die Distributoren?
Artikel 22 (2) der IVDR sagt aus: „Während des in Artikel 10 Absatz 7 genannten Zeitraums müssen die Wirtschaftsakteure der zuständigen Behörde gegenüber Folgendes angeben können:
a) alle Wirtschaftsakteure, an die sie ein Produkt direkt abgegeben haben;
b) alle Wirtschaftsakteure, von denen sie ein Produkt direkt bezogen haben;
c) alle Gesundheitseinrichtungen oder Angehörigen der Gesundheitsberufe, an die sie ein Produkt direkt abgegeben haben.“
Artikel 10 gibt vor „mindestens zehn Jahre, nachdem das letzte von der EU-Konformitätserklärung erfasste Produkt in Verkehr gebracht wurde“
Ist nur der Zeitraum von 10 Jahren zu berücksichtigen, oder mit der Einschränkung der Konformitätserklärung? Müsste ein Händler dies tatsächlich überwachen? Das scheint mir nicht nagemessen.
Absatz (1) des Artikel 22 selbst verweist auf ein „angemessenes Niveau der Rückverfolgbarkeit“
Die Distributionsaufzeichnung selbst ist ja auf ein spezifisches Produkt/Charge bezogen, es wäre also sinnvoll hier den Bezug zur Lebensdauer/shelf life des einzelnen Produktes herzustellen, analog zur Chargendokumentation. Dieser ist hier aber nicht vorgesehen.
Wie würden Sie das bewerten.
Viele Grüße, Kerstin Riemer
Sehr geehrte Frau Riemer,
ich teile Ihre Einschätzung, dass 10 Jahre u.U. ein etwas unangemessen langer Zeitraum sein können. Der Artikel 10 wendet sich auch an die Hersteller.
Die MDR gibt hier keine klare Antwort, eine Kommentierung durch die MDCG ist mir nicht gekannt.
Ihr Einschätzung, für die Aufzeichnungsdauer die Produktlebensdauer zu berücksichtigen, erscheint mir als pragmatisch.
Bei vielen Produkten mit unterschiedlichen Lebensdauern kann es aber auch einfacher sein, eine Datenbank zu pflegen, in der man beispielsweise abspeichert, an welchen Kunden man welche Produkt geliefert hat.
Bitte beachten Sie in dem Kontext auch andere Fristen z.B. aus dem Steuerrecht und Produkthaftungsrecht. Diese Unterlagen sind ggf. länger aufzubewahren.
Viele Grüße, Christian Johner
Sehr geehrtes Johner-Team,
gibt es mittlerweile Handlungsempfehlungen oder ein MDCG Paper zur Aufbewahrungspflicht für Händler? Ein Händler hat nicht zwingend Zugang zu der Produktlebensdauer des in Verkehr gebrachten Produkts. Im speziellen Fall geht es am Markt sicherlich vorrangig um MDD Produkte. Gemäß § 257 Abs. 4 HGB beträgt die Aufbewahrungsfrist 10 Jahre.
Ist es, auch aus Sicht des Medizinprodukteherstellers vertretbar, wenn der Händler die Rückverfolgung seiner Produkte (nur) über 10 Jahre aufrecht erhält?
Lieber Herr Wagner,
bei der Frage nach der Aufbewahrungspflicht von Dokumenten bei Händlern gibt es viele Gesichtspunkte. Sie schränken das Thema in Ihrer Frage auf die Dokumentation zur Produkt-Rückverfolgung ein.
Artikel 25 der MDR legt für alle Wirtschaftsakteure und damit auch für den Händler fest:
Der in Artikel 10 Absatz 8 genannte Zeitraum ist 10 bzw. 15 Jahre.
Interessanterweise berücksichtigt der Gesetzgeber hier die Lebensdauer des Produktes nicht.
Herzliche Grüße
Christian Rosenzweig
Hallo Herr Johner,
wir sind bislang, was das Archivieren von Entwicklungsartefakten betrifft, recht großzügig gewesen, um rechtlich auf der sicheren Seite zu sein.
