
Ein Vigilanz-System ist ein gesetzlich vorgeschriebenes System aus einem oder mehreren Prozesse, mit denen Hersteller (z.B. von Medizinprodukten) meldepflichtige Zwischenfälle mit ihren Produkten erfassen, bewerten auf Trends hin untersuchen und an die Behörden melden, um zur Sicherheit z.B. von Patienten und Anwendern beizutragen.
Welche regulatorischen Anforderungen Hersteller erfüllen müssen, wie sich ein Vigilanz-System von einem System für die Post-Market Surveillance unterscheidet und wie Firmen ein Vigilanz-System schnell und gesetzeskonform aufbauen und betreiben, erklärt dieser Beitrag.
1. Definition und Ziele der Vigilanz
Weltweit gelten gesetzliche Anforderungen an die Überwachung von Medizinprodukten im Markt. Damit sollen der Gesundheitsschutz und die Sicherheit von Patienten, Anwendern und Dritten gewährleistet bzw. verbessert werden. Ein wichtiger Aspekt ist dabei die Vigilanz. In einigen Rechtsbereichen, z. B. in den USA, spricht man von „adverse event reporting“.
Auch die Medizinprodukte-Verordnung MDR stellt umfangreiche Anforderungen an Vigilanz-Systeme. Sie definiert zwar den Begriff „Post-Market Surveillance“, aber nicht, was sie unter „Vigilanz“ versteht. Indirekt findet sich eine Definition in der inzwischen veralteten und überholten MEDDEV 2.12/1 (dazu später mehr):
„European system for the notification and evaluation of INCIDENTs and FIELD SAFETY CORRECTIVE ACTIONS (FSCA) involving MEDICAL DEVICEs, known as the Medical Device Vigilance System“
MEDDEV 2.12/1 Kapitel 2 (Einleitung)
Angelehnt an die globale IMDRF-Leitlinie, nennt das MEDDEV-Dokument auch die Ziele eines Vigilanz-Systems, nämlich:
Den Schutz der Gesundheit und die Sicherheit (Safety) von Patienten, Anwendern und Dritten dadurch zu verbessern, dass man die Wahrscheinlichkeit verringert, dass ein (negatives) Ereignis noch einmal auftritt
2. Abgrenzungen
a) Abgrenzung von Vigilanz und Post-Market Surveillance
Der Schutz der Gesundheit und die Sicherheit von Patienten, Anwendern und Dritten sind auch das Ziel der „Überwachung nach dem Inverkehrbringen“ (engl.: Post-Market Surveillance). Allerdings verfolgen Vigilanz-Systeme und Systeme für die Post-Market Surveillance unterschiedliche Ziele:
- Die Vigilanz ist ein reaktives System, das auf Zwischenfälle reagiert.
- Hingegen muss die Post-Market Surveillance proaktiv erfolgen. (Die MDR definiert die Post-Market Surveillance mit dem Begriff proaktiv.)
Lesen Sie hier mehr zum Thema Post-Market Surveillance (Überwachung nach der Inverkehrbringung).
‚Eine Verwechslungsgefahr besteht auch zwischen den Begriffen ‚Marktüberwachung‘ (Market Surveillance) und Überwachung nach der Inverkehrbringung‘ (Post-Market Surveillance). Für letztere sind, wie für die Vigilanz, die Hersteller zuständig. Die Marktüberwachung hingegen obliegt den Behörden.
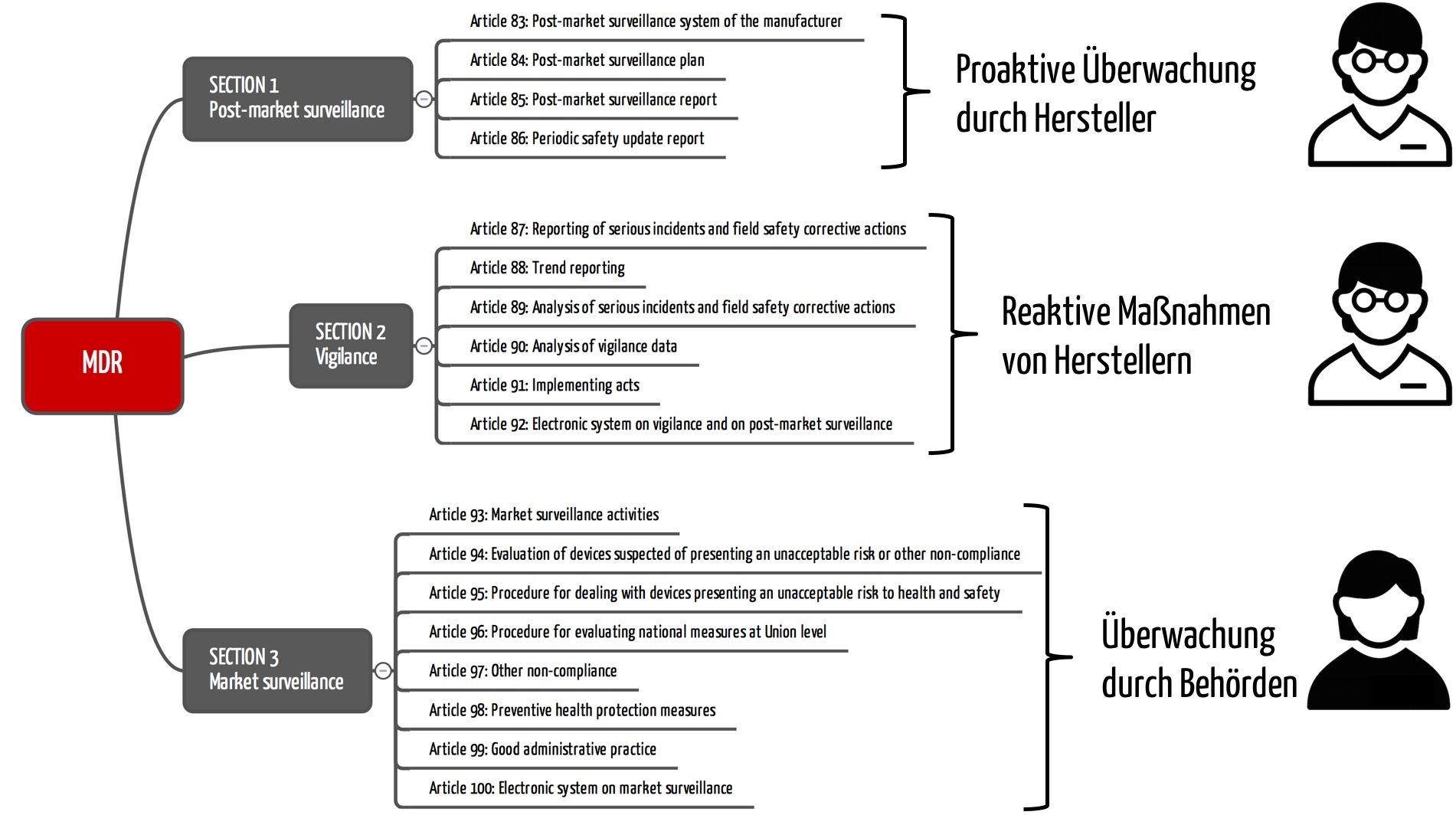
Auch die MDR unterscheidet präzise zwischen Vigilanz, Überwachung nach der Inverkehrbringung und Marktüberwachung. In Kapitel VII widmet sie jedem Begriff eine eigene Sektion (s. Abb. 1). Ähnlich unterscheidet die FDA Post-Market Surveillance in 21 CFR 822 und Medical Device Reporting in 21 CFR 803.
