
Um die Fallzahlplanung werden Sie nicht herumkommen, wenn Sie für Ihr Medizinprodukt oder Ihr In-vitro-Diagnostikum (IVD) eine klinische Prüfung bzw. klinische Leistungsstudie durchführen müssen. Bei der Berechnung der Fallzahl sollten Sie die beiden folgenden Situationen vermeiden:
- Situation 1: Sie wählen einen zu großen Stichprobenumfang. Dann haben Sie einen höheren Aufwand als nötig. Sie verschwenden Zeit, Geld und Ressourcen, wodurch sich der Zulassungsprozess verzögert.
- Situation 2: Sie wählen die Fallzahl zu gering. Dann ist die Wahrscheinlichkeit hoch, dass Sie entweder den vermuteten Effekt nicht nachweisen können und/oder die Benannte Stelle die Studie aufgrund fehlender Zuverlässigkeit und Robustheit der Daten nicht anerkennt.
Daher wollen Sie die richtige Fallzahl ermitteln, um weder zu viel Aufwand zu betreiben noch unzuverlässige Daten zu erheben und damit eine Wiederholung der Studie oder gar eine Gefährdung von Patienten zu riskieren.
Dieser Artikel stellt Ihnen sechs Fragen vor, die Sie beantworten müssen, um die passende Fallzahl zu bestimmen.
Hinweis zur Autorenschaft: Dr. Thomas Keller von ACOMED statistik ist Mitautor dieses Artikels. Er unterstützt die Kunden des Johner Instituts bei der Fallzahlberechnung für klinische Prüfungen von Medizinprodukten und Leistungsstudien von In-vitro-Diagnostika (IVD).
Der Biostatistiker Dr. Keller und die IVD-Spezialistin Dr. Bertram geben im Gespräch mit Professor Johner „Best Practices“ bei der Fallzahlplanung, die jeder Hersteller kennen sollte.
Diese und weitere Podcast-Episoden finden Sie auch hier.

1. Notwendigkeit der Fallzahlplanung in klinischen Studien
Die grundlegenden Sicherheits- und Leistungsanforderungen an Medizinprodukte bzw. In-vitro-Diagnostika (IVD) sind in Anhang I der MDR bzw. IVDR festgeschrieben. Hersteller weisen die Einhaltung dieser Anforderungen nach, indem sie das betreffende Produkt und seine Komponenten testen, verifizieren und validieren. Dieser Nachweis umfasst auch die klinische Bewertung für Medizinprodukte. Bei IVD spricht man von der Leistungsbewertung.
Wenn Hersteller ein Experiment planen, um valide Daten für ihr Produkt zu erheben, kommt immer wieder die Frage auf: Wie viele Proben, Probanden oder Datensätze müssen wir untersuchen?
Gelegentlich unterstützen Normen und Guidance-Dokumente dabei, den Stichprobenumfang einer Studie bzw. für einzelne Experimente zu ermitteln (z. B. die FDA Guidance Documents ). Jedoch sind solche Hilfestellungen nicht für alle Produkte und Tests verfügbar. Fehlen konkrete Vorgaben, müssen Hersteller eine eigene Fallzahlplanung für die beabsichtigte Studie und das zu erreichende Studienziel durchführen.
Mit einer guten Studienplanung wird es Ihnen als Hersteller gelingen, sowohl den Stichprobenumfang auf ein erforderliches Maß zu reduzieren als auch zuverlässige, robuste und qualitativ hochwertige Daten zu erheben.
Lesen Sie hier mehr zur klinischen Prüfung von Medizinprodukten und zur Leistungsbewertung von IVD.
2. Regulatorische Anforderungen an die Statistik in klinischen Studien
Sowohl die EU-Verordnung 2017/745 über Medizinprodukte (MDR) als auch die EU-Verordnung 2017/746 über In-vitro-Diagnostika (IVDR) fordern in den Definitionen in Artikel 2, dass ein klinischer Prüfplan für Medizinprodukte bzw. ein Leistungsstudienplan für IVD „statistische Erwägungen“ beschreibt.
Die IVDR hält weiterhin fest, dass eine Leistungsstudie für IVD eine „Studie zur Feststellung oder Bestätigung der Analyseleistung oder der klinischen Leistung eines Produkts“ darstellt (IVDR, Artikel 2 (42)). Die geforderten „statistischen Erwägungen“ betreffen bei IVD daher sowohl Experimente zum Nachweis der analytischen Leistungsparameter, als auch klinische Leistungsstudien.
a) Leistungsbewertung von IVD
Die IVDR fordert die IVD-Hersteller auf, sich schon frühzeitig mit den statistischen Methoden auseinanderzusetzen, die sie für die Leistungsbewertung nutzen. Bereits im Leistungsbewertungsplan sollen „geeignete statistische Instrumente“ beschrieben werden, „die zur Prüfung der Analyse- und klinischen Leistung des Produkts […] angewandt werden“ (s. IVDR, Anhang XIII, Abschnitt 1.1.).
Das detaillierte Vorgehen zum Nachweis der analytischen bzw. klinischen Leistung eines IVDs dokumentieren Hersteller in der Regel in separaten Plänen.
