
Unter Prozessvalidierung versteht man den Nachweis, dass ein Prozess die Anforderungen an seine Prozessergebnisse erfüllt.
Lernen Sie, wann Sie welche Prozesse (im Software-Kontext) validieren müssen und wie Ihnen das einfach gelingt. Finden Sie auch heraus, was Prozessvalidierung mit PQ, IQ und OQ zu tun hat.
Prozessvalidierung: Was ist das genau?
a) Definition des Begriffs „Validierung“
Die ISO 9000:2015 definiert eine Validierung wie folgt:
Bestätigung durch Bereitstellung eines objektiven Nachweises, dass die Anforderungen für einen spezifischen beabsichtigten Gebrauch oder eine spezifische beabsichtigte Anwendung erfüllt worden sind.
Quelle: ISO 9000:2015
Die Norm merkt an, dass der objektive Nachweis, der für eine Validierung notwendig ist, das Ergebnis eines Tests oder einer anderen Form der Bestimmung wie beispielsweise alternativer Berechnungen sei.
b) Definition des Begriffs „Prozess“
Die ISO 13485 greift bei der Definition des Begriffs Prozess erneut auf die ISO 9000:2015 zurück. Diese versteht unter einem Prozess:
Satz zusammenhängender oder sich gegenseitig beeinflussender Tätigkeiten, der Eingaben zum Erzielen eines vorgesehenen Ergebnisses verwendet.
Quelle: ISO 9000:2015
Beispiele für Prozesse sind:
- Entwicklungsprozess
- Sterilisationsprozess
- Produktionsprozess
- Einstellungsprozess
- Verkaufsprozess
Für das weitere sollten wir noch festhalten, dass Prozesse nicht nur Inputs („Eingaben“) und Outputs („Ergebnisse“) hat, sondern auch Ressourcen wie Menschen und Maschinen (einschließlich Software/IT) benötigt.
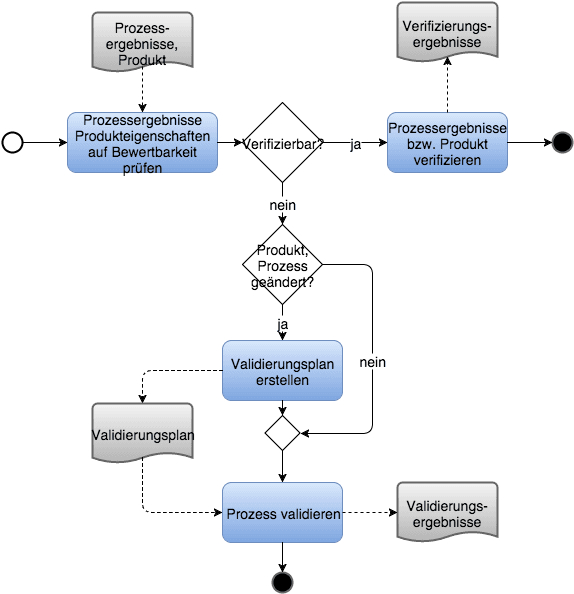
c) Prozessvalidierung
Setzt man beide Definitionen zusammen, ergibt sich, dass eine Prozessvalidierung eine Bestätigung durch Bereitstellung eines objektiven Nachweises ist, dass ein Prozess die beabsichtigten Prozessergebnisse liefert.
Beispielsweise würde man bei einem Entwicklungsprozess sicherstellen, dass das Entwicklungsergebnis die Anforderungen („Design Input“) erfüllt. Bei einem Sterilisationsprozess würde man sicherstellen, dass das zu sterilisierende Gut wirklich steril ist.
Die FDA definiert den Begriff direkt:
Process validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications.
Quelle: FDA
Praxistipps zum Validieren von Prozessen
a) Nur relevante Parameter validieren
Produkte verfügen meist über mehrere Eigenschaften, die die Hersteller sicherstellen müssen. Die Entscheidung, ob eine Validierung notwendig ist, müssen Sie nicht pro Produkt/Prozess treffen, sondern pro Eigenschaft (Parameter). Ein stark vereinfachtes Beispiel:
Eine Spritze muss steril sein und über eine bestimmte Länge verfügen. Den Parameter „Länge“ können Sie einfach verifizieren. Sie müssten den Prozess also nicht daraufhin validieren, ob er Spritzen mit der korrekten Länge produziert – es sein denn, Sie möchten die 100%-Prüfung der korrekten Länge nicht durchführen.
Den Parameter „Sterilität“ können Sie hingegen nicht durch eine „Endprüfung“ gewährleisten, weil das eine zerstörende Prüfung wäre. Eine Prozessvalidierung ist hier notwendig.
Viele Organisationen nutzen AQL (Acceptance Quality Level), um über die Güte/Akzeptanz von Prozessergebnissen zu entscheiden.
b) Verifizierungsprozess validieren?