b) Abgrenzung von Vigilanz in der Medizin
Der Begriff Vigilanz wird auch in der Medizin, beispielsweise in der Intensivmedizin verwendet. Hier hat er eine andere Bedeutung:
Die Vigilanz in der Medizin entspricht der Wachheit bzw. Aufmerksamkeit von Patienten bezeichnet. Eine verminderte Vigilanz reicht von einer leichten Benommenheit bis hin zur Bewusstlosigkeit.
Eine verminderte Vigilanz kann die Folge von Verletzungen insbesondere des Kopfes sein, von Medikamenten oder von neurologischen Erkrankungen.
Sowohl die Vigilanz in der Medizin als auch die Vigilanz bei Medizinprodukten haben ihren Ursprung im lateinischen Wort „vigilantia“, was als Fürsorge oder Wachsamkeit übersetzt werden kann.
Eine Vigilanzstörung in der Medizin ist ein medizinisches Problem, eine Vigilanzstörung bei Medizinprodukten ein rechtlicher Verstoß.
3. Regulatorische Anforderungen an die Vigilanz
a) EU
MDR
Im Gegensatz zu den alten Richtlinien hat die MDR ein einheitliches Meldesystem eingeführt mit EU-weit gleichen Bestimmungen. Somit entfällt unter der MDR die Pflicht zur nationalen Meldung über die jeweiligen landesspezifischen Datenbanken oder Meldewege. Stattdessen gibt es eine einheitliche „Meldeschnittstelle“ über die EUDAMED. Diese kann allerdings erst genutzt werden, wenn EUDAMED voll funktionsfähig ist. Bis dahin müssen Hersteller und sonstige meldepflichtige Akteure weiterhin die nationalen Verfahren durchlaufen.
Die Anforderungen an das Vigilanz-System finden sich in den Artikeln 87 bis 92 der MDR. Generell regelt das Vigilanz-System:
- Was muss gemeldet werden? (d. h. welche Art von Ereignis, z. B. schwerwiegende Vorkommnisse und Sicherheitskorrekturmaßnahmen im Feld)
- Wer muss melden? (d. h. welche Rolle oder welcher Wirtschaftsakteur)
- Wann muss gemeldet werden? (d. h. mit welchen Fristen)
- In welcher Form wird gemeldet?
- Welche Inhalte muss die Meldung enthalten?
Im folgenden Kapitel finden Sie dazu einen Vergleich mit den entsprechenden Anforderungen der FDA.
Zusätzlich regelt die MDR die Anforderungen an die nationalen Behörden bei der Analyse von Vigilanz-Daten und bei der Zusammenarbeit mit den zuständigen Behörden sämtlicher Mitgliedstaaten.
MDCG 2023-3
Die MDCG-Leitlinie aus dem Jahr 2023 mit dem Titel „Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices“ hat die bis dahin über viele Jahre anwendbare MEDDEV 2.12/1 abgelöst. In einem Q&A-Format liefert sie Hilfestellungen rund um das Thema Vigilanz.
So hilft das Dokument z. B. bei der Unterscheidung zwischen „incident“ und „serious incident“, der Interpretation der Meldefristen oder den generellen Meldekriterien. Für den Aufbau eines MDR-konformen Vigilanz-Systems sollten Hersteller und weitere Akteure dies unbedingt beachten.
MDCG 2024-1
Das MDCG-Dokument zur Vigilanz (MDCG 2024-1) ist nur sechs Seiten kurz. Ohne Einleitung, Wiederholung der regulatorischen Anforderungen und Referenzen bleibt sogar nur eine Seite. Diese Seite enthält ein Vigilanz-Formular.
Die Entscheidung darüber, in welchem Format eingereicht werden muss, liegt bei den EU-Mitgliedstaaten. So muss in Deutschland das Formular des BfArMs verwendet werden.
Die MDCG (ähnlich wie damals die MEDDEV) empfiehlt die IMDRF-Codes für die Klassifizierung von „Incidents“ und „Medical Device Problems“. Das ist hilfreich, weil das zur Harmonisierung der Vigilanz-Prozesse beiträgt.
Nationale Gesetze und Verordnungen
Zusätzlich zur MDR können Mitgliedsstaaten erweiterte, nationale Anforderungen an die Vigilanz stellen. In Deutschland finden sich diese beispielsweise in der Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV). Die nationalen Anforderungen dürfen den Anforderungen der MDR allerdings nicht widersprechen. So gibt es in Deutschland ausschließlich zusätzliche Meldeanforderungen, in diesem Fall an die Anwender von Medizinprodukten.
b) USA
Medical Device Reporting (21 CFR 803)
Part 803 des 21 CFR beschreibt Anforderungen an ein Verfahren zur Meldung von unerwünschten Ereignissen. Unglücklicherweise spricht die FDA hierbei von MDR Reportable Events, wobei dieser Begriff auch durch die EU-Medizinprodukteverordnung (MDR) belegt ist. Da sind Missverständnisse vorprogrammiert.
Auf keinen Fall sollten Hersteller, Importeure und Betreiber von Medizinprodukten die Anforderungen des Part 803 unterschätzen. Denn mangelhafte oder gar fehlende Verfahren zur Meldepflicht in den USA zählen zu den häufigsten Abweichungen bei FDA-Inspektionen.