Nun stellte sich hier ein Kollege – berechtigterweise, wie ich finde – die Frage, ob es in den heutigen Zeiten ökologisch eigentlich vertretbar ist, diese Politik zu fahren, schließlich kommen z.B. durch das Archivieren von Installern, die für offizielle Tests während der Entwicklung eingesetzt wurden (aber niemals ausgeliefert wurden), nicht unwesentliche Datenmengen zusammen, die auf WORM-Speichern nun ihr Dasein fristen, ohne vermutlich jemals wieder das Tagelicht zu sehen.
Wir denken, dass wir Installer, die für Tests genutzt wurden, ggf. auch wiederherstellen können. Je größer der Zeitraum zwischen dem originalen Build-Datum des Installers und „heute“ aber ist, desto aufwändiger und risikobehafteter wird der Prozess, da sich zwischenzeitlich die Buildumgebung ändert, die dann ebenfalls wieder hergestellt werden müsste.
Haben Sie einen Tip für uns, wie wir unsere Archivierungsstrategie diesbezüglich evtl. besser ausrichten können?
Viele Grüße
Anna Henke
Liebe Frau Henke,
bitte haben Sie noch ein wenig Geduld. Wir werden Ihnen zeitnah antworten!
Herzliche Grüße
Anja Segschneider | Redaktion
Liebe Frau Henke,
vielen Dank für diese spannende Fragen!
Wir sind uns im Team nicht ganz sicher, ob wir alle Fragen wirklich verstanden haben, daher hat unsere Antwort etwas gedauert.
Wir verstehen einen Installer als ein Software-Programme zum Installieren von Software-Programmen. Ist das auch Ihr Verständnis? Falls nicht würde ich Sie bitten, uns auf die richtige Spur zu bringen.
Es geht bei den Aufbewahrungsfristen darum, dass sie Ergebnisse wieder reproduzieren können. Ob ein Installer aufbewahrt werden muss, hängt davon ab, was Sie damit nachweisen wollen. Wenn Sie beispielsweise „nur“ das „Testobjekt“ (z.B. die installierte Software) wieder herstellen wollen, um einen Test zu reproduzieren, benötigen Sie nicht notwendigerweise Installer, sondern nur die entsprechende Software. Dieses können Sie auf verschiedene Weisen herstellen z.B. als
Eine Speicherung auf WORM-Speichern hat zwar den Charme, dass der Nachweis, dass der Inhalt unverändert ist, leicht zu führen ist. Aber dieser Ansatz ist, wie Sie anführen, ökologisch zunehmend fragwürdig. Daher können Sie auch andere Ansätze zur Archivierung wählen z.B.
Bei all diesen Varianten würde man Veränderungen mit ausreichend hoher Wahrscheinlichkeit bemerken. Die Maßnahmen sollten vom Risiko abhängen und z.B. in einer „offenen Umgebung“ (um den Begriff des 21 CFR part 11 zu nutzen) höher sein als in einer geschlossenen.
Ich kann nicht ausschließen, dass Sie sich viel Arbeit machen, die man ggf. durch Automatisierung reduzieren kann. z.B. könnte ein Skript einen finalen Test durchführen und direkt die Archivierung vornehmen. Der erste Impuls sollte auch bei der Archivierung sein, dass es kaum noch manuelle Arbeitsschritte gibt.
Es gibt jedoch ein „Aber“: Die Software, die Ihnen die Tests durchführt und die Artefakte archiviert fällt unter die Anforderungen der ISO 13485 z.B. Kapitel 4.1.6 (CSV).
Falls wir Ihre Fragen missverstanden haben sollten, dann melden Sie sich einfach.
Viele Grüße, Christian Johner
Hallo Herr Johner,
ich hoffe, meine Antwort wird richtig an der richtigen Stelle dargestellt – der Antworten-Link verhält sich etwas seltsam…
Zunächst mal – ja, ich glaube Sie haben meine Fragen im Grunde richtig verstanden. Wenn ich von einem Installer spreche, beinhaltet dieser alles, was wir im Buildsystem bauen, was notwendig ist, um unsere Software zu installieren, so dass sie nach der Installation lauffähig ist. Also, nicht nur den Installer, sondern auch die Software an sich. Die Größenordnung ist ~350MB pro archiviertem Installer. Somit können wir mit einem Installer auf einem entsprechenden System das Testobjekt (also die installierte Software) jederzeit wieder herstellen. Den Prozess des Archivierens von Builds, die wir für offizielle Tests erstellen, haben wir bereits automatisiert – jedoch erscheint es wiegesagt etwas … naja… „unüberlegt“ im Hinblick auf die erzeugten Datenmengen.