Allerdings suchen IVD-Hersteller in der IVDR vergebens nach Vorgaben für die Erstellung eines Plans zur analytischen Leistungsbewertung – die IVDR nennt lediglich die zu prüfenden Leistungsparameter in Anhang I, Abschnitt 9.1.a). Hingegen führt die IVDR in Anhang XIII, Abschnitt 2.3.2. die Anforderungen an den Inhalt eines klinischen Leistungsstudienplans detailliert auf. In Abschnitt 2.3.2, Unterpunkt j) finden Sie die Vorgaben, dass IVD-Hersteller
- das Design der klinischen Leistungsstudie begründen müssen,
- die wissenschaftliche Robustheit und Validität der Studie belegen sollen,
- das statistische Design und alle Maßnahmen zur Reduzierung von Verzerrungen (Bias) beschreiben sollen.
b) Klinische Prüfung für Medizinprodukte
Die Anforderungen an klinische Prüfungen für Medizinprodukte beschreibt die MDR in Anhang XV. Abschnitt 2.1. formuliert, dass „die klinischen Prüfungen […] eine angemessene Zahl von Beobachtungen umfassen [müssen], damit wissenschaftlich gültige Schlussfolgerungen gezogen werden können“.
Im Kapitel II des Anhangs XV geht die MDR in Abschnitt 3 auf die geforderten Inhalte eines klinischen Prüfplans ein. Dabei adressiert Unterabschnitt 3.6. das Design der klinischen Prüfung und den „Nachweis ihrer wissenschaftlichen Belastbarkeit und Aussagekraft“. Hier werden die zu berücksichtigenden Parameter und Einflussgrößen benannt. Abschnitt 3.7. konkretisiert, dass Hersteller statistische Erwägungen begründen und eine Power-Analyse für die Stichprobengröße durchführen müssen, wenn dies für die zugrunde liegende klinische Prüfung anwendbar ist.
c) Studienplanung gemäß Stand der Technik
ISO 20916:2019 für IVD
Konkretere Vorgaben zur guten Studienpraxis bei klinischen Leistungsstudien für IVD liefert die Norm ISO 20916:2019 „In vitro diagnostic medical devices – Clinical performance studies using specimens from human subjects – Good study practice“. Die IVDR referenziert den 2019 publizierten Standard in Erwägungsgrund 66. Die Norm liefert eine Art Prozessbeschreibung für die Planung und Durchführung einer klinischen Leistungsstudie. In Abschnitt 5.3 wird hervorgehoben, dass das Design einer Leistungsstudie von der Fallzahlberechnung und der geplanten statistischen Auswertung abhängt. Zusätzlich zur IVDR gibt die Norm den Inhalt des Leistungsstudienplans vor. Diese Vorgaben überlappen teilweise mit den Anforderungen der IVDR, enthalten darüber hinaus aber Ergänzungen und Konkretisierungen. So benennt die Norm Parameter und Einflussgrößen, die bei der Fallzahlberechnung eine Rolle spielen. Auf diese Faktoren gehen wir im nächsten Kapitel näher ein.
STARD 2015
Die „Reporting Guidelines“ gemäß STARD 2015 liefern IVD-Herstellern darüber hinaus wichtige Hinweise, die sie beim Studiendesign einer diagnostischen Leistungsstudie beachten sollten. Die Leitlinie beschreibt zwar, wie Ergebnisse von diagnostischen Studien zu berichten sind, sie ist aber auch ein gutes Tool zur Studienplanung.
CLSI-Guidelines für IVD
Wertvolle Hinweise und konkrete Anleitungen zur Planung der Experimente zur Analyseleistung und zur klinischen Leistung finden IVD-Hersteller zudem in den Guidelines des Clinical and Laboratory Standards Institute (CLSI). Die CLSI-Guidelines stellen den Stand der Technik für die Leistungsbewertung von IVD dar. Bereits die EU-Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD) referenziert die CLSI-Guidelines über die harmonisierte Norm EN ISO 18113-1:2011.
ISO 14155:2020 für Medizinprodukte
Die Norm ISO 14155:2020 „Clinical investigation of medical devices for human subjects – Good clinical practice“ beschreibt die gute klinische Praxis bei klinischen Prüfungen von Medizinprodukten am Menschen. Der Anhang A der Norm spezifiziert die Informationen, die in einem klinischen Prüfplan enthalten sein müssen. Genauere Hinweise zum statistischen Design, einschließlich der Fallzahlberechnung, gibt der Anhang A.7 der ISO 14155:2020. Die Norm ist in der MDR über den Erwägungsgrund 64 referenziert. Allerdings bezieht sich die MDR auf die Version ISO 14155:2011. Auch der Entwurf des Standardization Requests nennt die Version von 2011. In der älteren Version ist der Abschnitt über die statistischen Überlegungen jedoch deutlich kürzer gefasst. Wir empfehlen daher die Verwendung der aktuellen Version ISO 14155:2020.
