
Der Brexit wurde mit dem 01.01.2021 vollzogen. Was für manche ein Grund zum Feiern war, bedeutet für viele eine zusätzliche Last – auch für die Hersteller von Medizinprodukten.
Für Hersteller ist es wichtig zu verstehen, welche regulatorischen Anforderungen sie erfüllen müssen und von welchen Übergangsfristen sie profitieren, wenn sie ihre Produkte weiterhin in Großbritannien verkaufen wollen.
1. Regulatorische Anforderungen nach dem Brexit
a) Das britische Recht gilt und damit die EU-Richtlinien!
Mit dem 31.12.2020 endete die Übergangsphase, in der das EU-Recht in Großbritannien noch zur Anwendung kam. Seit dem 01.01.2021 gilt das Recht des United Kingdoms, das aber weiterhin auf altem EU-Recht basiert:
Für Medizinprodukte gelten die britischen Medical Devices Regulations 2002. Diese wiederum referenzieren weiterhin die (alten) EU-Richtlinien, also:
- Medizinprodukte-Richtlinie MDD (93/42/EEC)
- Richtlinie für aktive implantierbare Medizinprodukte AIMDD (90/385/EEC)
- In-vitro Diagnostik-Richtlinie IVDD (98/79/EEC)
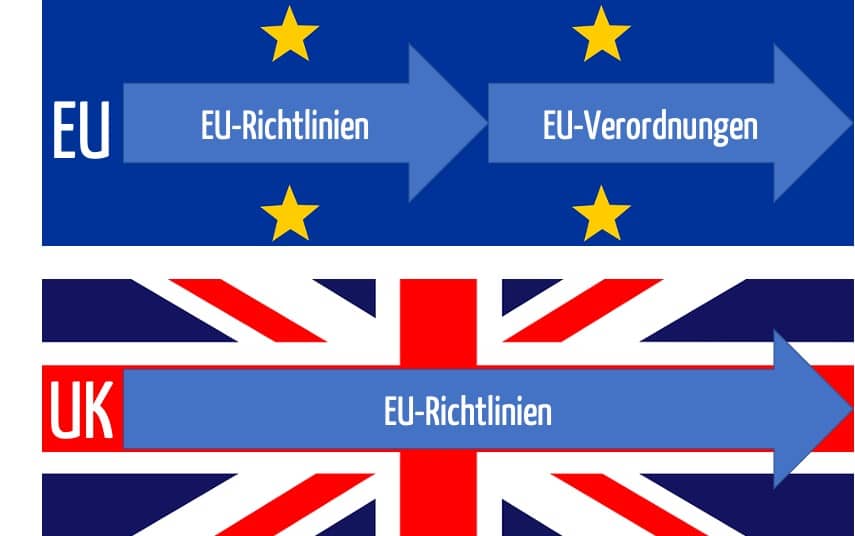
Hersteller, die gerade auf die MDR umgestellt haben, müssten also eine Rolle rückwärts machen. Die gute Nachricht besteht aber darin, dass die Anforderungen der MDR eher die Übermenge der Anforderungen darstellen, die die EU-Richtlinien formulieren.
b) Konformitätsbewertungsverfahren und CE-Kennzeichnung bleiben gültig – vorläufig
Mit den EU-Richtlinien bleiben im Vereinigten Königreich auch die Anforderungen an die Konformitätsbewertungsverfahren und die CE-Kennzeichnung bestehen.
Ursprünglich hat Großbritannien die Inverkehrbringung mit CE-Kennzeichnung nur bis zum 30.06.2023 akzeptiert. Inzwischen wurden diese Übergangsfristen aber verlängert (siehe Kapitel 2). Anerkannt werden dabei Konformitätserklärungen sowohl gemäß den alten EU-Richtlinien als auch der MDR und IVDR. Das heißt, dass Hersteller in dieser Zeit kein zusätzliches Konformitätsbewertungsverfahren durchlaufen müssen.
Nach den Übergangsfristen müssen Hersteller ein UK-spezifisches Konformitätsbewertungsverfahren durchlaufen. Dieses mündet aber nicht in eine CE-Kennzeichnung, sondern die UKCA-Kennzeichnung, d. h. Kennzeichnung mit dem UK Conformity Assessed Mark.
c) Den Benannten Stellen werden die UK Approved Bodies folgen
Für die UKCA -Kennzeichnung müssen die Hersteller einen UK Approved Body einbeziehen. Dieser entspricht den Benannten Stellen schon deshalb, weil das UK-Konformitätsbewertungsverfahren weiterhin auf den EU-Richtlinien beruht. Zumindest solange bis Großbritannien andere Vorgaben erlässt. Der Grundstein für eine grundlegende Überarbeitung des aktuell geltenden Rechts hat man durch die im Februar 2021 verabschiedeten Medicines and Medical Devices Bill gelegt.