Tipps zur Umsetzung der Anforderungen und zum Vermeiden von Abweichungen finden Sie weiter unten.
Reports of Corrections and Removals (21 CFR 806)
Die FDA verlangt die Meldung von sogenannten „corrections“ und „removals“, die den in der MDR geforderten Sicherheitskorrekturmaßnahmen im Feld ähneln. Sie dürfen nicht verwechselt werden mit den Recalls der FDA. Eine ausführliche Übersicht zu Rückrufen im FDA-Kontext und die Abgrenzung zum Part 806 finden Sie in diesem Artikel.
FDA-Guidance
Die FDA-Leitlinie „Medical Device Reporting for Manufacturers“ von 2016 liefert Herstellern und weiteren meldepflichtigen Personen auf über 50 Seiten umfangreiche Hilfestellungen für ein besseres Verständnis der Vigilanz-Anforderungen gemäß Part 803.
c) IMDRF/GHTF
Das International Medical Device Regulators Forum (IMDRF) hat Dokumente der nicht mehr bestehenden Global Harmonization Task Force (GHTF) übernommen. Sie enthalten vor allem globale Richtlinien für das Meldewesen (Webseite des IMDRF zu den Richtlinien zum Meldewesen).
Aufbauend auf den Arbeiten der GHTF beschäftigt sich das IMDRF mit der Kategorisierung von unerwünschten Ereignissen. So hat sie 2020 ein umfangreiches Vokabular mit zugehörigen Codes zu unerwünschten Ereignissen eingeführt, das kontinuierlich aktualisiert wird. Ziel ist es, negative Trends oder neuartige Risiken sowohl durch Hersteller als auch durch Behörden schneller erkennen und analysieren zu können.
4. Vergleich der Anforderungen EU und USA
Trotz der Harmonisierung durch das IMDRF existieren länderspezifische Unterschiede im Bereich der Vigilanz-Systeme. Dies zeigt schon der Vergleich der Definitionen eines meldepflichtigen Ereignisses gemäß MDR bzw. FDA.
a) Unterschiede bei der Definition
| EU | USA | Kommentar |
|---|---|---|
| Schwerwiegendes Vorkommnis: Ein Vorkommnis (= „eine Fehlfunktion oder Verschlechterung der Eigenschaften oder Leistung eines bereits auf dem Markt bereitgestellten Produkts, einschließlich Anwendungsfehlern aufgrund ergonomischer Merkmale, sowie eine Unzulänglichkeit der vom Hersteller bereitgestellten Informationen und eine unerwünschte Nebenwirkung“), das direkt oder indirekt eine der nachstehenden Folgen hatte, hätte haben können oder haben könnte: a) den Tod eines Patienten, Anwenders oder einer anderen Person, b) die vorübergehende oder dauerhafte schwerwiegende Verschlechterung des Gesundheitszustands eines Patienten, Anwenders oder anderer Personen, c) eine schwerwiegende Gefahr für die öffentliche Gesundheit (Artikel 2 65. MDR) „schwerwiegende Gefahr für die öffentliche Gesundheit“ bezeichnet ein Ereignis, das das unmittelbare Risiko des Todes, einer schwerwiegenden Verschlechterung des Gesundheitszustands einer Person oder einer schweren Erkrankung, die sofortige Abhilfemaßnahmen erfordert, bergen könnte, und das eine signifikante Morbidität oder Mortalität bei Menschen verursachen kann oder das für einen bestimmten Ort und eine bestimmte Zeit ungewöhnlich oder unerwartet ist; (Artikel 2 66. MDR) | (o) MDR reportable event (or reportable event) means: (1) An event that user facilities become aware of that reasonably suggests that a device has or may have caused or contributed to a death or serious injury or (2) An event that manufacturers or importers become aware of that reasonably suggests that one of their marketed devices: (i) May have caused or contributed to a death or serious injury, or (ii) Has malfunctioned and that the device or a similar device marketed by the manufacturer or importer would be likely to cause or contribute to a death or serious injury if the malfunction were to recur. (21 CFR 803.3) (w) Serious injury means an injury or illness that: (1) Is life-threatening, (2) Results in permanent impairment of a body function or permanent damage to a body structure, or (3) Necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. Permanent means irreversible impairment or damage to a body structure or function, excluding trivial impairment or damage. (21 CFR 803.3) | – In der EU sind erwartete unerwünschte Nebenwirkungen (falls akzeptabel gemäß Risikobewertung) nur im Falle von negativen Trends meldepflichtig. – In den USA sind sämtliche Nebenwirkungen meldepflichtig, wenn diese zum Tod oder „serious injury“ geführt haben. – Auch in den USA zählen zu meldepflichtigen Ereignissen Ursachen aufgrund von Anwendungsfehlern („user errors“) oder aufgrund der Kennzeichnung. |
b) Weitere Unterschiede und Gemeinsamkeiten
In der folgenden Tabelle sind weitere Unterschiede und Gemeinsamkeiten dargestellt:
| EU | USA | Kommentar | |
|---|---|---|---|
| Ist ein dokumentiertes Verfahren notwendig? | ja | ja | |
| Was ist meldepflichtig? | – Schwerwiegendes Vorkommnis – Trends von sonstigen Vorkommnissen – Sicherheitskorrekturmaßnahmen im Feld (FSCA) – Vom Produkt ausgehende schwerwiegende Gefahr (auch nur bei Grund zur Annahme) | – MDR reportable event – Meldepflichtige correction/removal | – EU: FSCA sind in der EU auch dann meldepflichtig, falls die Maßnahme nicht spezifisch anwendbar auf die Produkte im Drittland – USA: Die FDA fordert im Gegensatz zur EU-MDR die Meldung von Ereignissen, die im Ausland stattgefunden haben, falls das entsprechende Produkt auch in den USA vermarktet wird und das Ereignis der Definition des MDR-Events entspricht. – USA: Die FDA fordert keine Trend-Meldung. – EU: Schwerwiegende Gefahr (engl. „serious risk“) ist leider nicht definiert in der MDR. Allerdings liefert das MDCG 2023-3 eine Interpretation. |