d) Anforderungen aus Sicht der Benannten Stellen und Behörden – Genehmigung einer klinischen Prüfung bzw. einer Leistungsstudie
Für IVD ist bei bestimmten Leistungsstudien gemäß Artikel 58 der IVDR zunächst ein Antrag auf Genehmigung der Leistungsstudie erforderlich. Die IVDR beschreibt in Artikel 67, welche Kriterien bei der Prüfung dieses Antrags beachtet werden müssen. Dazu gehören unter anderem eine Bewertung der Zuverlässigkeit und Robustheit der durch die Leistungsstudie gewonnenen Daten. Die Prüfer sollen u. a. Folgendes bewerten:
- Statistischer Ansatz
- Studiendesign
- Methodische Aspekte
- Probenumfang
- Gewählte Komparatoren (Vergleichsprodukt/e)
- Wahl der Endpunkte
Für klinische Prüfungen von Medizinprodukten muss ein Genehmigungsantrag gestellt werden. Bei Produkten mit geringem Sicherheitsrisiko können sich Hersteller von der Genehmigungspflicht befreien lassen (s. Verordnung über klinische Prüfungen von Medizinprodukten (MPKPV)). Zur Bewertung eines Antrags auf Genehmigung einer klinischen Prüfung sollen die Prüfer gemäß Artikel 71 der MDR u. a. die Zuverlässigkeit und Belastbarkeit der im Rahmen der klinischen Prüfung gewonnenen Daten bewerten. Die in der MDR genannten Beurteilungskriterien entsprechen den bereits oben aufgelisteten Aspekten.
Fazit: Die zuständige Behörde kann eine klinische Studie nicht genehmigen, wenn eine systematische Studien- und Fallzahlplanung fehlt.
3. Die Fallzahlplanung
Während der Planung einer Leistungsstudie für IVD oder einer klinischen Prüfung für Medizinprodukte stellen sich Hersteller häufig die Frage, wie viele Patienten, Proben bzw. Datensets für die Studie erforderlich sind. Doch statt einer Antwort erhalten sie von einem Statistiker bzw. einer Statistikerin in der Regel zunächst einmal Gegenfragen.
Denn damit Statistiker:innen eine Fallzahlermittlung durchführen können, müssen sie bereits das Ergebnis der Studie kennen. Hingegen möchte der Hersteller das Ergebnis erst planen. Man spricht von einem Fallzahlparadoxon.
In diesem Abschnitt lernen Sie sechs Fragen kennen, die Sie für die Fallzahlberechnung in klinischen Studien beantworten können müssen. Lassen Sie sich bei der Beantwortung dieser Fragen von erfahrenen Statistiker:innen unterstützen, um schnell und ohne Reibungsverluste eine valide Fallzahlplanung zu erhalten.
Bei einem komplexen Studiendesign kommen weitere technische Fragestellungen hinzu, die Ihnen dieser Artikel ebenfalls aufzeigt.