Großbritannien ist allerdings nicht frei, diese Vorgaben beliebig zu ändern oder gar zu erhöhen. Der Brexit-Deal setzt dem Grenzen, weil man den weitgehend freien Warenverkehr nur dann erlaubt, wenn keine Seite die Regulierung nutzt, um den jeweiligen Markt abzuschotten.
Weil aktuell die UK-Vorgaben den EU-Richtlinien entsprechen, müssen Hersteller für Klasse-I-Produkte keinen UK Approved Body am Konformitätsbewertungsverfahren beteiligen.
d) Neu ist die Pflicht zur Registrierung
Allerdings müssen Hersteller, die Produkte nach Großbritannien importieren, diese bei der britischen Gesundheitsbehörde registrieren (der Medicines and Healthcare products Regulatory Agency, MHRA).
Die Registrierung darf nicht der Hersteller durchführen. Vielmehr ist dafür die UK Responsible Person verantwortlich.
Wir sind Ihr zuverlässiger Bevollmächtigter für Medizinprodukte im Vereinigten Königreich und sichern damit Ihren Marktzugang.
Weitere Informationen darüber finden Sie ebenfalls weiter unten.
Lesen Sie mehr zur Registrierung von Medizinprodukten auf den Seiten des MHRA.
Ebenfalls hilfreich ist die Seite Regulating Medical Devices in the UK.
e) Zusammenfassung: 3 Schritte zur Zulassung in Großbritannien
Hersteller aus der EU, die ihre Medizinprodukte in Großbritannien vermarkten wollen, müssen wie folgt vorgehen:
- UK-Bevollmächtigten (UK Responsible Person) finden und registrieren.
- Durch diesen Bevollmächtigten die eigenen Produkte registrieren lassen.
- Die Produkte vermarkten.
2. Übergangsfristen zur Anerkennung der CE-Kennzeichnung
CE-gekennzeichnete Produkte können aktuell weiterhin in Großbritannien vermarktet werden. Demnach bedarf es keiner zusätzlichen UKCA-Kennzeichnung. Es gelten dafür folgende Übergangsfristen:
| Anwendbare EU-Regularie | Übergangsfrist |
| Medizinprodukte mit gültiger CE-Kennzeichnung unter der MDD oder AIMDD | Maximal 30. Juni 2028 (oder bis Zertifikatsablauf) |
| In-vitro-Diagnostika mit gültiger CE-Kennzeichnung unter der IVDD | Maximal 30. Juni 2030 (oder bis Zertifikatsablauf) |
| Medizinprodukte (einschließlich Sonderanfertigungen) und In-vitro-Diagnostika mit gültiger CE-Kennzeichnung unter der MDR bzw. IVDR | Maximal 30. Juni 2030 |
Mit Ablauf der Übergangsfrist besteht die MHRA auf dem Durchlaufen der Konformitätsbewertungsverfahren nach britischem Recht mit Einbeziehung eines UK Approved Body.
Inzwischen gibt es Bestrebungen seitens der MHRA, auch über die oben genannten Übergangsfristen hinaus Produkte mit bestehenden Zulassungen in anderen Ländern anzuerkennen. Davon würden nach aktuellem Stand Hersteller mit Zulassungen in Australien, EU, Kanada oder den USA profitieren.
3. Notwendige Rollen
a) UK Responsible Person
Mit dem Brexit ist eine UK Responsible Person Pflicht geworden. Diese Person muss in Großbritannien ansässig und bei der MHRA registriert sein. Die UK Responsible Person muss vergleichbar dem Bevollmächtigten gemäß MDR und IVDR folgende Tätigkeiten durchführen:
- Produkte registrieren
- Als primärer Ansprechpartner der MHRA fungieren
- Informationen aus dem Feld an den Hersteller übermitteln inklusive Reklamationen und Berichten der Anwender, Patienten und Gesundheitseinrichtungen (insbesondere bezüglich möglicher schwerer Vorkommnisse)
- Bei der Umsetzung von Sicherheitskorrekturmaßnahmen im Feld und bei sonstigen Korrektur- und Vorbeugemaßnahmen unterstützen
- Überprüfen, ob der Hersteller ein Konformitätsbewertungsverfahren durchlaufen hat
- Das Vorhandensein der Konformitätserklärung sicherstellen und diese bereithalten
- Das Vorhandensein der technischen Dokumentation sicherstellen und diese bereithalten
- Der MHRA eine Liste der Importeure bereitstellen
Falls die UK Responsible Person den Verdacht hat, dass der Hersteller seine gesetzlichen Pflichten nicht erfüllt, muss sie ihr Mandat niederlegen und die MHRA und ggf. den UK Approved Body informieren.
Wir sind Ihr zuverlässiger Bevollmächtigter für Medizinprodukte im Vereinigten Königreich und sichern damit Ihren Marktzugang.
b) Importeure und Händler
Importeure müssen die UK Responsible Person über ihre Tätigkeit informieren. Sie können diese Rolle gleichzeitig auch übernehmen. Händler haben keine zusätzlichen Verpflichtungen zu erfüllen.
c) UK Approved Body
Nach Ablauf der Übergangsfristen müssen sämtliche Medizinproduktehersteller, die ihre Produkte in Großbritannien in Verkehr bringen möchten, einen UK Approved Body einbeziehen – zumindest, wenn ihre Produkte in die Klassen I*, IIa, IIb oder III fallen.