| Wer ist meldepflichtig? (behördliche Meldepflicht) | – Hersteller – Importeure – Händler – Anwender (kann national gefordert werden, in Deutschland beispielswese für berufliche/gewerbliche Anwender) | – Hersteller – Importeure – Device User Facility (z. B. ein Krankenhaus, aber keine(!) Arztpraxis) | – USA: Händler sind in den USA nicht meldepflichtig. Sie müssen aber Aufzeichnungen führen zu Vorkommnissen. – EU: Importeure und Händler in der EU müssen nur melden, wenn Sie Grund zur Annahme (oder auch Gewissheit) haben, dass von den bereitgestellten Produkten eine schwerwiegende Gefahr ausgeht. – USA: Device User Facilities müssen nur Ereignisse melden, die zum Tod geführt haben. Zusätzlich sind jährliche Sammelmeldungen, einschließlich der Fälle von „serious injuries“ gefordert. |
| Bis wann muss gemeldet werden? | – Schwerwiegendes Vorkommnis: max. 15 Kalendertage – Schwerwiegende Gefahr für die öffentliche Gesundheit: max. 2 Kalendertage – Tod oder Eintritt einer schwerwiegenden Verschlechterung des Gesundheitszustands: max. 10 Kalendertage – FSCA: Vor Ergreifen der Maßnahme | – MDR reportable event: max. 30 Kalendertage – MDR event which „necessitates remedial action to prevent an unreasonable risk of substantial harm to public health“: max. 5 Kalendertage – Correction/Removal: 10 Arbeitstage nach Ergreifung der Maßnahme | – Die Frist beginnt ab dem Bekanntwerden des Ereignisses. – Die MDR-Fristen sind wesentlich kürzer. – Die FDA unterscheidet bei den Fristen nicht, ob ein Todesfall oder ein „serious injury“ tatsächlich eingetreten ist oder nicht. |
| Wie muss gemeldet werden? | – Schwerwiegendes Vorkommnis: EUDAMED Vigilance Modul (sobald funktionsfähig) – Trends: EUDAMED Vigilance Module (sobald funktionsfähig) – FSCA: EUDAMED Vigilance Module (sobald funktionsfähig) – Vom Produkt ausgehende schwerwiegende Gefahr: Meldung an die jeweilige nationale zuständige Behörde | – MDR reportable event: elektronisch über das Electronic Submission Gateway (ESG), entweder als Formular oder per Maschine-zu-Maschine-Schnittstelle (HL7 XML per AS2-Gateway) – Correction/Removal: per ESG oder per E-Mail | – EU: Da die EUDAMED aktuell nicht voll funktionsfähig ist, müssen aktuell die nationalen Bestimmungen beachtet werden. |
| Welche Informationen sind gefordert? | Im EUDAMED Playground finden Sie ein User Guide mit einer Beschreibung der Eingabemasken und der geforderten Informationen. | Siehe Form 3500A. Eine Ausfüllanleitung finden Sie hier. | |
| IMDRF Kategorisierung verlangt? | ja | Nein, eigene Codierung | – USA: Die FDA bietet ein vollständiges Mapping der FDA Codes mit denen des IMDRF. |
5. Tipps zur Umsetzung
Die Anforderungen an die Vigilanz-Systeme unterscheiden sich landesspezifisch. Umso wichtiger ist es, dass Sie die Unterschiede und Gemeinsamkeiten kennen. Denn Vigilanz ist immer ein Thema im Audit und wird von den Benannten Stellen und Behörden wie der FDA streng geprüft.
Die folgenden Tipps mögen dazu beitragen, Abweichungen und Warning Letters der FDA zu minimieren.
- Erstellen Sie ein dokumentiertes Verfahren zur Vigilanz. Dies ist eine grundlegende Voraussetzung. Ein fehlendes oder nicht dokumentiertes Verfahren führt mit hoher Wahrscheinlichkeit zu großen Problemen bei einer FDA-Inspektion, bis hin zum Warning Letter.
- Beschreiben Sie in Verfahrens- und Arbeitsanweisungen genau, wer im Fall des Falles was, auf welche Weise und wie schnell erledigen muss. Denken Sie an eine Stellvertreterregelung.
- Versuchen Sie nicht, die Anforderungen der verschiedenen Länder zu kombinieren. Sowohl MDR als auch FDA und weitere Behörden weltweit haben eigene Begriffsdefinitionen, die sie nicht vermischen sollten. Nutzen oder referenzieren Sie daher unbedingt die jeweilige Definition im Gesetz.
- Stellen Sie sicher, dass Sie Begriffe wie „schwerwiegendes Vorkommnis“, „MDR reportable event“, „becoming aware“, „reasonably suggest“, „caused or contributed“ usw. präzise definieren (so wie in den Regularien). Erläutern Sie diese anhand von Beispielen, die für Ihre Produkte spezifisch sind.
- Aufgrund der landesspezifischen Unterschiede empfiehlt es sich, spezifische Arbeitsanweisungen pro Land festzulegen.
- Nutzen Sie Flussdiagramme und Checklisten. Sie sind schneller zu verstehen als lange Texte.
- Dokumentieren Sie sämtliche Kommunikation (auch mündliche) zum Vorkommnis.
- Schulen Sie alle Beteiligten immer wieder. Üben Sie das Meldewesen anhand von konkreten Beispielen.
Geben Sie uns Bescheid, wenn wir Sie beim Aufbau, Verbessern und Überprüfen Ihres Vigilanz-Systems unterstützen können, beispielsweise mit Templates, weiteren Tipps, Schulungen oder Mockup-Audits.
Änderungshistorie
- 2025-02-08: Kapitel 2.b) und daher Überschrift 2.a) eingefügt. Ziel: Missverständnisse zwischen medizinischem Personal und Regulatory Affairs Manager vermeiden
- 2024-02-25: Definition am Artikelanfang eingefügt
- 2024-02-09: Abschnitt zur MDCG 2024-1 ergänzt
- 2023-12-11: Artikel vollständig überarbeitet



Es sei die Bemerkung gestattet, dass die Personengrafik in „Abb. 1: Abgrenzung von Vigilanz von Post-Market Surveillance und Marktüberwachung“, doch etwas altbacken wirkt, da ausschließlich Männer abgebildet werden.
Insbesondere das Symbol für den Behördenvertreter, als Herr mit Brille und Schneuzer, wirkt nicht ganz zeitgemäßg. Gender-neutralere Darstellungen sind doch grundsätzlich wünschenswert, gerade im Lehrbetrieb mit implizierter Vorbildfunktion.
Sie haben absolut Recht: Das Bild habe ich ausgetauscht. Danke für Ihren Hinweis!
Ich würde mich über ein Austausch mit „offenem Visier“ freuen.
Guten Tag,
ich hätte einige Fragen, die möglicherweise mehrere Hersteller interessieren.