Falls Sie eine automatisierte 100%-Prüfung der Länge durchführen, ist diese automatisierte Prüfung ebenfalls ein Prozess oder Prozessschritt, den Sie zu validieren haben. Falls dieser Prozess oder Prozessschritt Software oder Computer-Systeme enthält, müssen Sie diese validieren. Wie Sie vorgehen können, beschreibt ein Artikel zur Computer System Validation CSV.
c) Statt validieren verifizieren
Im manchen Fällen ist zwar eine Endprüfung unmöglich, allerdings eine Prüfung des Produktparameters bereits in einem Zwischenschritt. Wenn Sie beispielsweise die korrekte Montage einer Komponente verifizieren können, was am Endprodukt nicht möglich ist, müsste der Prozess nicht notwendigerweise daraufhin validiert werden, ob die Komponentenmontage korrekt erfolgt.
| Produkt-Parameter | Prozess-Schritt 1 | Prozess-Schritt 2 | … | End-Test | Validierung notwendig? |
| Parameter 1 | Kein Test notwendig | Kein Test notwendig | Test durchgeführt | Nein | |
| Parameter 2 | Kein Test notwendig | Test durchgeführt | Kein Test möglich | Nein | |
| Parameter 3 | Kein Test möglich | Kein Test möglich | Kein Test möglich | Kein Test möglich | Ja |
| … |
d) Prozessvalidierung und PQ, IQ und OQ
Oft unterscheiden Firmen (besonders im Pharmaumfeld) folgende Phasen der Prozessvalidierung:
- IQ: Diese erste Prüfung beim Kunden während der Installation soll sicherstellen, dass das Gerät gemäß der Spezifikation geliefert, aufgebaut und installiert wurde, den Anforderungen der Nutzer entspricht und auch die Dokumentation vorliegt. Hier werden Grundlagen für Kalibrierung, Wartung und Reinigung gesetzt. Man sollte mal in der Anleitung schauen :-).
- OQ: Die umfangreichste Prüfung sollte prüfen, ob das Gerät spezifikationsgemäß arbeitet, v.a. auch an den Spezifikationsgrenzen, um zu wissen, was worst case passiert.
- PQ ist dann nur noch der Beweis, dass der Prozess auch unter normalen Bedingungen läuft. Diese Prüfung hat u.a. die Messgenauigkeit (inkl. Kalibrierung und Justierung) zum Ziel. Es geht auch um die Langzeit-Stabilität. In der Pharmawelt probiert man mindestens 3 Batches aus. Ein P-Diagramm zeigt, was beeinflussende Faktoren sind wie Umgebungsbedingung, Arbeitsschritte, Prozessparameter, Materialeigenschaften. Hier kommen Sampling-Plans ins Spiel.

Optimieren Sie Ihre Prozessvalidierung und vermeiden Sie Probleme bei Audits!
Wenige Themen führen zu so vielen Diskussionen und zu so vielen Problemen bei Audits wie die Prozessvalidierung. Mit unseren E-Learning-Videos und Vorlagen für eine konforme Prozessvalidierung vermeiden Sie diese Diskussionen und Probleme und glänzen in Ihrem nächsten Audit.
Regulatorische Anforderungen an die Prozessvalidierung
a) Forderungen der ISO 13485
Die ISO 13485:2016 fordert wie schon die ISO 13485:2010 die Prozessvalidierung unter folgenden Umständen:
- Bei dem Prozess handelt es sich um einen Produktions- oder Dienstleistungsprozess
- Die Ergebnisse dieses Prozesses können oder werden nicht verifiziert z.B. durch eine Messung.
- Die Defizite bei diesen Prozessergebnissen würden erst auffallen bei Verwendung des Produkts oder nachdem die Dienstleistung in Anspruch genommen wurde.
Falls diese Bedingungen zutreffen, ist die Prozessvalidierung vorgeschrieben. Dazu muss das Unternehmen Verfahren festlegen einschließlich
- Kriterien für die Bewertung des Prozesses
- Qualifikation des Personals
- Anwendung von Methoden und Verfahren
- Dokumentation/Aufzeichnungen
- Re-Validierung
- Freigabe von Änderungen an Prozessen.
Falls im Rahmen dieser Prozesse Software genutzt wird, ist diese Software zu validieren.
b) Weitere nationale und internationale relevante Vorschriften
Die inzwischen nicht mehr existente Global Harmonization Task Force GHTF hat ein Dokument GHTF SG 3 NB 99:10:2004 zur Prozessvalidierung veröffentlicht, das inzwischen vom IMDRF „verwaltet“ wird.
Die Zentralstelle der Länder ZLG hat ebenfalls ein Dokument veröffentlich, das die Nummer ZLG 3.9 B 18 und den Titel „Validierung von Prozessen der Produktion und der Dienstleistungserbringung (einschließlich Software)“ trägt.
Weiter sollten Sie die Good Manufacturing Practices und den GAMP5-Guide im Auge behalten.
c) Forderungen der FDA
Die FDA beschreibt in den Quality System Regulations, konkret im 21 CFR 820.75, die Anforderungen an die Prozessvalidierung:
Auch sie fordert diese nur, wenn die Prozessergebnisse nicht verifiziert werden können. Diese Validierungsaktivitäten müssen umfassen:
- Die durchgeführten Aktivitäten müssen mit Datum und Unterschrift dokumentiert werden.
- Verfahren müssen festgelegt werden, mit denen die Prozessparameter überwacht werden.
- Nur qualifizierte Personen dürfen die Prozesse validieren.