a) Sechs Fragen zur Fallzahlberechnung in klinischen Studien
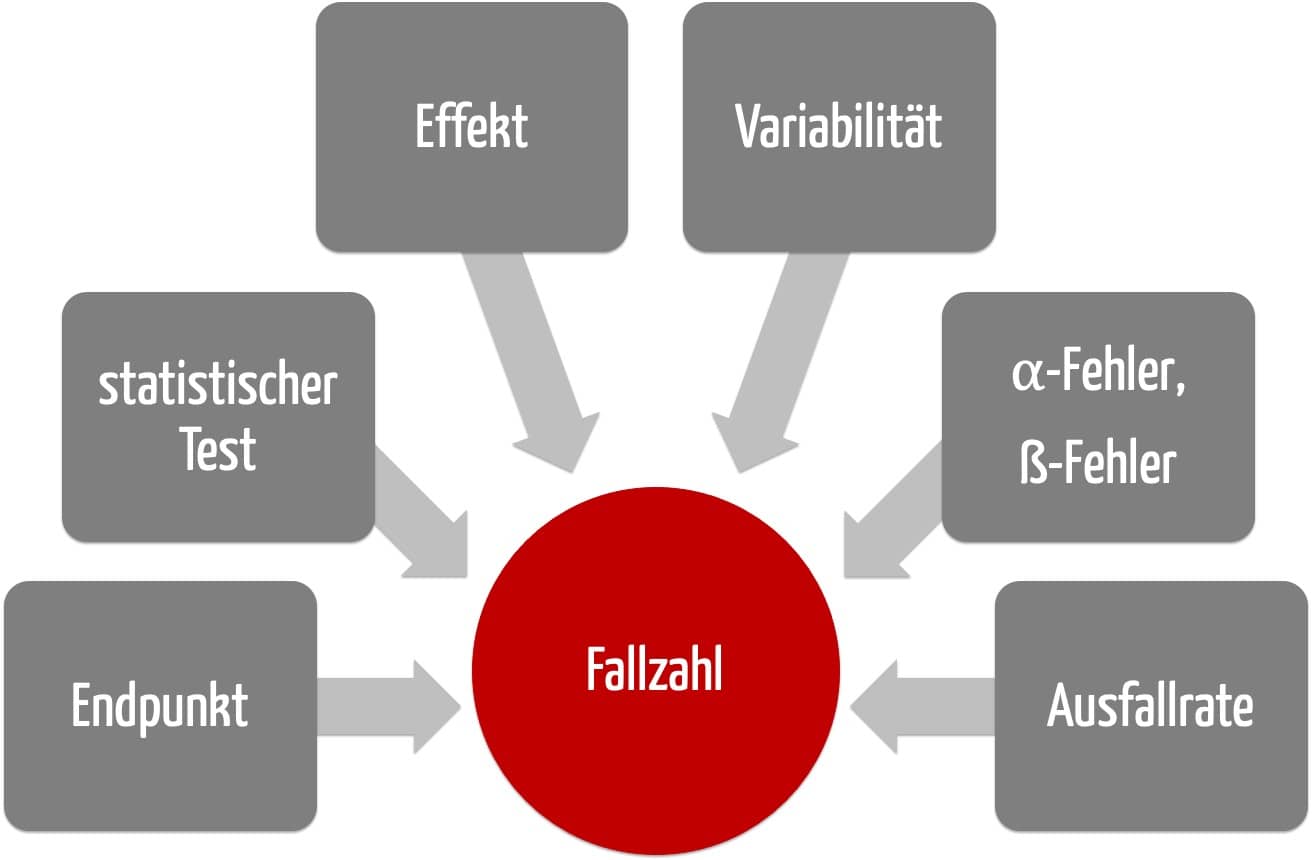
1. Frage: Mit welchem Endpunkt soll das Studienziel ermittelt werden?
Der Endpunkt ist ein statistisches Maß, an der das Erreichen des Studienziels gemessen wird. Je nach zu bewertendem Produkt und Art der Studie können die gewählten Endpunkte unterschiedlich sein.
Bei der analytischen wie auch der klinischen Leistungsbewertung von IVD sind die Endpunkte zu einem gewissen Grad vorgegeben durch die grundlegenden Sicherheits- und Leistungsanforderungen der IVDR in Anhang I, Abschnitt 9.1. Zum Nachweis der klinischen Leistung eines IVDs ist der Endpunkt in der Regel ein Anteil. Dies kann der Anteil an richtig-positiven Testergebnissen sein, also die diagnostische Sensitivität. Der Anteil richtig-negativer Testergebnisse entspricht der diagnostischen Spezifität. Der Endpunkt eines Experiments zum Nachweis der Wiederholbarkeit kann z. B. der Variationskoeffizient für wiederholt durchgeführte Tests unter denselben Bedingungen sein.
Bei Medizinprodukten sind Endpunkte zu wählen, die die Wirksamkeit bzw. den Nutzen einerseits und die Sicherheit des Produkts andererseits beschreiben. Der Endpunkt einer Studie zum Nachweis der Sicherheit eines Medizinprodukts kann z. B. der Anteil von Patienten mit einer Komplikation infolge der Produktanwendung sein.
Die Bewertung eines Medizinprodukts zur Wundversorgung kann z. B. folgende Endpunkte haben:
- Eine quantitative Größe, z. B. die Größe einer Wundfläche zu einem gewissen Zeitpunkt oder die Dauer bis zum Abheilen
- Anteil an Patienten mit einer Verringerung der Wundfläche auf mindestens 50 % zu einem definierten Zeitpunkt
- Ein patientenberichtetes Ergebnis (patient-reported outcome, PRO) zum subjektiv empfundenen Gesundheitszustand, z. B. basierend auf einem Score, der Schmerz, kosmetisches Resultat und Einschränkung der Lebensqualität beurteilt
- Ein Surrogat-Parameter wie das Auftreten bestimmter Zellen oder Biomarker in der Wunde
Wie das Beispiel zeigt, sind die Überlegungen zur Wahl des Endpunkts bei klinischen Prüfungen für Medizinprodukte häufig sehr umfassend.
2. Frage: Welcher statistische Test soll zum Nachweis verwendet werden?
Ein statistischer Test soll zeigen, dass die erhobenen Studiendaten die nachzuweisende Aussage (experimentelle Hypothese) belegen, z. B.: Test A hat eine höhere diagnostische Sensitivität als Test B.
Der statistische Test ergibt sich einerseits aus den zuvor gewählten Endpunkten, andererseits aus dem Design des Experiments. Das können statistische Tests zum Nachweis eines Mittelwertunterschieds, Tests zum Vergleich zweier Anteile, Tests zum Nachweis der Nichtunterlegenheit u. v. a. sein. Es können aber auch einfach Werte gemessen werden – in der Statistik heißt das Schätzen. Diese Werte müssen Hersteller zusammen mit der Unsicherheit präsentieren. Hierfür nimmt man meist das Konfidenzintervall.
Für Antigen-Tests zum Nachweis des Coronavirus SARS-CoV-2 gibt eine aktuelle Richtlinie der WHO, die neben den Grenzen (s. u.) z. B. auch das statistische Verfahren für den Nachweis vorgibt: Demgemäß soll die untere Grenze des Konfidenzintervalls idealerweise gleich oder größer als der Zielwert sein.
Sollten Sie bei der richtigen Anwendung eines geeigneten statistischen Tests oder bei der Beantwortung einer anderen hier aufgeführten Frage Unterstützung benötigen, können Sie sich über das Kontaktformular melden.
3. Frage: Welcher Effekt wird erwartet?
Die Angabe des erwarteten Effekts und dessen Variabilität (vgl. nächster Punkt) ist sicherlich die schwierigste. Hier wird noch einmal das oben genannte Paradoxon deutlich:
Während der Planung einer Studie soll bereits das Ergebnis benannt werden.
Dazu müssen Hersteller bei der Studienplanung Annahmen treffen, welchen quantitativen Effekt sie als Ergebnis der Experimente bzw. der Studie erwarten. Um je nach Produkt z. B. die erreichbare diagnostische Güte, den erwarteten Mittelwertunterschied oder die Komplikationsrate zu bestimmen, können Sie sich in der Praxis verschiedener Methoden bedienen:
- Recherchieren Sie in der Literatur und nutzen Sie Ergebnisse aus vergleichbaren Studien (systematische Literaturrecherche).