Im Hinblick auf die Wiederherstellbarkeit des Buildsystems sind wir im Grunde schon ganz gut aufgestellt, da wir SourceCode und Buildskripte bereits unter Versionskontrolle haben. Die Änderung der Buildumgebung (Update des Buildsystems, Betriebssystemupdates etc.) findet zwar kontrolliert, jedoch nicht zwingenderweise unter der Prämisse der Wiederherstellbarkeit über den Aufbewahrungszeitraum hinaus, statt.
Risikobasiert wäre es dann vermutlich sehr vertretbar, nur die ausgelieferten Installer auf die alt-herbgebrachte Art zu archivieren und lieber dahingehend zu investieren, die Buildumgebung an sich zu sichern, so dass sie auch nach Jahren noch wiederherstellbar ist. Ein gewiefter Kollege hat unserem Buildsystem bereits beigebracht, sich selbst täglich zu bauen und in ein Repository zu schreiben. Bleibt also lediglich das System an sich, auf dem Buildsystem läuft. Das werden wir aller Voraussicht nach über VM-Snapshots realisieren, so dass wir dann ab dem neuen Jahr unser Archiv entlasten und unseren okölogischen Fußabdruck deutlich verringern können.
Vielen Dank für Ihre Gedanken und ein Frohes Fest!
Anna Henke
Guten Tag Herr Johner
Wenn papierbasierte Dokumente digitalisiert werden (z.B. eingescannt und als PDF gespeichert werden), können dann die Originale auf Papier entsorgt werden?
Wo finde ich dazu verlässliche Anforderungen was wir einhalten müssen z.B. Validierung bzw. Qualitätskontrolle von eingescannten Inhalten?
Aktuell werden Dokumente bei uns sowohl eingescannt, weil einfacher verfügbar, aber auch als Papiere im Archiv abgelegt. Das ist ein doppelter Aufwand den wir gerne reduzieren möchten.
Beste Grüsse
Sandra Blakolmer
Sehr geehrte Frau Blakolmer,
besten Dank für Ihre Frage!
Eine Norm oder etwas Vergleichbares zum Digitalisieren von Unterlagen, die im Kontext des Medizinprodukterechts relevant sind, ist mir nicht bekannt.
Wie Sie sagen, ist die Validierung dieses Prozesses entscheidend. Dazu müssen Sie die Kriterien bestimmen, die dieser Prozess erfüllen muss. Und die hängen z.T. wieder von Ihrer speziellen Situation ab. Beispielsweise müssten wahrscheinlich für gewisse Zeichnungen höhere Auflösungen erreicht werden, als für einen Brief. Achten Sie auf die Anforderungen der MDR in Anhang II. Die Durchsuchbarkeit werden Sie nur mit OCR erreichen. Vergessen Sie auch nicht die Anforderungen an die Lenkung der digitalen Unterlagen. Dazu zählt auch deren Erhaltung. Themen wie Zugriffsrechte und Erprobung von Backups sind somit Aspekte Ihrer Validierung.
Viele Grüße
Christian Johner
Lieber Prof. Dr. Johner,
ich hoffe ich habe die Antwort in den vorherigen Kommentaren nicht überlesen. (Eine Frage war ähnlich aber ich konnte dich Antwort nicht sicher rauslesen)
Wie sieht es mit der Aufbewahrungsdauer der Abnahmeprotokolle (bzw. des ganzen „Device History Record) der ausgelieferten Produkte in Europa aus?
Wir nehmen hier bisher auch die 15 Jahre nach dem Inverkehrbringen des letzten Produkts an. Aber was ist gemäß MDR / AIMDD gefordert? Unter die oben genannten drei Kategorien fallen diese Aufzeichnungen nach meinem Verständnis nicht. Die FDA hingegen bezieht ihre Forderung ja auch auf das DHR.
Vielen Dank und beste Grüße
Nadine Langguth
Liebe Frau Langguth,
bitte entschuldigen Sie die ungewöhnlich lange Wartezeit. Sie bekommen zeitnah eine Antwort!