Derzeit gibt es gemäß der Webseite der britischen Regierung nur vier akkreditierte Firmen: SGS, BSI, DEKRA und UL.
Diese Liste wurde inzwischen erweitert, u. a. um den TÜV SÜD.
4. Weitere Anforderungen
a) Grundlegende Sicherheits- und Leistungsanforderungen
Weil die regulatorischen Anforderungen an Medizinprodukte, die in Großbritannien vermarktet werden, weiterhin in den EU-Richtlinien formuliert sind, gibt es keine anderen oder zusätzlichen grundlegenden Anforderungen. Dies wird sich mit Einführung der neuen Gesetzgebung (vermutlich 2025) ändern. Einen Ausblick erhalten Sie in diesem Dokument der MHRA.
Die MHRA hat inzwischen begonnen, ihre Gedanken zum Human Factors Engineering zu formulieren. Diese orientieren sich etwas stärker an den Vorgaben der FDA als an denen der alten IEC 62366. Hingegen dürften Hersteller, die die IEC 62366-1 befolgen, auch die Usability-Anforderungen der MHRA erfüllen.
b) UDI
Da die alten Richtlinien keine UDI verlangten, entfällt diese Pflicht vorerst in Großbritannien. Natürlich spricht nichts dagegen, wenn Hersteller Produkte in UK mit einer UDI in den Verkehr bringen.
c) Qualitätsmanagementsystem
Die Anforderungen der MHRA an das QM-System unterscheiden sich nicht von den entsprechenden Anforderungen in der EU. Auch hier ist die ISO 13485:2016 bzw. die Anforderungen z. B. des Anhangs II der MDD maßgebend.
d) Post-Market Surveillance & Vigilanz
Die Anforderungen an die Post-Market Surveillance und insbesondere an die Vigilanz sind schon immer spezifisch für die einzelnen Ländern gewesen, z. B. was das Format, die Fristen und den Empfänger von Vigilanzmeldungen betrifft.
Ähnlich wie bei Einführung der MDR werden trotz Übergangsfristen Anforderungen an die Post-Market-Surveillance (PMS) bereits vor Gültigkeitsbeginn der neuen Gesetzgebung anzuwenden sein. Dies ist zum 16. Juni 2025 mit der Ergänzung der UK Medical Devices Regulations durch den neuen Part 4A erfolgt. Die MHRA gibt auf ihrer Webseite umfangreiche Hilfestellungen zum PMS und weitere Hinweise.
5. Sonderfall Nordirland
Besonders kompliziert wird es für Produkte, die in Nordirland vermarktet werden sollen. Für diese greift nicht das britische Recht, sondern EU-Recht. Das heißt: In Nordirland gelten ab dem 26.05.2021 bzw. 26.05.2022 die MDR bzw. IVDR!
Damit bleibt nach jetzigem Stand in Nordirland auch nach den Übergangsfristen die Pflicht zur CE-Kennzeichnung bestehen. Allerdings bedarf es trotzdem einer UK Responsible Person, die u. a. für die Registrierung der Produkte in der MHRA-Datenbank verantwortlich ist.
Eine Ausnahme von der Pflicht zu einer UK-Verantwortlichen Person gilt bei Klasse-I-Produkten, bei Sonderanfertigungen und sonstigen IVDs, die bereits in der EU registriert wurden.
Eine weitere Sonderregelung betrifft die Kennzeichnung: Falls ein UK Approved Body in die Konformitätsbewertung einbezogen wird, dann bedarf es neben der CE-Kennzeichnung auch der UKNI-Kennzeichnung. Die Kombination beider Kennzeichnungen ist allerdings nur für den nordirischen Markt bestimmt und in diesem Fall verpflichtend. Innerhalb der EU darf allein die CE-Kennzeichnung genutzt werden. Eine zusätzliche Anbringung der UKNI-Kennzeichnung bei Inverkehrbringung in z. B. Deutschland ist unzulässig.
In addition to the CE marking, device manufacturers also need to apply the UKNI marking if they choose to use a UK Notified Body for mandatory third-party conformity assessment. Device manufacturers must never apply the UKNI marking on its own – it must always accompany a CE marking. To place goods on the EU market, manufacturers must use the CE marking on its own, without the UKNI marking. Goods bearing the “CE & UKNI” marking will not be accepted on the EU market.
Webseite des MHRA
Lesen Sie mehr zu den Sonderregelungen für Nordirland und zur UKNI-Kennzeichnung in Mitteilungen der britischen Regierung (PDF).