- Erheben Sie die Anforderungen des Marktes bzw. des Wettbewerbs (Stand der Technik).
- Bestimmen Sie die minimal interessierenden oder minimal relevanten Unterschiede im medizinischen Kontext (Stand der Technik). Bspw. ist für Fragebögen zur Erfassung patientenberichteter Endpunkte (patient-reported outcomes, PRO) die minimal interessierende Differenz eine vom Fragebogenentwickler anzugebende und zu validierende Eigenschaft.
- Nutzen Sie, soweit verfügbar, Vorgaben durch Richtlinien: Für Antigen-Tests zum Erregernachweis von SARS-CoV-2 zur Anwendung bei Verdacht auf COVID-19 bewertet die WHO-Richtlinie Werte der diagnostischen Sensitivität bzw. Spezifität von ≥ 80 % bzw. ≥ 97 % als akzeptabel. Die erwünschten Zielwerte liegen bei ≥ 90 % bzw. ≥ 99 %.
4. Frage: Wie groß ist die Variabilität des erwarteten Effekts?
Zur Angabe der erwarteten Variabilität (Standardabweichung) der Daten innerhalb der untersuchten Population können Hersteller Informationen aus der Literatur zu ähnlichen Untersuchungen sowie statistische Abschätzungen nutzen.
Häufig gewinnen Hersteller bei internen Voruntersuchungen Daten, aus denen sie Rückschlüsse auf die Standardabweichung ableiten. Als eine erste Näherung können Sie die Standardabweichung z. B. aus dem zugrunde liegenden Messbereich als ein Viertel oder Sechstel des Bereichs herleiten.
Bei Proportionen ist die Variabilität bereits durch die Proportion selbst gegeben. Für diese Endpunkte (z. B. Komplikationsraten, Maße der diagnostischen Güte) ist die Information zur Variabilität daher bereits implizit vorhanden.
5. Frage: Wie groß sind der alpha-Fehler und der beta-Fehler?
Die sogenannten alpha- und beta-Fehler werden auch als Fehler 1. und 2. Art bezeichnet. Sie geben die Wahrscheinlichkeit für ein falsch-positives bzw. ein falsch-negatives Studienergebnis an. Diese Fehler können Sie auch als Risiken interpretieren.
- Der alpha-Fehler steht für das Risiko der Allgemeinheit, dass in der Studie ein Effekt gezeigt wird, der in Wahrheit gar nicht existiert. Das heißt, dass die Studienergebnisse für das Produkt eine bessere Wirkung bzw. Leistung aufzeigen, als es tatsächlich der Fall ist.
- Der beta‑Fehler hingegen beschreibt das Risiko des Herstellers, dass die Studie einen vorhandenen Effekt nicht aufdeckt. Die Ergebnisse der Studie stellen das Produkt schlechter dar, als es eigentlich ist.
Für diese beiden Fehler gibt es „übliche“ Werte: Der beta-Fehler als „Hersteller-Risiko“ liegt typischerweise zwischen 10 % und 20 %. Den alpha-Fehler, also das Risiko für die Allgemeinheit, sollten Sie deutlich geringer wählen, z. B. 5 %. So gibt die Norm ISO 14155:2020 in Annex A.7 für klinische Prüfungen an, dass Werte des alpha-Fehlers von 5 % bei 2-seitigem Testen keiner weiteren Begründung bedürfen.
6. Frage: Welche Ausfallrate erwarten Sie während der Studie?
Die sechste Frage, mit der Sie sich während der Fallzahlplanung für klinische Studien befassen sollten, adressiert die erwartete Ausfallrate. Sie müssen erwägen, ob während der Studie Verluste an Probanden bzw. Proben oder Testergebnissen zu erwarten sind.
Diese sogenannten Dropouts werden quantifiziert und fließen in die Berechnung der Fallzahl ein. Auf diese Weise wird sichergestellt, dass die berechnete Stichprobengröße zuverlässige und robuste Studienergebnisse liefert.
b) Zusätzliche technische Fragen bei einem komplexen Studiendesign
Je nach zugrunde liegendem Studiendesign und Ziel der Studie müssen Sie weitere Aspekte bei der Fallzahlplanung berücksichtigen.
Dazu gehört u. a. das Allokationsverhältnis. Es beschreibt das Verhältnis, mit dem Patienten bzw. Proben den Untersuchungsgruppen zugeordnet werden. Optimal ist in der Regel ein Verhältnis von 1:1, aber es gibt auch Gründe für eine andere Verteilung. Die Wahl der Verteilung ist abhängig von der geplanten Studie zu evaluieren.
Werden mehrere Endpunkte gemeinsam untersucht oder sollen mehr als zwei Gruppen verglichen werden, hat das damit verbundene multiple Testen eine fallzahlerhöhende Konsequenz. „Multiples Testen“ bedeutet die gleichzeitige Betrachtung von mehreren Endpunkten, wobei mindestens ein Endpunkt nachgewiesen werden muss. In einem solchen Fall muss der Wert für den alpha-Fehler (s. Frage 5 in Kapitel 3) reduziert werden, um noch akzeptabel zu sein. Das erreicht man am einfachsten über eine Division des alpha-Fehlers durch die Zahl der Vergleiche (Bonferroni-Korrektur).