- Die Methoden und Daten zur Kontrolle und Überwachung der Prozesse, das Datum der Durchführung und die durchführenden Personen sowie das relevante Equipment sind zu dokumentieren.
- Bei Änderungen muss der Hersteller bewerten, ob eine Re-Validierung notwendig ist, und diese dann bei Bedarf durchführen.
Die Anforderungen an die Validierung von Software nennt die FDA im „Guidance Document Software Validation “.
Professor Johner geht in dieser Episode den Fragen nach, welche Prozesse und Prozessschritte die Hersteller im Auge behalten sollten, welche Risiken die Hersteller im Blick behalten und beherrschen müssen, an welche regulatorischen Vorgaben (Gesetze, Normen usw.) sie dabei gebunden sind, welche typischen Fehler Hersteller machen und wie sie vorgehen sollten, um Probleme bei Audits und Inspektionen oder im schlimmsten Fall bei der Anwendung der Produkte am Patienten zu vermeiden.
Diese und weitere Podcast-Episoden finden Sie auch hier.



Sehr geehrter Herr Professor Johner,
mir geht es bei meinem Beitrag nur um Fertigungsprozesse. In dem von Ihnen zitierten ZLG Dokument ist der Fertigungsprozess Löten genannt. Ist es richtig, dass es hier ausschließlich um automatisches Löten in einer Lötstraße geht, oder muss der Prozess oder wohl besser die Tätigkeit Handlötung auch validiert werden? Hier hängt das Ergebnis ja ausschließlich von der Fertigkeit der Person ab, wenn Lötgerät und Lot vorgegeben sind. Wie sehen Sie das?
Mit freundlichen Grüßen,
Werner Neumüller
Sehr geehrter Herr Neumüller,
ich glaube nicht, dass die ZLG die Anforderungen an die Prozessvalidierung auf automatisierte Prozesse beschränken will. Allerdings könnte man bei dem manuellen Löten argumentieren, dass der Vorgang immer mit einer direkten Überprüfung des Ergebnisses einhergeht. Man sieht i.d.R. der Lötstelle an, ob sie etwas taugt – zumindest wann man sein Fach beherrscht. Dann bedürfte es dieser Validierung nicht.
Prozesse, deren Prozessergebnisse man nicht (ausreichend) prüft oder prüfen kann, bedürfen der Validierung unabhängig davon, ob der Prozess manuelle Tätigkeiten umfasst.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
wenn es bei einem Fertigungsprozess mehrere Anlagen gibt (die gleichen Anlagen vom gleichen Hersteller und haben den gleichen Aufbau) und das Worst-Case Produkt wird nur auf einer davon hergestellt, da es nur ein Werkzeug dafür genügt. Muss man hier nur die betreffende Anlage qualifizieren dann den Prozess validieren und die anderen Anlagen nur qualifizieren oder muss man für jede Anlage separat das Worst-Case Produkt definieren.
Beste Grüsse, Ferhad Mussa
Sehr geehrter Herr Mussa,
eine Beschränkung auf den Worst Case reicht i.d.R. nicht aus. Sie müssen im Risikomanagement für jedes Produkt (und damit jede Anlage, die es produziert,) die Risiken identifizieren, die in diesem Fall bei der Produktion entstehen können. Sie müssen dann abwägen, ob diese Risiken akzeptabel sind. Entsprechende Maßnahmen müssen Sie ergreifen, entsprechend streng müssen Sie den Prozess beherrschen und validieren.
Es geht auch deshalb nicht nur um den Worst-Case, weil der Worst-Case mit einem hohen Schweregrad korreliert. Hier geht es aber um Risiken. D.h. ein weniger schlimmer Fall könnte zu höheren Risiken führen, weil dessen Wahrscheinlichkeit höher ist.
Beste Grüße, Christian Johner
Hallo,
wir führen einen Wärmebehandlungsprozess durch, ist das Schreiben und prüfen eines Glühdiagramms eine Verifizierung?
Validiert wurde die Parameter des Glühen durch die Ermittlung der mechanischen Werte des Produktes.
Gruß
T. Siemon
Sehr geehrte Frau Siemon,
danke für Ihre Frage!
Das Schreiben eines Glühdiagramms ist eher keine Verifizierung. Es kann aber sein, dass das Diagramm die Ergebnisse einer konkreten Messung darstellt, die eine Verifizierung sein könnte.
Es wäre darauf zu achten, dass die Werte in dem Diagramm nachweisen, dass eine spezifizierte Eigenschaft nachweisen. Den eine Verifizierung ist als eine objektive Prüfung definiert, die nachweist, das spezifizierte Eigenschaften erfüllt sind.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
erst einmal vielen Dank für die interessanten und hilfreichen Beiträge.
Zur Definition eines „Prozesses“ im Sinne der DIN EN ISO13485:2016, in Kapitel 0.3 findet man folgendes:
„Jegliche Tätigkeit, die eine Eingabe erhält und diese in ein Ergebnis umwandelt, kann als Prozess
angesehen werden.“
Folglich wären nach meinem Verständins lediglich „Herstellprozesse“, also wo eine Eingabe in ein Ergebnis/Output umgewandelt wird, im Scope einer Prozessvalidierung, ist das richtig?