Herzliche Grüße
Anja Segschneider | Redaktion
Liebe Frau Langguth,
die MDR bezieht sich in der Angabe der Aufbewahrungspflicht in Artikel 10 (8) auf die Technische Dokumentation. Diese enthält nach Anhang II Abschnitt 3 (b) der MDR auch die Produktionsdokumentation z.B. zur finalen Produktprüfung. In meinen Augen ist damit auch der DHR gemeint, der ja neben der Identifikation und Konfiguration des Produktes auch dessen Prüfprotokolle aus der Produktion umfasst. Demnach muss also für jedes gefertigte individuelle Produkt die entsprechende Aufzeichnung 10 (bzw. bei Implantaten 15) Jahre aufbewahrt werden. Das ist für mich logisch, denn es geht darum, für jedes im Markt befindliche Produkt die komplette Technische Dokumentation rekonstruieren zu können, um im Bedarfsfall die Produktkonformität mit allen regulatorischen und abgeleiteten technischen Anforderungen zu belegen.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Prof. Dr. Johner,
beim Thema Aufbewahrungsfristen stellt sich uns die Frage bezüglich der Lebensdauer eines Produktes.
– Handelt es sich bei der spezifizierten Lebensdauer um das Haltbarkeitsdatum?
– Bedeutet Lebensdauer bei einem Implantat die uneingeschränkte Funktionalität? (unbegrenzt?)
Welche Dokumente (TD, Beschaffung, Wareingangsprüfung, Herstellberichte, Sterilisation) müssen demgemäß 10 oder 15 Jahre aufbewahrt werden?
Für eine Erläuterung vielen Dank im Voraus!
Mit freundlichen Grüßen
MHMedical-Tec GmbH
Detlev-Sven Neumann
Lieber Herr Neumann,
was genau Lebensdauer ist und wie sich diese von „Haltbarkeitsdauer“ abgrenzt, lesen Sie in unserem Blogbeitrag: Was macht die Lebensdauer von Medizinprodukten aus?.
Kurz zusammengefasst: Lebensdauer ist die Zeitspanne, für die der Hersteller Sicherheit und Leistungsfähigkeit des Produktes nachweist und die er in der Technischen Dokumentation explizit angibt und damit im Risikomanagement berücksichtigt. Beim Implantat wäre das sicher die Funktionsfähigkeit, aber eben auch andere „Grundlegende Sicherheits- und Leistungsanforderungen“.
Welche Dokumente Sie aufbewahren müssen, ist vielleicht am ehesten mit dieser Aussage ausgedrückt: „Alle Vorgabedokumente und Aufzeichnungen, mit denen Sie die regulatorische Konformität bezogen auf ein individuelles Produkt im Markt nachweisen können“. Das bezieht sich auf das QMS und entsprechende Aufzeichnungen genauso, wie auf produktspezifische Dokumentationen.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrtes Team vom Johner Institut,
Etwas weiter oben im Block stellte Frau Weinbrenner diese Frage:
„Wo kann ich etwas über die Aufbewahrungsfristen von Klinischen Studien sowie des dabei verwendeten Implantate finden?“
welche hier im Blog leider nicht weiter berücksichtigt wurde.
Meine Frage geht in ähnliche Richtung. Die MDR, Anhang III „Weitere Pflichten des Sponsors“ beschreibt die aufzubewahrenden Dokumente und nennt die üblichen 10(15) Jahre nach Studienende oder Inverkehrbringen. Zu den genannten Dokumenten gehört auch der Studienbericht mit „Ergebnisanalyse in Bezug auf die ausgewählten Endpunkte“.
Mir ist nun unklar ob auch die Rohdaten ggf. inkl. (e)CRF dazu gehören oder diese Aufbewahrungsfristen aus anderer Gesetzgebung unterliegen.
Herzlichen Dank bereits vorab für Ihre Antwort.
Viele Grüße,
Joachim Schneider
Lieber Herr Schneider,
ich würde aus meiner jetzigen Perspektive die Rohdaten genauso wie die Klinische Dokumentation behandeln und sie ebenfalls mindestens 10/15 Jahre lang aufbewahren.
Das gilt dann auch für die eCRF-Daten und -Systeme.