Das multiple Testen ist auch relevant, wenn Sie während der Studie eine Zwischenanalyse wünschen. Da sich das multiple Testen fallzahlerhöhend auswirken kann, sollten Sie die Erforderlichkeit einer Interimsanalyse und die daraus abzuleitenden Konsequenzen zuvor genau prüfen.
4. Beispiele für die Fallzahlberechnung
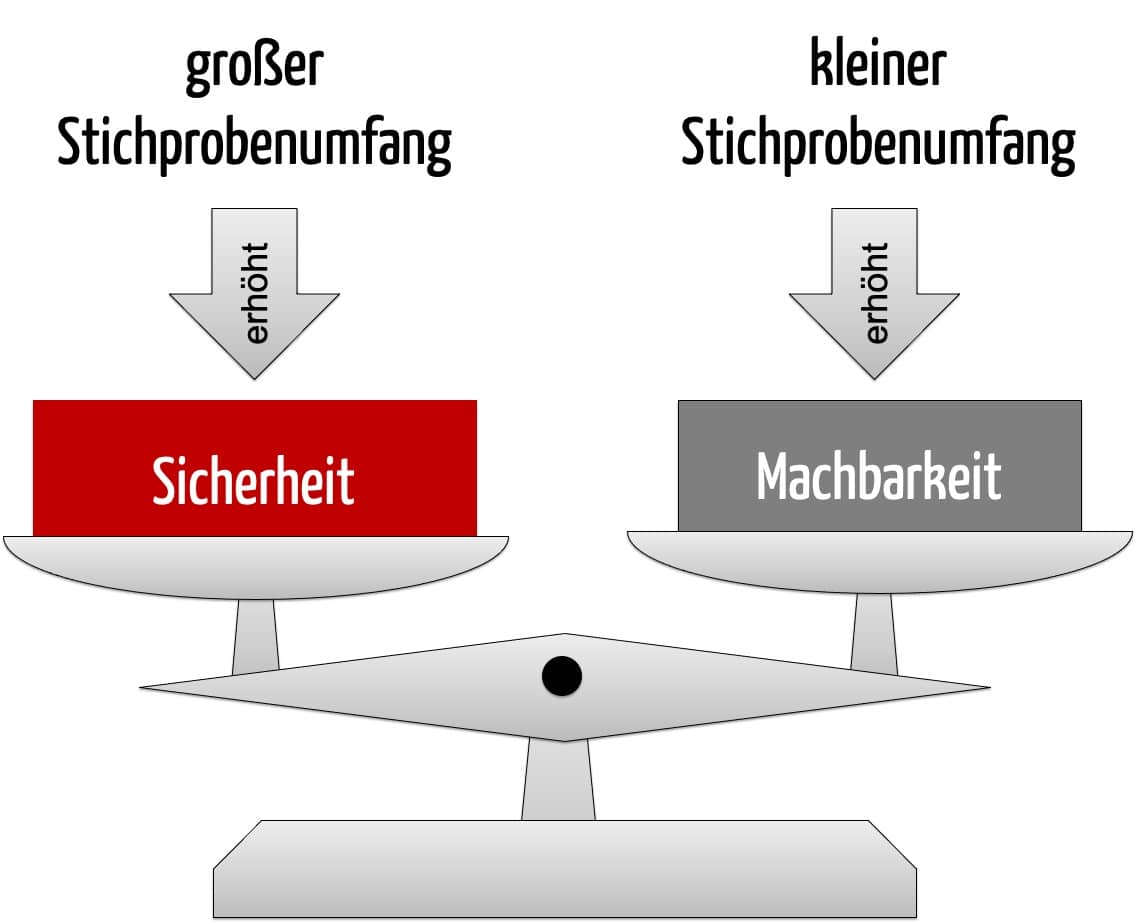
Haben Sie die zuvor aufgeführten Überlegungen zur Fallzahlplanung für die beabsichtigte Studie adressiert, folgt die Berechnung der Fallzahl. Diese wird in der Regel in Sekundenschnelle durch den Computer ausgeführt. Die Berechnung liefert jedoch meist nicht eine einzige Fallzahl, sondern verschiedene Szenarien. Diese Szenarien tabellieren Fallzahlen in Abhängigkeit von variierenden Faktoren. Gemeinsam mit dem Statistiker bzw. der Statistikerin können Sie die verschiedenen Szenarien bewerten, indem Sie zwischen der resultierenden Unsicherheit eines Szenarios einerseits und der Machbarkeit einer solchen Studie andererseits abwägen.
a) Beispiel 1: Fallzahlplanung für die klinische Leistungsstudie eines IVDs
Das nachfolgende Beispiel veranschaulicht die iterative Vorgehensweise bei der Fallzahlplanung einer klinischen Leistungsstudie für ein IVD:
Das zu bewertende IVD sei ein Test zur Diagnose einer Krankheit mit geringer Prävalenz, z. B. ein Screening-Test für Krebs. Die klinische Studie soll einen möglichst unverzerrten Nachweis führen, dass der Test für seinen Einsatzzweck zum Screenen der Gesamtpopulation ab einem bestimmten Alter geeignet ist. Dabei ist darauf zu achten, dass die Studienpopulation das gleiche klinische Spektrum aufweist wie die Population, in welcher der Test angewendet werden soll. Es darf kein sogenannter Spektrum-Bias auftreten (z. B. nur schwere Fälle). Als Einschlusskriterium gilt die beabsichtigte Anwendung des Tests, jedoch nicht der bekannte Erkrankungsstatus des Patienten.