„Verifizierungsprozesse“ haben für mich nichts mit Prozessvalidierung zu tun. Hier geht es meiner Meinung nach, je nachdem, eher in Richtung CSV oder Qualifizierung eine Prüfstands oder Testmethodenvalidierung, oder bringe ich da etwas durcheinander?
Generell verwirrt das die Kollegen, nicht nur außerhalb QA/QS oft sehr und das, meiner Meinung nach, zu Recht.
Ich wäre Ihnen sehr dankbar, wenn Sie zu den Begriffen Klarheit schaffen könnten: Equipment-Qualifizierung, Prozessvalidierung, CSV, TMV; wann ist was notwendig?
Vielen Dank im Voraus.
Mit freundlichen Grüßen
M. Koch
Sehr geehrte/r M. Koch,
Ihre Kritik hat mich erreicht.
Besten Dank für Ihre wichtigen und hilfreichen Hinweise.
Ihre Einschätzung, dass Prozesse ein Prozessergebnis haben müssen, teile ich. Der möglichen Schlussfolgerung, dass nur Herstellungsprozesse dazu zählen, kann ich noch nicht ganz folgen. Auch ein Audit kann und sollte als Prozess durchgeführt werden. Das Ergebnis sind dabei u.a. Aufzeichnungen und eine Bewertung.
Ein weiterer Typ an Prozessen sind die Prozesse, die der Überprüfung dienen. z.B. das Usability-Testing oder die Überprüfung der Software wären als Prozesse abbildbar. Auch Prozesse selbst können Gegenstand von Überprüfungen sein. Und auch diese Überprüfungen können und sollten in Form von Prozessen stattfinden. Dann sind wir bei der Prozessvalidierung.
Der Begriff „Verifizierungsprozess“ ist in der Tat etwas unglücklich. Ich werden Ihrer Aufforderung gerne nachkommen, bei Gelegenheit, diesen Absatz zu überarbeiten und die Links zu verwandten Begriffen besser herausarbeiten.
Nochmals danke für diese Hinweise.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
vielen Dank für die schnelle Antwort auf meinen Kommentar.
Die Folgerung, dass nur Herstellungsprozesse dazu zählen, mache ich an dem unscheinbaren Wort „umwandeln“ fest, denn nur bei solchen wird ja etwas, in dem Fall Material in ein Produkt, umgewandelt.
Beim Verifizieren oder Überprüfen folge ich einem bestimmten Ablauf/Prozess, aber wandle ja nichts in dem Sinne um. Deshalb würde ich persönlich hier nicht den Prozess des Überprüfens validieren, sondern das Prüfequipment qualifizieren, also dessen prinzipielle Eignung nachweisen.
Es ist gut möglich, dass ich mich da in etwas verzettelt habe, da ich versuche die angesprochenen Themen „Prozess-, Computersystem-, Testmethodenvalidierung und Equipmentqualifizierung“ sinnvoll und klar voneinander zu trennen und deren Anwendungsbereiche herauszuarbeiten.
Deshalb bin ich Ihnen und Ihrem Team sehr dankbar über die bereits bestehenden interessanten Beiträge zu Inhalten der 13485 und auch die Möglichkeit, diese kommentieren zu können. Ich freue mich über weitere solcher Beiträge.
Beste Grüße,
M.Koch
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihre zahlreichen, hilfreichen Blog Einträge. Ich stöbere gern mal nach Tipps und Tricks. Auf diesen Beitrag bin ich mit dem Hintergedanken gestoßen, einen PMS Prozess zu validieren. Die MDR drückt immer wieder klar und deutlich aus, dass die PMS Prozesse „effective and appropriate“ sein sollen. Haben Sie einen Tipp, wie ich eine Prozessvalidierung demnach für PMS gestalten kann?
Dabei habe ich im Kopf, dass der Prozess auch outsourced sein kann an den EC-Rep bzw. Importeure/ Distributoren.
Ganz liebe Grüße! Und vielen Dank im Voraus.
Josefine Wagener
Sehr geehrte Frau Wagener,
das ist wirklich eine großartige Frage!
Für die Validierung des PMS-Prozess hätte ich folgende Ideen:
Soweit meine ersten Gedanken.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
für die Validierung des Aufbereitungsprozesses unserer Medizinprodukte nach ISO 17664 stellt sich für uns die Frage nach einem angemessenen AQL zur Festlegung unserer Stichprobengröße nach ISO 2859. Für Gesichtsmasken, zB., legt die DIN EN 14683:2019-10 sich mit einem AQL von 4% eindeutig fest. Was sind ihre Erfahrungen für akzeptierte AQLs?
Besten Dank,
Fabian Kaiser
Sehr geehrter Herr Kaiser,
danke für Ihre Frage!
Meines Wissen gibt es keine allgemeingültigen Festlegung für die AQLs. Wenn die Benannten Stellen diese überhaupt ansprechen (nicht alle Auditoren sind Expert:innen in Statistik), orientieren sie sich am Stand der Technik. Der wiederum hängt vom jeweiligen Kontext (u.a. Risiko) und den zu prüfenden Werten ab bzw. deren Relevanz für die Konformität und die Sicherheit des Medizinprodukts.