6. Anforderungen für britische Hersteller
Für die britischen Hersteller, die Produkte in der EU in den Verkehr bringen wollen, gelten die europäischen Regularien wie die MDR. Damit benötigen sie bereits ab dem 01.01.2021 eine EU-Benannte Stelle und einen EU-Bevollmächtigten. Produkte, die bereits vor dem 01.01.2021 in der EU in Verkehr gebracht wurden, dürfen weiterhin dort bereitgestellt und in Betrieb genommen werden.
Falls die britischen Hersteller ihre Produkte in Nordirland vermarkten wollen, benötigen sie einen EU-Bevollmächtigten mit Sitz in Nordirland.
7. Fazit
Durch den Brexit gibt es zumindest in der Welt der Medizinproduktehersteller keine Gewinner., sondern (fast) nur Verlierer.
Verlierer 1: Die britischen Patienten
Wenn MDR und IVDR ihr Versprechen erfüllen würden, im Vergleich zu den bisherigen Richtlinien die Sicherheit, Leistungsfähigkeit und den Nutzen von Medizinprodukten zu erhöhen, dann würden die britischen Patienten verlieren. Denn in Großbritannien müssen die Produkte nur die „alten“ Anforderungen der EU-Richtlinien erfüllen.
Verlierer 2: Das britische Gesundheitssystem
Die Inverkehrbringung wird für EU-Hersteller aufwendiger.
- Eine zusätzliche Rolle, die UK Responsible Person, muss geschaffen werden.
- Mit Ablauf der Übergangsfristen müssen die EU-Hersteller ein zweites Konformitätsbewertungsverfahren durchlaufen.
- Sie benötigen dann (abhängig von der Klasse) zusätzlich zu den Benannten Stellen auch noch einen UK Approved Body.
Dieser zusätzliche Aufwand muss bezahlt werden – letztendlich vom britischen Gesundheitssystem. Oder die Hersteller entscheiden sich, ihre Produkte nicht mehr in Großbritannien zu vermarkten. Dann fehlen diese im britischen Gesundheitssystem.
Verlierer 3: Die EU-Hersteller
Für die EU-Hersteller bedeutet dies den oben geschilderten zusätzlichen Aufwand für die Registrierung, das zusätzliche Konformitätsbewertungsverfahren und den zusätzlich notwendigen UK Approved Body.
Verlierer 4: Die Behörden (EU und UK)
Gleich ob Zollbehörden, MHRA oder europäische Behörden, die für die Marktaufsicht verantwortlich sind: Alle haben zusätzlichen bürokratischen Aufwand, der keine zusätzliche Sicherheit bringt. Im Gegenteil: Redundante oder zusätzliche und unnötige Aufgaben halten die Behörden davon ab, ihre eigentliche Pflicht zu erfüllen. Eine Pflicht, die schon jetzt offensichtlich mehr als herausfordernd ist.
Zusammenfassung
„Take Back Control“ war ein der Slogan der Brexit-Befürworter. Das Ergebnis ist, dass Großbritannien mit veralteten Regularien arbeitet. Dass diese EU-Regularien sind, erscheint wie ein Treppenwitz der Geschichte.
Gute Medizinprodukte sind gute Medizinprodukte. Daher sollte das Ziel sein, weltweit einheitliche Regularien zu schaffen. Der Brexit war leider ein Weg in die entgegengesetzte Richtung. Die MDR mit dem Konzept der Common Specifications und den bis heute fehlenden harmonisierten Normen aber auch. Schade.
Nutzen Sie die Hilfe des Johner Instituts, um die UK-Anforderungen an Medizinprodukte gesetzeskonform und mit minimalen Aufwänden zu erfüllen.
Wir unterstützen Sie bei der Registrierung Ihrer Produkte bei der MHRA und übernehmen für Sie die Rolle der UK Responsible Person.
Unsere Expert:innen begleiten Sie auch bei der Erstellung und beim Review aller notwendiger Unterlagen.
Änderungshistorie
- 2025-06-20: Information über die Einführung der neuen PMS-Anforderungen in Kapitel 4 d)
- 2025-04-07: Hinweis in Kapitel 1 und 3 ergänzt
- 2024-10-15: Ergänzung zur geplanten Anerkennung von internationalen Zulassungen
- 2023-12-04: TÜV SÜD ergänzt
- 2023-06-22: Artikel grundlegend überarbeitet mit Referenz auf die neuen Übergangsfristen und geplante Gesetzgebung



Hallo Herr Salvatore,
danke für den ausführlichen und informativen Beitrag.
Ich habe eine Frage zum Labelling als deutscher Hersteller, der seine Produkte auf dem UK Markt bereitstellen möchte. Ich habe in der Leitlinie „Regulating medical devices in the UK“ gelesen, dass auch der Name und die Adresse UKRP auf dem Label angebracht werden muss. Gilt das für nur für Produkte, die bereits das UKCA Zeichen bzw. CE und UKCA Zeichen haben? Oder bereits auch für Legacy Produkt in der jetzige Übergangszeit?