Allerdings kann es sein, dass mit diesen Daten auch andere Rechtsbereiche tangiert werden, z.B. Haftungsrecht, Ärztegesetze, Arzneimittelgesetz, Krankenhausgesetze, Röntgenschutzverordnung, spezifische nationale Gesetze. Unter Umständen sind dadurch andere bzw. höhere Aufbewahrungszeiten verknüpft.
Bei einer kurzen Recherche habe ich diesen Hinweis gefunden:
Es scheint deshalb sinnvoll, sich eingehender mit dem Thema zu befassen und im Zweifelsfall Juristen zu befragen, z.B. Frau Sonja Seubert, die uns in solchen Fragen auch unterstützt.
Herzliche Grüße
Christian Rosenzweig
Lieber Herr Johner,
gilt die Aufbewahrungsfrist auch für die Produkte in der Übergangsphase und „noch“ unter der Rechtlinie 98/79 fallen.
Denn bis lang war für diese Produkte eine Frist von 5 Jahren vorgesehen
Liebe Grüße
M
Liebe M,
ich würde interpretieren: ab dem Geltungstag der IVDR (26. Mai 2022) müssen alle Produkte, die unter die neue IVDR fallen, deren Aufbewahrungsfristen (zehn Jahre) erfüllen. Das gilt dann für alle Dokumente, die ab diesem Tag neu erstellt werden, oder diejenigen, die noch existieren und von der Fristverlängerung betroffen sind.
In Zeiten der Digitalisierung sollte das hoffentlich keine übermäßige Belastung für Hersteller mehr darstellen.
Herzliche Grüße
Christian Rosenzweig
Lieber Herr Prof. Johner,
Hat Deutschland die von Ihnen geschilderte Verpflichtung für den Konkursfall erfüllt? Ggfls mit welchem Gesetz ? Von einem Benannten Unternehmen wurde uns mitgeteilt wir müssten für unseren Konkursfall selbst vorsorgen.
Vielen Dank
Lieber Herr Schöfer,
die MDR fordert in Anhang IX, Absatz 8 von den Mitgliedstaaten:
Entsprechend wurde es im Medizinprodukterechtdurchführungsgesetz (MPDG) in Deutschland umgesetzt:
Das heißt, Sie müssen sich im Falle Ihrer Geschäftsaufgabe tatsächlich selbst um die Übergabe der Dokumente kümmern.
Herzliche Grüße
Christian Rosenzweig
Vielen Dank und einen schönen Arbeitstag.
Lieber Herr Rosenzweig,
Darf ich noch ergänzend fragen,ob auch die Art und Weise geregelt ist,wie diese Sicherstellung erfolgen muss.
Vielen Dank
Lieber Herr Schöfer,
man würde vermutlich keinen Prozess dafür in Ihrem QMS erwarten. Wichtig ist die Umsetzung dieser Anforderung im Bedarfsfall. Die zuständige Behörde wäre in Ihrem Fall diejenige, die in Ihrem Bundesland die Überwachung der Medizinproduktehersteller durchführt (z.B. Regierungspräsidium). Ich kenne keinen Fall bisher aus der Praxis, aber ich empfehle, mit dieser Behörde einfach Kontakt aufzunehmen und zu erfragen, wie die dort festgelegten Prozeduren sind.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrtes Team vom Johner Institut,
Beim Punkt 3c) Hersteller geht in Konkurs ist nur von der MDR die Rede, gibt es diesbezüglich auch Anforderungen von der Schweiz, UK oder USA?
Vielen Dank
Alisia
Liebe Alisia,
vielen Dank für diese interessante Zusatzfrage!
Bei der FDA gibt es eine Forderung im Zusammenhang mit dem PMS:
Section 822.27: „If I go out of business, what must I do?“
In der Schweiz sind mir im Moment keine besonderen Bestimmungen bekannt.
In UK wird vermutlich dazu etwas in der neuen kommenden Verordnung geregelt sein. Die wird allerdings erst im Herbst 2023 erwartet.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrtes Johner Team,
vielen Dank, für den aufschlussreichen Artikel.