Beste Grüße, Christian Johner
Meine Frage wäre folgende zur Validierung eines Lasergerätes für Medizinprodukte:
– gibt es eine Vorgabe wieviel Teile aus verschiedenen Chargen validiert werden müssen?
– würden zum Beispiel 20 Teile aus zwei verschiedenen Chargen ausreichen die gelasert werden?
Mit frdl. Grüßen
X. Matthies
Lieber Herr Matthies,
ich empfehle Ihnen, zunächst zu prüfen, ob es spezifische Normen bzgl. der Akzeptanzkriterien des zugrundeliegenden Laserprozesses gibt. Diese Normen würden Ihnen bestenfalls die Stichprobengröße je Chargengröße vorgeben. Ihren Risikomanagement-Prozess können Sie nutzen, um das Akzeptanzlevel je Prozessparameter zu begründen. Mithilfe der Norm 16269-6 können Sie die Stichprobengröße für OQ und PQ herleiten. Ein Ansatz, drei Chargen für die PQ zu nutzen, leitet sich vermutlich aus der Arzneimittelherstellung ab. Dieser Ansatz gilt jedoch nicht für Medizinprodukte. Bei Medizinprodukten muss der Hersteller die Anzahl an Chargen und auch die Stichprobengröße unter Berücksichtigung der relevanten Einflussparameter für den zugrundeliegenden Prozess bestimmen und begründen.
Mit den allerbesten Grüßen,
Catharina Bertram
Hallo,
im Beitrag oben steht zum Thema „Praxistipps zum Validieren von Prozessen“ das Beispiel mit der Spritzenlänge. Es wird gesagt, dass der Prozess zur Herstellung der Spritzenlänge validiert werden muss, wenn die Prüfhäufigkeit reduziert werden soll. Aber ist es tatsächlich so, dass eine Reduzierung einer Prüfhäufigkeit immer eine Validierung zur Folge hat? Oder reicht nicht vielmehr aus eine reduzierte Prüfhäufigkeit zu begründen, z.b. mithilfe von Statistik und/oder Risikoabschätzungen. In der 13485 heißt es im Abschnitt 7.4.3 „Das Ausmaß der Verifizierungstätigkeiten“, ich interpretiere dies so, dass Verifizierung eben sehr wohl weniger als 100%-Prüfung bedeuten kann. Irre ich mich hier? Falls ja, könnten Sie mir bitte erklären, wo genau definiert ist, dass Verifizierung immer gleich 100% Prüfung bedeutet?
Liebe Frau Walter, vielen Dank für Ihre Frage,
Der Praxistipp zur Spritzenlänge bezieht sich im wesentlichen auf den Grundsatz: Wenn keine 100% Kontrolle durchgeführt werden kann, muss der Prozess validiert werden. Es geht also nicht darum, ob die Prüfhäufigkeit reduziert wird, sondern dass entschieden wird, keine 100% Kontrolle mehr durchzuführen. Was die Betrachtung aller Prozesse angeht, ergibt sich aus ISO 13485, 7.5.6. Dort steht:
Die Organisation muss sämtliche Prozesse der Produktion und Dienstleistungserbringung validieren, deren Ergebnis nicht durch nachfolgende Überwachung oder Messung verifiziert werden kann oder verifiziert wird, wodurch sich Unzulänglichkeiten erst zeigen, nachdem das Produkt in Gebrauch genommen oder die Dienstleistung erbracht worden ist.
Daraus leitet sich indirekt die Argumentation mit 100% Kontrolle ab. D.h. wenn Sie alle Schritte in der Herstellung Ihres Produkts auflisten und betrachten, bewerten Sie, ob das Ergebnis zu 100% geprüft wird, oder nicht. Und wenn Sie das verneinen müssen, ist der Prozess zu validieren. Bei Umfang und Planung der entsprechenden Prozessvalidierung dürfen Sie natürlich Risikoüberlegungen anstellen. Überlegungen dazu stellen wir in unserem Artikel zur Prozessrisikoanalyse an: https://www.johner-institut.de/blog/iso-14971-risikomanagement/pfmea/. Der Umfang an zu prüfenden Teilen kann z.B. auf einem risikobasierten Vertrauensintervall festgelegt werden (z.B. nach ISO 16269-6).
Der von Ihnen referenzierte Abschnitt 7.4.3 bezieht sich auf die Beschaffung. Der Begriff Verifizierung ist grundsätzlich so belegt, dass die Einhaltung von Spezifikationen geprüft wird, wohingegen Validierung das Erreichen/Erfüllen eines Ziels meint – wobei das auch verifizierende Tätigkeiten beinhalten kann. Die Definitionen der Begriffe in unserem ISO Umfeld finden Sie z.B. in der ISO 9000. Bei der Kontrolle von beschafften Produkten werden sie deren Einhaltung von Spezifikationen prüfen und verifizieren. Wareneingangsprüfungen können m.E. durchaus auf Stichprobenplänen beruhen (Stichwort: AQL), die durch diese Stichprobenpläne definierten Teile werden Sie verifizieren. So gesehen: Wenn Sie eine Prozessvalidierung durchführen und dabei Teile vermessen, wäre das explizite Messen im Prinzip auch eine verifizierende Tätigkeit bezogen auf diese Teile im Rahmen der Prozessvalidierung. Ich würde bzgl. Diskussionen zu Prozessvalidierung empfehlen, lediglich Abschnitt 7.5.6 anzuziehen. Verbeissen Sie sich dabei nicht in die Deutung und Interpretation der Begriffe Verifizierung und Validierung und wenden obige Überlegungsideen an.