Mit freundlichen Grüßen
C.Rockstroh
Guten Tag,
die Antwort auf Ihre Fragen finden Sie im genannten Dokument:
“ UK Responsible Person details do not need to be included on labelling for CE marked devices.“
Somit ist die Angabe des UK Rep auf Legacy-Produkten nicht notwendig.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
Medizinproduktehersteller mit Sitz außerhalb der EU, die ihre Produkte innerhalb der EU in Verkehr bringen möchten, benötigen einen Bevollmächtigten mit Sitz in der Union. Das bedeutet doch, dass seit dem Brexit Medizinproduktehersteller, die mit einem Bevollmächtigten innerhalb des Vereinigten Königreichs zusammenarbeiten, ihre Produkte nicht mehr in der EU vertreiben dürfen, oder?
Vielen Dank im Voraus!
Mit freundlichen Grüßen
Sarah Dreßen
Sehr geehrte Frau Dreßen,
Ihre Annahme ist korrekt. Der Bevollmächtigte muss seinen Sitz in der EU haben. Andernfalls ist eine Inverkehrbringung in der von Ihnen dargestellten Konstellation nicht möglich.
Herzliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
wäre es regulatorisch falsch das UKCA bereits jetzt neben dem CE Zeichen aufzubringen auf dem Labeling?
Ich kann mir vorstellen, dass die Briten noch nicht soweit sind mit eigenen Verordnungen und deshalb das CE erstmal weiter akzeptieren. Somit könnte das UKCA auf keine britische Verordnung verweisen und auch auf ein „noch“ nicht existierendes Konformitätsbewertungsverfahren.
Beste Grüße
Jens Meyer
Hallo Herr Meyer,
es ist erlaubt, bereits jetzt das UKCA-Marking zusätzlich zur CE-Kennzeichnung anzubringen, wenn ein entsprechendes Konformitätsbewertungsverfahren durchlaufen wurde.
Aktuell basiert diese auf den „alten“ EU-Richtlinien.
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore! Ich habe folgendes Problem: Mein Lieferant aus England liefert Medizin-Produkte zu uns nach Deutschland. Er hat bisher nur ein MDD-Zertifikat auf dem noch kein EU-Bevollmächtigter steht. Darf er noch ohne EU-Bevollmächtigten Medizin-Produkte in die einführen?
Beste Grüße
Wolfgang Maier
Guten Tag Herr Maier,
in diesem Fall muss der Hersteller mit Sitz in England einen EU-Bevollmächtigten benennen. Ansonsten wäre der Import nicht rechtens.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
ich habe eine Frage zur Rolle des Wirtschaftsakteurs in UK.
Ein Unternehmen in Deutschland produziert in UK in einer Niederlassung ein Medizinprodukt. Das dt. Unternehmen ist CE-verantwortlich= Legal Manufacturer.
Die Niederlassung in UK ist die UK Responsible Person für das dt. Unternehmen.
Der Vertrieb (europaweit) soll von der Produktionsstätte aus erfolgen.
Welche Rolle als Wirtschaftsakteur hat die produzierende Niederlassung des deutschen Unternehmens dann in UK?
Ist sie Importeur?
Ist sie Händler? (was ich nicht vermute)
Vielen dank im Voraus für Ihre Unterstützung.
Mit freundlichen Grüßen
C.G.
Sehr geehrte Frau Graß,
bezüglich der Inverkehrbringung in UK, würde die Niederlassung in UK die Rolle Importeur einnehmen. Bezüglich der EU nicht, da der Importeur immer seinen Sitz im Zielland bzw. Rechtsbereich hat.
Herzliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
vielen Dank für den informativen Artikel.
Bedeutet das, dass Hersteller von „Sonstigen IVDs“ auch nach dem 30.06.2023 weiterhin KEINEN UK approved body brauchen und das UKCA Kennzeichen selbst anbringen können?
Vielen Dank im Voraus für die Antwort.
Christina Pfaff
Guten Morgen Frau Pfaff,
aktuell findet eine Überarbeitung der UK Medical Device Regulations statt, die am 01.07.2023 in Kraft treten soll. Es wird allerdings Übergangsfristen geben, auch für IVDs. Welche Fristen vorgesehen sind, können Sie hier nachlesen: https://www.gov.uk/government/consultations/consultation-on-the-future-regulation-of-medical-devices-in-the-united-kingdom/outcome/chapter-15-transitional-arrangements
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore,
in meiner Frage geht es um Anbringung des UKCA-Zeichen und in diesem Zusammenhang um die Angabe der UK Responsible Person (mit Namen und Adresse) für Produkte der Klasse I.
Ab wann muss die UK Resp.Pers. angegeben werden? Sobald das UKCA-Zeichen aufgebracht (auf Produktlabel/Verpackung/IFU) wurde?
Oder ist die Angabe erst ab dem 01.07.2023 verbindlich?
Vielen Dank im Voraus für Ihre Hilfe!
Mit freundlichen Grüßen
C. Graß
Guten Tag Frau Graß,
die UK Responsible Person muss angegeben werden, sobald Sie Ihr Produkt UKCA-kennzeichnen. Das ist also unabhängig vom 01.07.2023.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
vielen dank für die schnelle Antwort! Sie haben mir damit sehr geholfen.