Das Ziel der Studie ist, nachzuweisen, dass die diagnostische Sensitivität des Tests über einer gewissen, vorgegebenen Grenze von z. B. 75 % liegt. Für die diagnostische Spezifität werden 90 % gefordert. Diese Mindestanforderungen leiten sich z. B. aus dem medizinischen Stand der Technik für den hier beispielhaft dargestellten Screening-Test ab. Der Hersteller des IVDs erwartet zudem, dass sein IVD eine diagnostische Güte von 85 % Sensitivität und 95 % Spezifität aufweist.
Aufgrund der geringen Prävalenz bei einem Screening-Test (meist ≤ 10 %) bestimmt das Akzeptanzkriterium der diagnostischen Sensitivität (hier: 85 %) in der Regel die erforderliche Fallzahl für die Studie. Nach Beantwortung der oben aufgeführten sechs Fragen zur Fallzahlberechnung erheben Hersteller (ggf. zusammen mit einem/einer Statistiker:in) zunächst die Fallzahl an Erkrankten (s. Tabelle 1, Spalten 1 bis 3). Denn sie bilden die Grundlage für die Ermittlung der diagnostischen Sensitivität. Die Fallzahl für die Nichterkrankten ergibt sich entsprechend aus der Zahl der Erkrankten und der Prävalenz (s. Tabelle 1, Spalten 4 bis 6).
Welches Ergebnis erhält man für die Fallzahlberechnung für die klinische Leistungsstudie des IVD-Tests?
Zunächst erhält der IVD-Hersteller von dem Statistiker bzw. der Statistikerin z. B. eine Tabelle, die unterschiedliche Szenarien auflistet. In Abhängigkeit verschiedener Werte für die tatsächliche diagnostische Sensitivität (z. B. 80 %, 85 %, 90 %) zeigt die Tabelle die resultierenden Fallzahlen für die Erkrankten auf (hellgrau schattiert). Für die Nichterkrankten lässt sich eine gleichartige Berechnung angeben. In diesem Beispiel haben wir 92,5 %, 95 % und 97,5 % für die tatsächliche diagnostische Spezifität angenommen (Tabelle, rechts, weißer Hintergrund).
Wie komplex die Fallzahlberechnung und die unterschiedlichen Szenarien werden können, zeigt das Beispiel für ein IVD bei geringer Prävalenz. In einem solchen Fall wird die Anzahl der zu rekrutierenden Nichterkrankten fallzahlbestimmend (s. Tabelle 1, Spalte 4 bis 6). Daher geben Statistiker:innen stets Szenarien an, die sich aus unterschiedlichen Annahmen für die Prävalenz ergeben.
Das Beispiel zeigt, dass die Fallzahlen je nach zugrunde liegenden Annahmen erheblich variieren. Aus diesen Szenarien wählt der Hersteller letztlich eines aus, das die Fallzahl für die klinische Leistungsstudie des IVDs vorgibt. Bei dieser Auswahl wägt er ab zwischen der Sicherheit einer erfolgreichen Studie mit verlässlichen Daten und der Machbarkeit einer zielführenden Umsetzung der Studie. Seine Auswahl unterlegt der Hersteller mit quantifizierbaren Kriterien (z. B. Annahmen, Prävalenz) und begründet sie im Kontext der Zweckbestimmung des IVDs.
Ist sich der Hersteller sicher, dass sein IVD eine deutlich höhere diagnostische Leistung aufweist als z. B. gemäß Stand der Technik gefordert, wird die Studie mit geringeren Fallzahlen auskommen. Ist der Hersteller wenig risikobereit, wird er einen geringeren beta-Fehler sowie konservativere Annahmen für die tatsächliche Leistung und damit höhere Fallzahlen wählen.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Beta-Fehler | Diagnostische Sensitivität | Anzahl der Erkrankten | Anzahl der Nichterkrankten bei 5 % Prävalenz | Anzahl der Nichterkrankten bei 10 % Prävalenz | Anzahl der Nichterkrankten bei 15 % Prävalenz | Beta-Fehler | Diagnostische Spezifität | Anzahl der Nicht- erkrankten |
| 20 % | 80 % | 563 | 10.697 | 5.067 | 3.191 | 20 % | 92,5 % | 1.049 |
| 10 % | 80 % | 742 | 14.098 | 6.678 | 4.205 | 10 % | 92,5 % | 1.371 |
| 20 % | 85 % | 133 | 2.527 | 1.197 | 754 | 20 % | 95 % | 239 |
| 10 % | 85 % | 171 | 3.249 | 1.539 | 969 | 10 % | 95 % | 301 |
| 20 % | 90 % | 54 | 1.026 | 486 | 306 | 20 % | 97,5 % | 93 |
| 10 % | 90 % | 68 | 1.292 | 612 | 386 | 10 % | 97,5 % | 111 |
Spalten 1-3: Fallzahlen für Erkrankte bei verschiedenen Annahmen für die tatsächliche diagnostische Sensitivität des Tests (Binomialtest, zweiseitig, alpha = 5 %, beta = 20 % bzw. 10 %); Spalten 4-6: Fallzahlen für Nichterkrankte, wie sie sich korrekt aus der Zahl der Erkrankten (Np) und der Prävalenz (prev) ergeben (Np x (1-prev)/prev)); Spalten 7-9: Fallzahl für Nichterkrankte basierend auf der diagnostischen Spezifität.