Wenn ich in einem EU Land, in dem ein Produkt in Verkehr gebracht wurde, ein Vorkommnis zu melden habe, melde ich das in dem Land in dem ich als Hersteller registriert bin, in dem Land, in dem das Vorkommnis statt fand, oder in beiden Ländern?
Zusätzlich würde mich interessieren, wie die Kommunikation der Behörden untereinander in den EU Ländern funktioniert? Ist es die Pflicht des Herstellers in jedem Land, das von dem Vorkommnis betroffen ist, eine Meldung an die nationale Behörde zu tätigen, oder kommunizieren die Behörden diesbezüglich auch untereinander?
Freundliche Grüße!
Sehr geehrte Frau Kedwani,
die nationalen Regelungen unterscheiden sich. Beispielsweise gibt es in Deutschland die MPSV. Identische Anforderungen gibt es nicht in allen Ländern.
Allerdings kann man generell sagen, dass man in dem Land melden muss, in denen das Vorkommnis stattfand. Im Fall von Deutschland schreibt das MPSV:
Die Behörden informieren sich ggf. gegenseitig. Das ist nicht immer ganz transparent. Die MDR und die EUDAMED werden diesen Mangel (hoffentlich) beheben.
Viele Grüße, Christian Johner
Hallo Herr Johner,
mich beschäftigt auch gerade diese Thematik. Ihre Antwort auf diese Frage stammt aus 2018, bisher wurde das MPSV mit dem 25.05.21 außer Kraft gesetzt un durch MPAMIV abgelöst. Die EUDAMED ist noch nicht voll funktionsfähig. Wo entnehme ich nun die regulatorische Vorgabe schwerwiegende Vorkomnisse und FSCA auch in EU-Ländern zu melden, wenn ich als Hersteller in DE ansässig bin.
Guten Tag,
gemäß Übergangsbestimmungen der MDR, gelten die Vigilanz-Pflichten gemäß MDR seit Gültigkeitsbeginn, d.h. 26.05.2021. Dies allerdings ausgenommen der EUDAMED-betreffenden Pflichten, da diese noch nicht voll funktionsfähig. Generell müssen Sie ein schwerwiegendes Vorkommnis in dem Mitgliedsstat melden, in dem es aufgetreten ist (siehe auch MDCG-Guidance https://health.ec.europa.eu/system/files/2023-02/mdcg_2023-3_en_0.pdf, Frage 16). FSCAs müssen in allen betroffenen Mitgliedstaaten gemeldet werden sowie immer der zuständigen Behörde des Landes, in dem der Hersteller seinen Sitz hat. In EUDAMED werden die Meldungen dann automatisch an die zuständigen Behörde(n) weitergeleitet.
Freundliche Grüße
Luca Salvatore
Hallo Herr Prof. Johner,
die Meldefristen laut MDR sind nicht korrekt übersetzt.
es dürfte nicht heissen: ’spätestens nach 15 Tagen‘. In der originalen englischen Version heisst es : ’not later than‘ , müsste also
‚ nicht später als‘ ALS X Tagen heißen.
Viele Grüße
Peter Postert
Besten Dank, Herr Postert!
Ich habe „spätestens nach“ durch „nicht später als nach“ übersetzt. Ich hoffe, dass ich Sie nicht missverstanden habe.
Mit herzlichem Dank und vielen Grüßen, Christian Johner
Sehr geehrter Herr Dr. Johner,
können Sie sagen, ob die die MDR im Zusammenhang mit den Meldefristen mit „Tagen“ Kalendertage oder Arbeitstage meint?
Beste Grüße,
Nadine Langguth
Sehr geehrte Frau Langguth,
Sie haben Recht, die MDR spezifiziert das nicht. Da sie nur von Tagen spricht, dürften Kalendertage gemeint sein, weil sonst die Sicherheit der Patienten von den Feiertagsregelungen abhängen würde, was nicht sein darf.
Das Medizinproduktedurchführungsgesetz schafft keine weitere Klarheit. Möglicherweise werden noch nationale Verordnungen erlassen, die explizit von Kalendertagen sprechen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
eine große benannte Stelle hat über das Mailingsystem verbreitet, dass eine Vorkommnismeldung auch an die benannte Stelle verpflichtend ist. Ich kann aus dem Mailing nicht entnehmen ob dazu das Formular des BfArM verwendet werden kann oder nicht. Zudem wird darauf hingewiesen, dass für die Bearbeitung der Meldung eine pauschale Gebühr von 400.–€ erhoben wird.
Wissen Sie dazu näheres oder können Sie mir sagen, wie es sich da mit der rechtlichen Grundlage verhält?
Vielen Dank für Ihre Bemühungen.
Viele Grüße,
Werner Neumüller
Sehr geehrter Herr Neumüller,
die Vorgabe, nach denen Sie fragen sind nur tw. im MPG bzw. der MPSV geregelt. Es gibt bezüglich der Risiken den Artikel 22 MPSV.
D.h. dass diese Formen und auch Gebühren letztlich eine zivilrechtliche Vereinbarung sind, die normalerweise über die AGBs bzw. über den Vertrag mit der BS geregelt sind.
Aus Gründe der Bequemlichkeit bietet es sich an, das gleiche Formular an die Benannte Stelle zu schicken und kein zusätzliches Dokument zu nutzen. Das sieht natürlich anders aus, wenn Ihre Benannte Stelle besondere Vorgaben hat.
Viele Grüße, Christian Johner
Hallo,
wann genau wird eigentlich das „schwere Vorkommnis“ als „Gefahr für die öffentliche Gesundheit“ eingestuft?
Ist es der systemische Charakter der Fehlerursache, damit also z.B. der Design-Fehler oder die Tatsache, daß der Fehler in einer Batch mehrfach auftaucht?
Viele Grüße,
Jörg Bigalke
Lieber Herr Bigalke,
ich freue mich, von Ihnen zu hören!
Die MDR liefert die Definition:
Da dieser Definition von „Menschen“ (Plural) spricht, ist es wahrscheinlich, dass es sich um einen systemischen Fehler handeln muss. Das kann ebenso ein Design- wie Produktionsfehler sein oder ein Problem mit der Gebrauchstauglichkeit.
Viele Grüße, Christian Johner
Sehr geehrtes Johner Team
Als Berater in der IVD Branche kommt vermehr immer wieder folgende Thematik auf:
Muss ich als *nur* Händler jegliche Meldung innerhalb der zeitlichen Frusten einhalten bzw überhaupt melden?