Mit freundlichen Grüßen
C. Graß
Guten Tag Herr Salvatore,
so wie ich Ihren Artikel verstanden habe und auch einen Teil der offiziellen UK Goverment Seite, wird es möglich sein Produkte, die nur über ein MDD Zertifikat verfügen, auch noch unter UKCA zu zertifizieren.
Allerdings hebelt für mich der Absatz, von der gleichen UK Seite, das oben genannte aus und fordert MDR ab 1.7.2023:
Registrations in Great Britain
All medical devices, including IVDs, custom-made devices and systems or procedure packs must be registered with the MHRA before being placed on the Great Britain market. In Great Britain (England, Wales and Scotland), devices must conform to the UK MDR 2002, the EU MDR (until 30 June 2023), or the EU IVDR (until 30 June 2023) in order to be registered with the MHRA. In addition, devices that have been CE marked under the EU MDD, EU AIMDD or EU IVDD will continue to be accepted on the Great Britain market until 30 June 2023 if their certificates remain valid for the EU market under the transitional arrangements in the EU MDR and EU IVDR.
Wie interpretieren Sie das?
Über ein Feedback würde ich mich sehr freuen.
Viele Grüße
Marina Hillen
Guten Tag Frau Hillen,
Sie haben Recht. Gemäß den aktuellen Übergangsbestimmungen ist die CE-Kennzeichnung nur noch bis zum 30.06.2023 anerkannt. Allerdings wird an der Revision des Gesetzes von 2002 gearbeitet. Geplant ist, die neuen UK Medical Device Regulations in 2023 einzuführen. Geplant sind in diesem Zusammenhang erweiterte Übergangsbestimmungen für CE-gekennzeichnete Produkte. Nähere Informationen dazu finden Sie hier: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1085333/Government_response_to_consultation_on_the_future_regulation_of_medical_devices_in_the_United_Kingdom.pdf (ab Seite 150).
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore
Was ist bei einem Import von Sonderanfertigungen nach UK zu berücksichtigen? Gibt es schon Informationen hinsichtlich der Änderungen, welche 2023 in Kraft treten werden?
Besten Dank und freundliche Grüsse
Manon Rordorf
Guten Tag Herr Rordorf,
auch für den Import von Sonderanfertigungen benötigen Sie eine UK Responsible Person, welche die Produktregistrierung übernimmt (siehe https://www.gov.uk/government/publications/custom-made-medical-devices/custom-made-devices-in-great-britain). Aktuell werden Erklärungen zu Sonderanfertigungen nach MDD/MDR noch anerkannt.
Geplant ist eine gesetzliche Änderung ab dem 01.07.2023. Erste Hinweise finden Sie dazu hier ab Seite 140: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1085333/Government_response_to_consultation_on_the_future_regulation_of_medical_devices_in_the_United_Kingdom.pdf
Die Anforderungen an Sonderanfertiger werden demnach verschärft.
Beste Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
haben Sie Informationen darüber, dass die verpflichtende Verwendung des UKCA-Symbols für Medizinprodukte vom 01.07.2023 auf den 01.07.2024 verschoben wurde?
Wenn ja, hätten Sie einen Link dazu?
Vielen Dank im Voraus .
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
schauen Sie hier: https://www.gov.uk/government/publications/implementation-of-the-future-regulation-of-medical-devices-and-extension-of-standstill-period
Dort wird die Verlängerung der Übergangsperiode erklärt.
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Salvatore,
ich habe noch eine Zusatzfrage zu dem Kommentar vom 10. August.
Wo muss die UK Personen angegeben werden? Auf dem Produktlabel, der Verpackung und/oder der IFU?
In dem MHRA document steht es wie folgt geschrieben:
„The name and address of the UK Responsible Person, where applicable, must be included on the product labelling or the outer packaging, or the instructions for use in cases where the UKCA marking has been affixed. UK Responsible Person details do not need to be included on labelling for CE marked devices, unless the device bears both the CE and UKCA markings.“
Also würde eine der drei Möglichkeiten ausreichen?
Vielen Dank
Klaus Steger
Guten Tag Herr Steger,
ohne UKCA-Marking muss die Angabe der UK Responsible Person nicht ergänzt werden gemäß den von Ihnen referenzierten Angaben der MHRA. Dies wäre nur notwendig, falls Ihr Labeling bereits das UKCA-Mark enthält. In diesem Fall stehen die drei genannten Möglichkeiten zur Auswahl.
Freundliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
ich habe eine Frage zu den Übergangsfristen in Zusammenhang mit dem EU-Zertifikat. Die von der MHRA genannten „Transitional arrangements“ sind gebunden an das MDD- bzw. MDR-Zertifikat (z.B: MDD-Produkte: either until the certificate expires or a maximum of 3 years after the new regulation comes into force“). Nun hat die EU eine Verlängerung der Übergangsfristen zur MDR veröffentlicht, die Notified Bodies werden allerdings ihre Zertifikate nicht verlängern (dürfen). Die meisten Zertifikate laufen am 26.05.2023 ab, sodass die Deadline für das UKCA also bei Mitte 2023 bleibt. Gibt es evtl. Hinweise darauf, dass die MHRA auf die verlängerten Übergangsfristen reagiert und die transitional arrangements noch einmal anpasst?