Bringt Sie das so schon einmal weiter?
Freundliche Grüsse
Urs Müller
Hallo Herr Mueller,
vielen Dank für Ihre zügige und umfangreiche Antwort.
Nicht das, was ich hören wollte ;-), aber zumindest habe ich es jetzt schwarz auf weiß vom Experten. Ich hatte immer ein Knoten im Kopf, weil die Verknüpfung von Verifizierung = 100%-Kontrolle wohl nicht wahr haben wollte, weil ich das so explizit in der Norm nicht herausgelesen habe.
Ich würde das gern trotzdem nochmal an einem konkreten Beispiel durchspielen. Nehmen wir einen CNC-Drehprozesses, bei dem z.B. der Durchmesser am Hüftschaftkonus hergestellt wird. Nach ihren Ausführungen oben würde es also nicht ausreichen, eine Stichprobenprüfung einzuführen, nachdem ich für dieses Prüfmerkmal eine Prozessfähigkeitsanalyse durchgeführt habe (sprich die Kennwerte für Cp/Cpk ermittelt)? Stattdessen müsste ich auch den Drehprozess für die entsprechende Maschine grundsätzlich validieren?
Liebe Frau Walter,
Ich fasse mich hier kurz und melde mich bei Ihnen direkt. Das von Ihnen erwähnte Beispiel lässt durchaus Spielraum bzgl. 100% Kontrolle zu: Wann ist das Ergebnis eines Prozesses ausreichend überprüft? Im Zusammenspiel mit Qualifizierungsaktivitäten bestehen bzgl. Prüfung des Ergebnisses durchaus Spielräume, genügend Sicherheit zu schaffen. Können Sie im Rahmen der Prozessvalidierung nicht ausreichend Vertrauen schaffen und ist eine Messung von Ergebnissen nur durch zerstörende Prüfung möglich, könnten Sie z.B. in Abhängigkeit der Chargengrösse eine Anzahl an zu prüfenden Einheiten pro Batch festlegen. In einem aktuellen Projekt haben wir beispielsweise die Infrastruktur für einen Crimp Prozess qualifiziert und anschliessend für jeden Batch an hergestellten Kabelverbindungen festgelegt, wo wir Prüfmuster ziehen und diese anschliessend zerstörend bzgl. Abzugskraft gegen empfohlene Vorgaben des Werkzeugherstellers prüfen (so gesehen, Designvorgaben). Der Samplingplan folgt Empfehlungen des Werkzeugherstellers und ist statistisch begründet. Die Beurteilung des Prozesses und die Wahl des Vorgehens zur Kontrolle des Prozesses ist abgestimmt mit der Produktrisikoanalyse. Wir haben also den Prozess als solches nicht validiert sondern haben festgelegt, dass wir den Prozess wie oben erwähnt kontrollieren.
Ohne detaillierte Kenntnis Ihrer Herstellverfahren, würde ich einmal folgende Überlegungen anstellen: Ich würde bzgl. Ihres Herstellungsprozesses die Schritte zur Fertigung des Hüftschaftkonus listen. Darin würde ich das Drehen und den Einfluss auf mein Produkt betrachten und festlegen, ob und wann ich das Ergebnis als verifiziert betrachten kann oder ob ich den Prozess bzgl. weiterer Eigenschaften in einer Validierung betrachten muss. Die ganzen Betrachtungen würde ich dabei immer mit Bezug zur Produktrisikoanalyse anstellen. Am Ende muss jeder Schritt/Prozess bewertet sein. Übrigens: Die Prozessfähigkeitsanalyse alleine wird ggf. bzgl. Prozessvalidierung nicht mehr als ausreichend betrachtet.
Freundliche Grüsse
Urs Müller
Hallo Herr Müller,
Ich habe eine Frage bezüglich der Risikoanalyse bei der Prozessvalidierung.
Wenn mehrere Maschinen den gleichen Prozess durchführen (z.B. Beschriften oder Fräsen), muss dann für jede Maschine eine Risikoanalyse erstellt werden und die Risikopunkte in den einzelnen Validierungsphasen abgearbeitet /getestet werden oder reicht hier eine Risikoanalyse für den Prozess aus?
Falls eine Risikoanalyse ausreicht, wie verhält es sich dann mit der Versionierung bei verschiedenen Maschinen? Normalerweise hat man zu Beginn der Validierung Version 1 und am Ende Version 2.