Leider verstehen viele Firmen als *nur* Händler es wie folgt: wenn jemand an mich mit einer Nebenwirkung (oder allgemein medical inquiery) herantritt, bin ich als Händler nicht verantwortlich.
Ich sehe das als sehr kritisch an.
Sobald ein Händler die Ware in Umlauf bringt: welche Verpflichtung Zwecks Meldung von Events sind einzuhalten und gibt es Unterschiede zwischen Hersteller und Händler?
Bsp1: eine Firma (Händler) bekommt über ihre LinkedIn Seite an einem Samstagabend einen Kommentar, dass nach benutzen eines über den Händler bezogenen Produktes eine schwerwiegende allergische Reaktion aufgetreten ist (Ehemann ins Krankenhaus gekommen).
Was muss der Händler hier (in zeitlicher Chronologie) sicherstellen?
Bsp2: eine Firma bekommt über eine Email an info@ am Samstagabend einen Kommentar, dass alle über den Händler bezogene Ware (10K – Großbestellung) kontaminierten Puffer beinhaltet und zu Reizungen bei öffnen geführt hat.
Was muss der Händler hier (in zeitlicher Chronologie) sicherstellen?
Vielen Dank
Schollmeier
Liebe:r Frau/Herr Schollmeier,
mein Kollege Alexander Thern hat mir darauf die folgende Antwort gegeben:
Wenn ein Unternehmen keinen 24/7-Support hat, arbeitet es am Wochenende nicht. Montagmorgen fangen die Mitarbeitenden dann spätestens um 9:00h an zu arbeiten und erfüllen Ihre Pflicht nach Art. 14(4), Art. 14(5) IVDR (oder MDR ), indem sie den Hersteller unverzüglich informieren.
Unverzüglich bedeutet: Ohne schuldhafte Verzögerung, also gleich, spätestens am selben Tag.
Bei Gefahr im Verzug würden Sie natürlich auch am Wochenende bzw. sofort handeln.
Z. B. Unternehmen, die Herzschrittmacher in Verkehr bringen, bieten daher meist auch einen rundum-die-Uhr-Support an, durch den sie schnell reagieren können.
Herzliche Grüße
Anja Segschneider | Redaktion
Bezüglich der Meldung von Trends (TrendR) wollte ich gerne fragen ob schon klar ist, ob es hier abweichende Meldefristen gibt oder ob hier auch die max. 15 Tage gelten.
Wenn ja, ab wann beginnen die 15 Tage?
Da die Ermittlung von Trends retrospektiv erfolgt, ist eine Meldung innerhalb von 15 Tagen ja nicht möglich.
Hier könnte dann die Frist beginnen „spätestens 15 Tage nach Erkennung des Trends“ oder ähnlich…
Vielen Dank für den Austausch!
Andrea Hagel
Liebe Frau Hagel,
mein Kollege Philipp Malsch schrieb mir dazu folgende Antwort:
Liebe Frau Hagel,
zunächst einmal möchte ich mich für die verspätete Antwort vielmals entschuldigen, ich hatte einfach Probleme mit dem Browser…
Vielen Dank aber für Ihre tolle Frage! Mir sind bis jetzt auch keine expliziten Fristen bekannt. Allerdings hatte ich die Vorgaben daher soweit interpretiert, dass Trends sofort gemeldet werden sollten, wenn sie entdeckt werden. Das BfArM sieht hierfür zurzeit eine recht formlose Meldung per Email vor: https://www.bfarm.de/DE/Medizinprodukte/Antraege-und-Meldungen/Uebersicht-Meldewege/_node.html
Nun gibt es laut MDR Artikel 91 die Möglichkeit, dass die Meldung von Trends durch Durchführungsakte präzisiert wird. Ganz explizit „d) Fristen für die Meldung von Sicherheitskorrekturmaßnahmen im Feld und für die Vorlage von periodischen Sammelmeldungen und Trendmeldungen seitens der Hersteller unter Berücksichtigung der Schwere des zu meldenden Vorkommnisses gemäß Artikel 87“; außerdem kündigt die MDCG an, dass es in Q3 ein Q&A Dokument geben soll (https://ec.europa.eu/health/system/files/2022-06/mdcg_ongoing_guidancedocs_en.pdf). Ich denke, wir können gespannt sein. Falls Sie mehr wissen, freue auch ich mich jederzeit über den weiteren Austausch!
Mit besten Grüßen
Philipp Malsch
Sehr geehrte Herr Johner,
wir sind ein ein Unternehmen das Produkte noch in der Übergangsfrist verkauft aber auch einige die keine Übergangsfristen haben.
In der IVDR Art. 87 steht, dass alle Meldung durch das elekronische System laufen sollten.
Zurzeit ist uns klar, dass dies mit dem MIR Dokument geschieht bis zur vollen Funktionalität der Eudamed ungeachtet von Fristen. Korrekt?
Frage: der spätere Ablauf können wir jedoch nicht ganz klar von der IVDR 2017/746 entnehmen.
Sollte der Hersteller (wir) zuerst die Behörden kontaktieren und von dem schwerwiegendes Vorkommnis berichten und dann das erst in der Eudamed eintragen?
Der Rückruf erfolgt auch dann direkt in die Eudamed ohne die Behörden zu erst zu kontaktieren ?
Außerdem in der 98/97 EG war die Meldefrist 30 tage nun laut IVDR 2017/746 15 tage?
(3) Die Hersteller melden jedes schwerwiegende Vorkommnis im Sinne des Absatzes 1 Buchstaben a unverzüglich, nachdem sie einen Kausalzusammenhang oder einen durchaus möglichen Kausalzusammenhang zwischen dem Vorkommnis und ihrem Produkt festgestellt haben, spätestens jedoch 15 Tage, nachdem sie Kenntnis von dem Vorkommnis erhalten haben.
Gilt die neue Meldefrist auch für die Produkte mit der Übergangsfrist (also 15 Tage), denn in der Bfarm Startseite sind immer noch die 30 tage erwähnt.
Liebe Maria,
mein Kollege Philipp Malsch hat mir die folgende Antwort gegeben:
Liebe Maria,
vielen Dank für Ihre Frage!
Die Vigilanz-Meldungen sollen zukünftig über EUDAMED geschehen, wie Sie selber schon sagen.
Die IVDR und MDR beschreiben den Meldeverlauf in Artikel 92. 5-9 MDR und Artikel 87.5-9 IVDR.