Viele Grüße
Kim Heimhuber
Liebe Frau Heimhuber,
laut aktueller Informationen der MHRA können CE-gekennzeichnete Medizinprodukte mindestens bis Juli 2024 ohne zusätzliches UKCA-Marking in Großbritannien in Verkehr gebracht werden. Danach sollen die neuen (noch nicht finalisierten) Übergangsbestimmungen gelten. Vermutlich haben Hersteller dann weitere 3-5 Jahre Übergangsfrist. Diese neuen Übergangsbestimmungen sollen in Q2/2023 veröffentlicht werden. Laut den bald in Kraft tretenden MDR-Übergangsbestimmungen werden die MDD-Zertifikate unter gewissen Voraussetzungen gültig bleiben, auch wenn das Datum auf dem Zertifikat nicht mehr aktualisiert werden darf. Die Zertifikate behalten aber dennoch ihre Gültigkeit. Somit gehen wir davon aus, dass diese auch weiterhin für eine Inverkehrbringung in UK unter den geplanten Übergangsbestimmungen („sooner of 3 years for general medical device or when certificate expires“) genutzt werden können.
Viele Grüße
Luca Salvatore
Sehr geehrte Salvatore,
In UK gibt es bisher keine UDI-Verpflichtung.
Ich vermute das gilt auch für Produkte die MDR zertifiziert sind und unter den entsprechenden Übergangsbestimmungen in UK in den Handel gebracht werden?
Vielen Dank
Sehr geehrter Herr Flötotto,
UK fordert aktuell noch keine UDI-Vergabe und -Registrierung für Medizinprodukte. Wenn Sie allerdings die Konformität mit der MDR erklären, müssen Sie für Ihre Produkte UDIs vergeben, um die Produkte rechtmäßig mit CE-Kennzeichnung in den Verkehr bringen zu können.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
anhand Ihres Blogeintrags habe ich bereits einige wissenswerte Informationen erhalten.
Da dieser jedoch von März 2021 ist, möchte ich Sie um einige Updates bitten:
• Wie sind aktuell die Übergangsfristen für die Registrierung von Medizinprodukten bei der MHRA?
• Wie ist aktuell die „Frist“, die in Ihrer Zusammenfassung genannt wird? Ist dies immer noch der 30.06.2023? Ist mit „UK-Bevollmächtigter“ die „UK Responsible Person“ gemeint?
• Wie ist aktuell die Übergangsfrist für die Registrierung bei der MHRA von Klasse I-Produkten? In Ihrem Blogeintrag ist weiterhin die Rede von „Mit Ablauf des Jahres 2021 müssen alle in Großbritannien vermarkteten Produkte auch dort registriert sein.“ Ich schätze, dass das Jahr 2021 nicht mehr dem aktuellen Stand entspricht?
• Ab welchem Datum muss man als Medizinproduktehersteller z.B. der Klasse I einen
UK Approved Body einbeziehen?
• Meines Wissens ist für Produkte in Nordirland die CE-Kennzeichnung weiterhin ausreichend wenn eine Selbsterklärung durch den Hersteller erfolgt. Können Sie das bestätigen?
Sehr geehrter Herr Braun,
aktuell fehlt noch die Anpassung des Gesetzes bezüglich der erweiterten Übergangsfristen. Geplant ist folgendes (siehe https://www.gov.uk/government/publications/implementation-of-the-future-regulation-of-medical-devices-and-extension-of-standstill-period/implementation-of-the-future-regulations):
– Akzeptanz von MDD/AIMDD-konformen Produkten bis 30.06.2028, Akzeptanz von IVDD/MDR/IVDR-konformen Produkten bis 30.06.2030
Zu Ihren weiteren Fragen:
– Die Übergangsfristen für die Registrierung sind abgelaufen, somit muss vor Inverkehrbringung eine Registrierung abgeschlossen sein durch die UK Responsible Person.
– Mit UK-Bevollmächtigter ist die UK Responsible Person gemeint
– Einen UK Approved Body benötigen Sie, wenn Sie das UKCA-Marking anstreben. Im Rahmen der Übergangsfristen ist dies allerdings nicht notwendig.
– In Nordirland gilt die MDR/IVDR. Somit ist die CE-Kennzeichnung notwendig.
Freundliche Grüße
Luca Salvatore
–
Guten Tag,
bin ich in der Anzahl der UK Responsible Person auf eine beschränkt? Oder darf ich die Pflicht für mehrere Produkte auf unterschiedlich UK Responsible Persons verteilen?