Beste Grüße und vielen Dank im Voraus
Lieber Herr Kohler, vielen Dank für diese spannende Frage! Lassen Sie mich bzgl. Antwort kurz ausholen: Idealerweise wird es eine Produktrisikoanalyse nach ISO 14971 und eine PFMEA geben. Mit der PFMEA begutachten Sie die einzelnen Schritte in der Herstellung und identifizieren ggf. kritische Prozessparameter bzw. legen damit den Umfang/Aufwand für allfällige Validierungen fest. D.h. das nötige Equipment würde dahingehend nicht individuell betrachtet. Akzeptanzkriterien ergeben sich aus Produktmerkmalen und damit damit verbundenen Risiken. Ein Prozess für die Prozessvalidierung kann aber auch so aussehen, dass die Risikoanalyse als Teil der Prozessvalidierung aufgesetzt wird, im Verbund mit den Qualifizierungstätigkeiten für ein darin verwendetes Equipment. Es gibt schlussendlich unzählige Möglichkeiten, die verschiedenen Tätigkeiten für die Prozessvalidierung zusammenzufassen, z.B. pro Prozess, pro Produkt, für jedes Equipment – mit allen Vor- und Nachteilen. Das hängt grundsätzlich von Ihren Grundvoraussetzungen ab.
So gesehen würde ich das so handhaben: wenn der gleiche Prozess auf mehreren, gleichen Maschinen durchgeführt wird, ist der Prozess an diese Maschinen geknüpft und eine einmalige Beschreibung der Validierung und zugehörige Risikoanalyse würde m.E. gesehen ausreichen, da der Umfang und Einfluss der Maschinen bzgl. Produktqualität beschrieben ist und damit verbundene Validierungstätigkeiten herausgearbeitet sind. Als Beispiel kommen mir diesbezüglich Lötstationen bzw. Lötprozesse in den Sinn: Lötzinn, Temperatur des Kolbens, Dauer und Fähigkeiten der Bediener könnten Parameter sein, die relevant sind für das Löten. Die Lötstationen werden Sie je nachdem IQ, OQ und PQ unterziehen – der Lötprozess, die Risikoanalyse und die Akzeptanzkriterien bleiben gleich. Mit IQ/OQ/PQ werden Sie sicherstellen, dass die Lötstationen das für die Produktqualität relevante Ergebnis liefern – den Prozess und die relevanten Parameter kennen Sie. Ich möchte aber betonen: das heisst nicht, dass Sie nicht eine RA pro Equipment machen dürfen – vielleicht wollen Sie auf einzelnen Maschinen zusätzliche Schritte fahren (so ein Projekt hatte ich auch einmal – 6 Anlagen, 6 Risikoanalysen, die initial genau gleich waren).
Bzgl. Versionierung der Risikoanalyse kann ich Ihnen nicht ganz folgen – das dürfte ebenfalls daran liegen, dass es verschiedene Möglichkeiten gibt, die Risikoanalysetätigkeit in Prozessen zu verorten. Darum allgemein: Mein Ort der Wahl um alle Abweichungen oder Besonderheiten festzuhalten, wäre der Validierungsbericht. Ggf. fassen Sie die Validierung von mehreren Anlagen bzgl. eines Prozesses in einem Validierungsplan zusammen, dann haben Sie einen abschliessenden Validierungsbericht für alle Anlagen und können Erkenntnisse bzgl. Risiken in Bezug auf die verschiedenen Anlagen im abschliessenden Validierungsbericht begründen. Oder Sie halten das in Ihrer Liste der validierten Prozesse, oder Equipmentliste oder VMP fest und bringen das bzgl. Dokumentation bei der nächsten Revalidierung entsprechend auf Stand.
Bringt Sie das so schon einmal weiter?
Freundliche Grüsse
Urs Müller
Hallo Herr Müller,
ich bin gerade dabei ein neues Validierungskonzept für unterschiedlichste CNC geführte Laserschweißteile in der Medizintechnik zu erarbeiten.
Das Problem was sich mir aktuell stellt, ist die große Anzahl (1300 Artikel) an unterschiedlichen Artikel mit unterschiedlichsten Geometrien, Schweißarten, Schweißtiefen, Materialien, Schweißparameter.
Zu dem aktuellen geschilderten Problem hätte ich folgende Fragen:
1. Bekomme ich alle Artikel über einen Worst-Case Ansatz abgedeckt?
2. Macht es Sinn sich unterschiedlichste Dummys zu erstellen, die gewisse Worst-Case abdecken ( Geometrie, Schweißnahtart, Schweißtiefe, Wandung)? Oder sollte besser für die Validierung reale Worst-Case Artikel herangezogen werden. Diese Artikel decken aber nicht zu 100% den Worst-Case ab.
3. Es gibt verschiedene Auswertungsverfahren wie z.B. Schliffbilduntersuchung, Zugprüfung, Dichtigkeitsprüfung, Sichtprüfungs usw.
Reicht es in der Prozessvalidierung aus die unterschiedlichen Prüfparameter abzuprüfen (auf in Toleranz) oder sollte eine statische Auswertung erfolgen? Oder kann es auch ein Mix daraus sein.
4. Gibt es eine Vorgabe, wieviel Teile ich für die 3 PQ Runs heranziehen muss?
5. Wie gehe ich mit, den aus der Validierung ermittelten Schweißparameter um? Für die unterschiedlichen Artikel müssen dementsprechend unterschiedliche Programme mit unterschiedlichem Parameter erstellt werden. Reicht es aus, zu sagen: Die Parameter müssen innerhalb des ermittelten Worst-Case- Parameter ( MIN-MAX) liegen. Oder muss ich jedes Programm nochmal einzeln Betrachten, da das Zusammenspiel der Parameter Einfluss auf die Schweißnaht hat. Könnte ich hier auf einen Erstmusterprüfbericht verweisen?