So heißt es z.B.:
„MDR Artikel 92 (5) Die Meldungen schwerwiegender Vorkommnisse gemäß Artikel 87 Absatz 1 Buchstabe a werden nach ihrem Eingang über das elektronische System gemäß Absatz 1 des vorliegenden Artikels automatisch an die zuständige Behörde des Mitgliedstaats übermittelt, in dem das Vorkommnis aufgetreten ist.“
Es gibt noch die Möglichkeit für weitere Durchführungsakte und es sind/ waren noch MDCG Guidance Dokumente für den Themenbereich Vigilanz geplant; jedoch aus dem zitierten Artikel würde ich davon ausgehen, dass die Meldung aus EUDAMED automatisch an die Behörden geht.
Wie Sie zurzeit (da EUDAMED noch nicht funktionsfähig ist) melden müssen, hängt von den nationalen Vorschriften ab; in Deutschland hat das BMG dazu folgende Bekanntmachungen herausgegeben:
MDR:
https://www.bundesanzeiger.de/pub/publication/IGrRaNuJBWgwK7Giz8Y/content/IGrRaNuJBWgwK7Giz8Y/BAnz%20AT%2028.05.2021%20B6.pdf?inline
IVDR: https://www.bundesanzeiger.de/pub/publication/qOIieZMq3dnFbUZs734/content/qOIieZMq3dnFbUZs734/BAnz%20AT%2027.05.2022%20B4.pdf?inline
Und das BfArM greift dieses Thema ebenfalls hier auf:
https://www.bfarm.de/DE/Medizinprodukte/Antraege-und-Meldungen/Vorkommnis-melden/Hersteller-und-Bevollmaechtigte/_node.html
Sie haben aber sehr wohl Fristen für die Meldungen. Diese sind in Artikel 87 IVDR beschrieben. Sie müssen immer unverzüglich melden, aber spätestens nach 2, 10 oder 15 Tagen.
Dies gilt auch für Produkte in der Übergangsfrist, da hier MDR bzw IVDR schon voll gelten (siehe z.B. IVDR Artikel 110 (3)).
Rückrufe sind in IVDR und MDR mittlerweile anders definiert, und sollten nicht mit den Sicherheitskorrekturmaßnahmen im Feld gleichgesetzt werden. Bitte beachten Sie dazu Artikel 2 der MDR und IVDR.
Sicherheitkorrekturmaßnahmen im Feld müssen aber durchaus vor der Umsetzung den Behörden gemeldet werden.
So sagt z.B. Artikel 87 MDR:
„8) Außer in dringenden Fällen, in denen der Hersteller unverzüglich eine Sicherheitskorrekturmaßnahme im Feld ergreifen muss, meldet der Hersteller ohne ungebührliche Verzögerung die Sicherheitskorrekturmaßnahme im Feld gemäß Absatz 1 Buchstabe b, bevor er die Sicherheitskorrekturmaßnahme im Feld ergreift.“
Ich hoffe, ich konnte Ihnen weiterhelfen!
Mit besten Grüßen
Philipp Malsch
Liebes Johner-Team,
ich habe eine Frage zur Rolle des Bevollmächtigten im Vigilanz-System. Der Bevollmächtigte ist laut MDR unter anderem zu folgenden verpflichtet:
– Kooperation mit den zuständigen Behörden bei allen Präventiv- oder Korrekturmaßnahmen zur Abwendung oder, falls dies nicht möglich ist, Minderung von Gefahren, die mit Produkten einhergehen;
– Unverzügliche Unterrichtung des Herstellers über Beschwerden und Berichte seitens Angehöriger der Gesundheitsberufe, der Patienten und Anwender über mutmaßliche Vorkommnisse im Zusammenhang mit einem Produkt, für das der Bevollmächtigte benannt wurde;
Es gibt hier keine direkten Vigilanzverpflichtungen an die Bevollmächtigten, außer als Bindeglied zum Hersteller zu agieren.
Das „EUDAMED Summary Document“, welches vom Hersteller in EUDAMED bei der Registrierung eingereicht werden muss, beinhaltet aber auch den Punkt „Mandated to Vigilance – yes/no“. Wenn ich es richtig verstehe, dann würde bei einer Beantwortung mit „Nein“ weiterhin der Hersteller den Vigilanzpflichten nachkommen müssen. Bei einer Antwort mit „Ja“ müsste es dann der Bevollmächtigte. Was wäre hier der Umfang der Verpflichtungen für den Bevollmächtigten, wenn dies mit „Ja“ beantwortet werden würde?
Mit freundlichen Grüßen,
Michael
Hallo Michael,
im Regelfall werden bei einer Delegation die gesamten Vigilanzpflichten gemäß MDR an den Bevollmächtigten übertragen. D.h. das gesamte Handling der incidents. Dieser wäre dann auch für die Meldung an die Behörden bzw. zukünftig über EUDAMED zuständig.
Freundliche Grüße
Luca Salvatore
Sehr geehrte Herr Johaner,
ich habe eine Frage zur Meldepflicht der Händler von IVD Produkten:
Artikel 14 spricht von unverzüglicher Meldung bei der Annahme von schwerwiegenden Gefahr etc.
Im MPAMIV steht
§ 3 Meldepflicht:
Wer Produkte beruflich oder gewerblich betreibt oder anwendet, hat dabei aufgetretene mutmaßliche schwerwiegende Vorkommnisse unverzüglich der zuständigen Bundesoberbehörde zu melden. Satz 1 gilt entsprechend für Ärzte und Zahnärzte, denen in Ausübung ihrer beruflichen Tätigkeit mutmaßliche schwerwiegende Vorkommnisse bekannt werden.
Nichtamtliches Inhaltsverzeichnis
Mir ist nicht ganz klar wie ich die Meldefristen als Händler implementieren soll, gelten denn die oben genannten Fristen auch für Händler oder nur Hersteller.
Guten Tag,
das MPAMIV richtet sich an Anwender, nicht an Händler. Somit ist für Sie Artikel 15 der IVDR relevant. Die Meldung erfolgt gemäß §81 MPDG an das BfArM. Dies erfolgt über ein Online-Formular: https://www.bfarm.de/DE/Medizinprodukte/Antraege-und-Meldungen/Vorkommnis-melden/Anwender-Betreiber-Haendler/_node.html
Freundliche Grüße
Luca Salvatore
Guten Tag,
Frohes Neues Jahr!