MfG
Dominik Sammer
Guten Tag Herr Sammer,
gemäß MHRA Guidance dürfen Sie nur eine UK Responsible Person benennen: „if you are a medical device manufacturer based outside the UK and wish to place a device on the Great Britain market, you need to appoint a single UK Responsible Person for all of your devices, who will act on your behalf to carry out specified tasks, such as registration.“
Freundliche Grüße
Luca Salvatore
Guten Tag,
ich habe gehört, dass bis 30 Juni 2024 Produkte mit CE Kennzeichnung in den britischen Markt gebracht werden können. Wir sind ein Hersteller von Klasse 1 Produkten.
Was gilt nun?
2023 oder 2024?
Mit freundlichen Grüßen
S. Schunk
Guten Tag Frau Schunk,
die Antwort auf Ihre Frage finden Sie direkt im Artikel im Kapitel 2. Die Spanne reicht von 30.06.2028 bis 30.06.2030.
Freundliche Grüße
Luca Salvatore
Hallo Mr.Sammer,
according to Britian regulation, is it mandatory to add manufacturer address on label or outerpackaging? or the information of UKRP is only needed?
Thanks and Regards
Navya Varkey
Dear Navya,
the name and address of the manufacturer must appear on the label.
Best regards
Luca
The list of approved notified bodies is now far larger than 4. Moreover, this list contains TÜV SÜD as UK assessment body.
Gratulations! Thanks for the information!
I updated the article right away.
Best regards, Christian Johner
What does Brexit mean for the use of IH-IVDs in UK? The MHRA has a definition of Health Institution significantly different from the IVDR. Would the only option be to UKCA mark IVD assays which are used as IH-IVD in the EU?
Thanks and best,
F. Kopp
Dear Franziska,
I’m not sure if I understood your question correctly. However, it is true that the definition of a health institution in the UK is much stricter than under the IVD. In general, only the larger NHS-affiliated institutions are considered health institutions. But the MHRA guidance does not say that independent laboratories cannot be considered HI as long as they meet very specific requirements. If qualification as an in-house IVD is not possible, placing on the market as an IVD would be required (with a conformity assessment accepted by MHRA such as UKCA or CE mark). To better answer your question, I recommend contacting our IVD team (www.johner-institut.de/kontakt/) and referring to this article.
Best regards
Luca Salvatore
Hallo Herr Salvatore,
wir sind Hersteller von Medizinprodukten mit Sitz in der EU. Wir verkaufen unsere Produkte in der EU sowie an unsere Niederlassung in UK. Diese vertreibt die Medizinprodukte in UK und liefert sie an einen weiteren Händler in Irland. Wenn unsere Niederlassung in UK die Medizinprodukte von UK nach Irland an einen Händler weiterverkauft, ist der Händler in Irland ein Importeur in die EU? Wenn ja, muss der Importeur in EUDAMED registriert und am Labelling angegeben werden?
Gibt es dazu ein Q&A, ein Guidance Dokument oder ähnliches, wo das definiert ist?
Vielen Dank im Voraus!
MfG, Maximilian
Guten Tag Herr Fürhauser,
der Sinn eines Importeurs gemäß MDR und weiterer EU-Rechtsvorschriften ist die Übernahme „wichtiger und eindeutig festgelegter Aufgaben, die zu einem großen Teil auf der Art der Aufgaben eines in der EU niedergelassenen Herstellers aufbauen.“ (siehe EU Blue Guide 2022, 3.3). Man könnte nun die Sinnhaftigkeit dieser Regelung in Frage stellen im Falle eines Re-Imports, denn der Hersteller ist in diesem Fall ja in der EU niedergelassen. Die Definition des Importeurs der MDR hilft hier leider nicht weiter: „jede in der Union niedergelassene natürliche oder juristische Person, die ein Produkt aus einem Drittland auf dem Unionsmarkt in Verkehr bringt“. Es heißt hier nicht, dass der Hersteller seinen Sitz im Drittland haben muss.
Man könnte versuchen, in Ihrem Fall die Frage umzukehren. Wenn die Person in Irland ein Händler gemäß MDR wäre, dann müsste bereits eine Inverkehrbringung in der EU stattgefunden haben. Falls nicht, trifft in diesem Fall die Definition des Händlers nicht zu, denn dieser bringt definitionsgemäß keine Produkte in den Verkehr. Somit bliebe nur die Rolle des Importeurs übrig (vorausgesetzt das Produkt bzw. Kennzeichnung wird nicht entsprechend geändert, so dass dieser die Herstellerverantwortung übernehmen würde). Sinnvoll wäre die Importeursverantwortung ohnehin auch in dieser Konstellation, denn es ist nicht klar, ob der Hersteller das Produkt ausschließlich für die Vermarktung in UK vorgesehen hatte und ggfs. keine vollständige Konformität mit den anwendbaren EU-Rechtsvorschriften vorliegt.
Ein Q&A oder Leitfaden, welcher genau diesen Fall beleuchtet ist uns leider nicht bekannt.
Freundliche Grüße
Luca Salvatore