Beste Grüße und vielen Dank im Voraus
Lieber Herr Böhler, vielen Dank für diese spannende Fragensammlung. Ich versuche, jeden Punkt kurz erörtern.
Bringt Sie das so schon einmal weiter?
Freundliche Grüsse
Urs Müller
Sehr geehrtes Johner-Team,
ich hätte noch eine Frage bzgl. der ISO 16269-6:
Mit welcher Begründung legt man die C/P-Level fest? Gibt es State-of-the-Art Festlegungen für Medizinprodukte oder auch Risikoklassen, wann man bspw. 90/90 oder 95/95 wählt?
Über jede Hilfe wäre ich sehr dankbar.
Viele Grüße
Nicolai Pottin
Lieber Herr Pottin, vielen Dank für Ihre Frage
Die wenigen Ausnahmen, in denen die Werte für Vertrauensintervall und Wahrscheinlichkeit für Produkte vorgegeben werden, sind im Artikel verlinkt. Die Werte sollten Sie über das Risikomanagement pro Prozess bzw. zu validierendem Bearbeitungsschritt begründen, beispielsweise so:
Nachfolgend ein paar Beispiele, wie ich das argumentieren würde, ohne Anspruch auf Richtigkeit:
Freundliche Grüsse
Urs Müller
Sehr geehrtes Johner – Team,
an welcher Stelle im Entwicklungsprozess bzw. im Design – Transfer wird sichergestellt, dass nachgelagerte Fertigungsprozesse keinen Einfluss auf mein betrachtetes Prozessergebnis haben? Bzw. müssen nachgelagerte Prozesse, z.B. Wärmebehandlungen, die später in der Prozesskette durchgeführt werden aufgrund von anderen Prozessen in einer Validierung mit berücksichtigt werden, da sie evtl. Einfluss auf das Prozessergebnis haben könnten oder muss dies durch die Spezifikation an die Klebestelle abgefangen werden?
Ein gutes Beispiel ist vielleicht ein abschließender Autoklavierzyklus, der auf viele vorherige Prozessergebnisse einen Einfluss haben könnte. Müsste dieser dann in jeder Validierung mit berücksichtigt werden?
Über eine Rückmeldung würde ich mich sehr freuen.
Mit freundlichen Grüßen
Andrea Meyer
Liebe Frau Meyer,
vielen Dank für diese wichtige und spannende Frage!
Die Bewertung von potenziellen Einflüssen durch nachgelagerte Prozesse kann an mehreren Stellen im Entwicklungsprozess erfolgen:
Wenn man einen Zeitpunkt benennen möchte, an dem alle Überlegungen zusammenlaufen, dann würde ich dafür Prozessvalidierung inklusive Prozess-FMEA wählen. Dort betrachten Sie alle Prozessschritte im Zusammenhang – sowohl deren Einfluss aufeinander als auch auf das Endergebnis.
Wenn Sie z. B. im Rahmen der Produktentwicklung bereits gezeigt haben, dass die Klebung autoklavierbar ist, kann der Einfluss des Autoklavierens ggf. als vernachlässigbar bewertet werden. Andernfalls sollte dies über eine kombinierte Validierung geprüft werden – z. B. durch Tests an bereits autoklavierten Teilen.
Fragen Sie gerne nach, wenn Sie das nicht weiterbringt.
ehr geehrtes Johner‑Team,
ich habe eine Frage zu einem Herstellungsprozess eines Schlauches.
Bei der Herstellung wird ein Schlauchteil verklebt. Anschließend erfolgt an 100 % der Teile ein Dichtigkeitstest.
Benötigt man in diesem Fall zusätzlich eine Validierung des Klebeprozesses?
Die Funktionsfähigkeit wird ja bereits durch den Dichtigkeitstest vollständig überprüft. Wie kann man gegenüber der Benannten Stelle argumentieren, dass keine zusätzliche Klebevalidierung erforderlich ist? Lässt sich dies über das Risikomanagement begründen?
Viele Grüße
Sehr geehrter Herr Quand,
mein Kollege Urs Müller schätzt das wie folgt ein:
—
Nein, wenn eine 100% Kontrolle der Dichtigkeit gemacht wird, ist keine Prozessvalidierung mehr nötig (ISO 13485 7.5.6: Prozessvalidierung ist nur erforderlich, wenn eine Überprüfung des Ergebnisses nicht möglich ist).
Die Ermittlung der Prozessparameter (z.B. Klebstoffmenge, Aushärtezeit) erfolgt unter ISO 13485 7.5.1, Beherrschbarkeit des Prozesses ergibt sich über Schulung, SOPs und qualifiziertes Equipment sowie natürlich die 100% Prüfung.
Dass eine 100% Kontrolle nach der Klebung durchgeführt wird, wäre ein Ergebnis der Risikoanalyse bzw. Prozessrisikoanalyse (undichte Klebestelle – Verunreinigung oder keine Versorgung des Patienten mehr – Schaden am Patient (Sepsis, Tod,…) und die 100% Kontrolle die Massnahme um sicherzustellen, dass dieser Qualitätskriterium für das Produkt eingehalten werden.
—-
Beste Grüße, Christian Johner