Meine Frage richtet sich an die Meldefristen für Händler.
Für Hersteller sind diese klar in Artikel 82 IVDR definiert und hängen von der schwere des Vorkommnisses ab
0/2
bis10 Tage
bis15 Tage,
Jedoch finde ich keine klare Definition für Händler, dass klar definiert in wie viele TAGEN ein Händler ein Vorkommnis melden muss . Artikel 14 IVDR spricht von unverzüglich bei einem schwerwiegenden Vorfall. Hätte man dann als Händler nur die Option von bis max 2 Tage einen Schwerwiegenvorfall an die Bundesbehörden zu melden.
Guten Tag Noor,
ich wünsche Ihnen ebenso ein frohes neues Jahr.
In der Tat definiert weder die IVDR noch das MDCG eine Frist für die Meldung im Falle von einer vom Produkt ausgehenden schwerwiegenden Gefahr („serious risk“). Diese ist nicht zu verwechseln mit dem (schwerwiegenden) Vorkommnis, welches nur die Hersteller melden müssen. Eine Definition liefert das MDCG 2023-3: „For the purpose of this document, a ‘serious risk’ is defined as a situation where a serious harm resulting from the use of a device that might affect patients, users or the public is likely to happen. A serious risk may include situations where the effects of the risk are not immediate“.
Da keine konkreten Fristen definiert sind, würde ich empfehlen, sich eher an den max. 2 Tagen zu orientieren. Generell bedeutet „immediately“, ohne absichtliche Verzögerung oder Verzögerung durch Fahrlässigkeit. Sicherlich muss seitens des Händlers eine Risikobewertung stattfinden („…likely to happen“). Eventuell sind dafür Informationen vom Hersteller notwendig, so dass eine gewisse nicht-beabsichtigte Verzögerung möglich sein kann.
Freundliche Grüße
Luca Salvatore
Hallo, ich habe da mal eine Abgrenzungsfrage. Wir diskutieren hier im Hause, ob wir einen Vigilanzprozess überhaupt qualifizieren müssen, da wir nur Entwicklung und Produktion jedoch nicht die Inverkehrbringung von Medizinprodukten und auch nicht die Instandsetzung von am Markt befindlichen Medizinprodukten durchführen sondern nur Entwicklungsleistungen erbringen, Messdienstleistungen erbringen und Messtechnische Dienstleistungen erbringen im Rahmen der Entwicklung von Medizinprodukten. Die Inverkehrbringung ist Sache der Kunden, die als Hersteller auftreten.
Besten Dank für Ihre spannende Frage!
Die Vigilanzpflichten (MDR Artikel 87 ff.) wenden sich an die Hersteller, nicht an Dienstleister.
Allerdings ist es üblich, dass die Hersteller Ihre Dienstleister im Rahmen einer QSV dazu verpflichten, Post-Market-Informationen zu liefern. Aber das ist eine Verpflichtung im „Innenverhältnis“.
Beste Grüße, Christian Johner
Liebes Team des Johner Instituts,
ich habe mir jetzt die Artikel zu Vigilanz, CAPA und Nichtkonformitäten durchgelesen.
Ich hoffe, dass meine Frage am besten zu diesem Artikel passt:
In der der IVDR existieren die Definitionen Produktmangel und Vorkommnis (Begriffe 62 bzw. 67).
Bezieht sich ein Produktmangel nur auf Produkte in Leistungsstudien und Vorkommnis auf Produkte nach dem Inverkehrbringen?
Würde das bedeuten, dass vormalige Produktmängel mit Inverkehrbringen zu Vorkommnissen werden würden?
Würde dies auch bedeuten, dass ein Produktmangel niemals zu einer Korrekturmaßnahme führen kann, da das Produkt noch nicht im Markt ist und daher wenn überhaupt eine Vorbeugungsmaßnahme durchgeführt werden kann, um eine potenzielle Nichtkonformität zu verhindern?
Eine Nichkonformität kann ja vor der Konformitätserklärung gar nicht vorliegen.
Guten Tag Herr Handt,
die Definitionen von Produktmangel und Vorkommnisse ähneln sich in der Tat sehr. Wie Sie richtig schreiben, redet die IVDR von „Produktmangel“ nur im Rahmen von Leistungsstudien und von „Vorkommnis“ im Rahmen der Inverkehrbringung (ausgenommen für Leistungsstudien). Somit würden im Regelfall Produktmängel aus Leistungsstudien zu Vorkommnissen werden, wenn diese vor der Inverkehrbringung nicht behoben werden.
Eine Korrekturmaßnahme („bezeichnet eine Maßnahme zur Beseitigung der Ursache eines potenziellen oder vorhandenen Mangels an Konformität oder einer sonstigen unerwünschten Situation;“) sollten Sie immer, auch für Produktmängel in Leistungsstudien, in Betracht ziehen. Eine Vorbeugungsmaßnahme wird hingegen definitionsgemäß für potentielle Nicht-Konformitäten ergriffen. Sicherlich kann eine Nichtkonformität vor der Konformitätserklärung vorliegen, auch während der Entwicklung, z.B. gegen Ihre Spezifikationen oder angewandte Normen.
Herzliche Grüße
Luca Salvatore
Sehr geehrtes Johner-Team!
Wir hatten gerade intern eine Vigilanz-Schulung und eine Sache bleibt weiterhin unklar. Ein schwerwiegendes Vorkommnis, wie z.B. eine sehr starke allergische Reaktion – ein Risiko auf das auch in der IFU hingewiesen wird – führt zu einer starken körperlichen Einschränkung oder auch bis zum Tod. Muss dieser Einzelfall als schwerwiegendes Vorkommnis gemeldet werden, oder – weil darauf in der IFU hingewiesen wird – wird es „nur“ ins Trend Reporting mitaufgenommen? Wir finden es sehr schwer hier eine Linie zu ziehen, da es ja dennoch ein schwerwiegendes Vorkommnis wäre, selbst wenn davor gewarnt wird.
Mit freundlichen Grüßen,
Kathrin
Sehr geehrte Kathrin,
ich habe aus meinem Team folgende Rückmeldung bekommen:
Es gibt somit zwei Überlegungen: Schweregrad und die Frage, ob es eine „erwartete Nebenwirkungen“ ist, wie das die MDR im Artikel 87(1) formuliert.
Bei Ihnen scheint es so zu sein, dass die starke allergische Reaktion eine solche „erwartete Nebenwirkung“ ist.
Beste Grüße, Christian Johner