Wir sind Hersteller von Sonderanfertigungen. Wir machen OP-Planungen per Software und stellen die zugehörigen Bohrschablonen her. Die digitale OP-Planung, sowie die Bohrschablone wird dem Kunden zur Verfügung gestellt. Welche Aufbewahrungsfrist gilt dann für die digitale Planung? Wir sehen diese als Produktionsbezogene Informationen (für die Bohrschablone) an, welche eine Frist von mindestens zehn Jahren haben. Allerdings ist die zu speichernde Datenmenge enorm. Jetzt stellt sich uns die Frage, ob wir die digitale OP-Planung überhaupt archivieren müssen, da der Kunde diese ja auch bekommt? Können Sie uns dazu eine Einschätzung geben?
Vielen Dank schon mal im Voraus
Jula Knieps
Liebe Frau Knieps,
für die Aufbewahrungsfrist der Daten und Schablone sehe ich wie Sie prinzipiell die 10 Jahre als geltend.
Eine kleine Unsicherheit ergibt sich aus der Formulierung der MDR. In Anhang XIII heißt es
„4. Die in der Einleitung von Abschnitt 1 genannte Erklärung wird für einen Zeitraum von mindestens zehn Jahren nach dem Inverkehrbringen des Produkts aufbewahrt. Bei implantierbaren Produkten beträgt dieser Zeitraum mindestens 15 Jahre.“
Hier scheint sich die Frist also nur auf die Erklärung zu beziehen.
In Abschnitt 2 steht:
„2. Der Hersteller verpflichtet sich, für die zuständigen nationalen Behörden die Dokumentation bereitzuhalten, die seine Fertigungsstätte bzw. Fertigungsstätten angibt und aus der die Auslegung, die Herstellung und die Leistung des Produkts, einschließlich der vorgesehenen Leistung, hervorgehen, sodass sich beurteilen lässt, ob es den Anforderungen dieser Verordnung entspricht.“
Das bezieht sich konkret auf die Fertigungsunterlagen, also Ihre digitalen Planungsdaten und die Bohrschablone. Hier wird aber keine Aufbewahrungsdauer angegeben!
Ob das ein Fehler der MDR ist oder eine bewusste Unschärfe, kann ich nicht sagen.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Professor Johner,
für den gesetzten Fall, dass ein medizinisches Softwareprodukt abgekündigt wird, aber noch einige Monate Updates angeboten werden gelten die zehn Jahre mit der Abkündigung oder erst mit dem letzten Softwareupdate?
Herzlichen Dank
Georg Bilow
Lieber Herr Bilow, eine sehr gute Frage! Es kommt (wie immer) darauf an. Wenn ein Update im Grunde eine neue Produktversion ist (neue Features, neues Major-Release), dann ist das die Inverkehrbringung eines Produktes und würde die 10-Jahres-Frist neu starten. Wenn es sich bei dem Update nur um ein Bugfix/Patch handelt, dann eher nicht.
Herzliche Grüße
Christian Rosenzweig
Mit einem Hallo in die Runde würde ich gerne auch noch eine Frage stellen:
Da wir nicht Hersteller eines Produktes sind, sondern das hergestellte Produkt bei uns bearbeitet wird ( anodisieren ) ist nicht ganz klar, wie lange unsere Dokumente aufbewahrt werde müssen.
Meiner Ansicht nach, bewegt sich die Vorgabe max. 30 Jahre oder die lebenslange Dauer des Implantates.
Man kann ja nicht sagen, wir machen nur die Schicht, alles Andere geht mich nichts an, oder liege ich hier völlig falsch.
Vielen Dank für Ihre Antwort / Mithilfe..
Lieber Herr Sigg,
die gute Nachricht: aus Sicht der Regulatorik für Medizinprodukte müssen Sie zunächst in Ihrer Lieferanten-Rolle keine gesetzliche Anforderung an die Aufbewahrungsdauer beachten. Die wird Ihnen vielmehr durch den Medizinproduktehersteller als Auftraggeber mitgeteilt. Meist steht sie im Vertrag oder in der Qualitätsvereinbarung. Dort wird dann auch die Verpflichtung genannt, jederzeit ein Lieferantenaudit oder ein Audit durch die Benannte Stelle des Herstellers zu ermöglichen. In diesem Rahmen könnten dann solche Dokumente geprüft werden.
Sie müssen aber auch auf andere Gesetze achten, wie das Handelsrecht, Finanzrecht oder Steuerrecht mit entsprechenden Dokumentenaufbewahrungspflichten.
Herzliche Grüße
Christian Rosenzweig