Achtung: Diese Zahlen sind nicht maßgeblich und nur zur Information. Für geringe Prävalenzen sind die Fallzahlen für Nichterkrankte, die anhand der Prävalenz der Erkrankten berechnet wurden (Spalten 4-6), höher und daher maßgeblich.
Die Fallzahlplanung ist ein iterativer Prozess, währenddessen der Hersteller prüfen kann, welchen Effekt jeweils variierte Parameter auf die Fallzahl haben. Gültige Akzeptanzkriterien und die Festlegung eines Studienziels sind Voraussetzungen hierfür. Die Auswahl des Stichprobenumfanges beruht auf quantifizierbaren Kriterien für die Erfolgswahrscheinlichkeit und Machbarkeit der Studie.
b) Beispiel 2: Fallzahlberechnung für eine klinische Prüfung zum Nachweis der Sicherheit des Medizinprodukts
Bei einer klinischen Prüfung von Medizinprodukten zum Nachweis der Sicherheit wird als Endpunkt z. B. die Komplikationsrate untersucht. Diese Raten weisen meist sehr geringe Werte zwischen 0 und 5 % auf. Die zugehörigen Konfidenzintervalle (CI) sind entsprechend schmal. Um einigermaßen sichere Vorhersagen über den Anteil an Patienten mit Komplikation treffen zu können, resultieren oft Fallzahlen im drei- bis vierstelligen Bereich.
Folgendes Beispiel soll die resultierenden Fallzahlen in Abhängigkeit vom Endpunkt – der akzeptablen Komplikationsrate – veranschaulichen: Die erwartete Komplikationsrate beträgt 2 % und der Hersteller möchte für sein Medizinprodukt eine Rate von < 5 % nachweisen (entspricht der oberen Grenze des Konfidenzintervalls). Diese Anforderung leitet der Hersteller z. B. aus dem medizinischen Stand der Technik oder gesundheitsökonomischen Betrachtungen (Kosten-Nutzen-Betrachtungen) ab. Im vorliegenden Beispiel werden ca. 330 Patienten benötigt. Würde die Komplikationsrate 3 % betragen, so müssten bei gleicher oberer Grenze des CI von 5 % 815 Patienten in die Studie eingeschlossen werden [1], [2]. Berücksichtigt man bei der Fallzahlberechnung noch, dass 5 % der Patienten in der Nachverfolgung nicht zugänglich sind (5 % Dropout-Rate), so erhöht sich die Fallzahl entsprechend um den Faktor 1/(1-0,05) = 1,053.
[1] Fallzahlberechnung: Binomialtest, 2-seitig, alpha-level = 5 %, beta-level = 20 %, Software PASS 20.0
[2] Eine Idee zur Fallzahlsenkung ist die prospektive Verwendung 1-seitiger Tests für derartige Fragestellungen. Für das Beispiel würden sich 253 (2 %) bzw. 631 Fälle (3 %) ergeben. In der klinischen Forschung wird für 1-seitige Tests jedoch alpha = 2,5 % festgelegt, um einem Missbrauch 1-seitiger Tests vorzubeugen. Damit verschwindet der Fallzahlvorteil. Inhaltlich spricht jedoch nichts gegen 1-seitige Tests bei Fragestellungen, bei denen die ideale Rate 0 % beträgt. Dies ist gegebenenfalls mit den Behörden vorab zu diskutieren.
5. Fallzahlplanung: Fazit und Zusammenfassung
Die regulatorischen Anforderungen von IVDR und MDR geben keine konkreten Vorgaben zu erforderlichen Fallzahlen für eine klinische Prüfung für Medizinprodukte bzw. für eine Leistungsstudie für IVD. Vielmehr darf und muss der Hersteller den Stichprobenumfang einer Studie produktspezifisch je nach Studienziel festlegen und begründen.
Die frühzeitige Einbindung eines Statistikers bzw. einer Statistikerin in die Studienplanung ist für die Abschätzung der Fallzahl und die geplante statistische Auswertung hilfreich. So stellen Hersteller die wissenschaftliche Robustheit und Validität der erzeugten Daten sicher und vermeiden unnötig hohen Aufwand für klinische Prüfungen oder Leistungsstudien.
Die Fallzahlberechnung ist eine Art „Verhandlungssache“ im Rahmen gültiger Akzeptanzlimits. Sie liefert meist nicht eine einzige anzuwendende Fallzahl, sondern verschiedene Szenarien. Diese Szenarien bewerten Statistiker:innen gemeinsam mit dem Hersteller hinsichtlich der Unsicherheit und Machbarkeit einer Studie.
Erfahren Sie, wie Sie mit einer korrekten Fallzahlberechnung unnötige Aufwände bei klinischen Studien ebenso vermeiden können wir Ärger bei der Zulassung.
Diese und weitere Podcast-Episoden finden Sie auch hier.
Das Team des Johner Instituts unterstützt Sie gerne bei der
- Erstellung der klinischen Bewertung für Ihr Medizinprodukt,
- Planung der klinischen Prüfung Ihres Medizinprodukts,
- Strategie zur Leistungsbewertung Ihres IVDs,
- Planung der klinischen Leistungsstudie für Ihr IVD und
- Fallzahlberechnung für Ihre klinische Prüfung bzw. klinische Leistungsstudie.
Kontaktieren Sie uns über unser Webformular.


