
Die Anforderungen an klinische Prüfungen zur Bewertung eines Produkts sind unter der MDR enorm gestiegen. Erfahren Sie hier das Wichtigste, was es über den regulatorischen Weg der klinischen Prüfungen im Rahmen der MDR zu wissen gibt.
Die klinische Prüfung ist nicht der einzige Weg zur Konformität. Alternativ lässt sie sich durch richtiges Vorgehen bei der klinischen Bewertung vermeiden. Das Johner Institut berät Sie gern in Bezug auf klinische Bewertungen und prüft z. B., ob ausreichend klinische Daten für Ihr Medizinprodukt vorliegen.
1. Klinische Prüfungen von Medizinprodukten
a) Definition und Ziele
Laut MDR ist eine klinische Prüfung
„eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird“
Quelle: MDR, Artikel 2, Absatz 45
Die Medizinproduktehersteller müssen bei den klinischen Prüfungen beweisen, dass ihre Produkte sicher sind und die versprochene klinische Leistung und den versprochenen Nutzen bringen – einen Nutzen, der die Risiken überwiegt. Sie sind verpflichtet, diesen Nachweis anhand klinischer Daten zu führen.
Ob eine klinische Prüfung notwendig ist, zeigt sich in der Regel während der klinischen Bewertung. Hier führen Hersteller eine GAP-Analyse (Lückenanalyse) durch, um einen etwaigen Mangel an klinischen Daten aufzudecken. Sind diese klinischen Daten nicht in ausreichender Menge oder Güte vorhanden (z. B. in der wissenschaftlichen Literatur), müssen die Hersteller diese Daten im Rahmen klinischer Prüfungen erheben.
Manchmal lässt sich durch das richtige Vorgehen bei der klinischen Bewertung eine aufwändige und teure klinische Prüfung vermeiden. Lesen Sie mehr hierzu in unserem Beitrag zur klinischen Bewertung oder kontaktieren Sie das Johner Institut für eine Beratung.
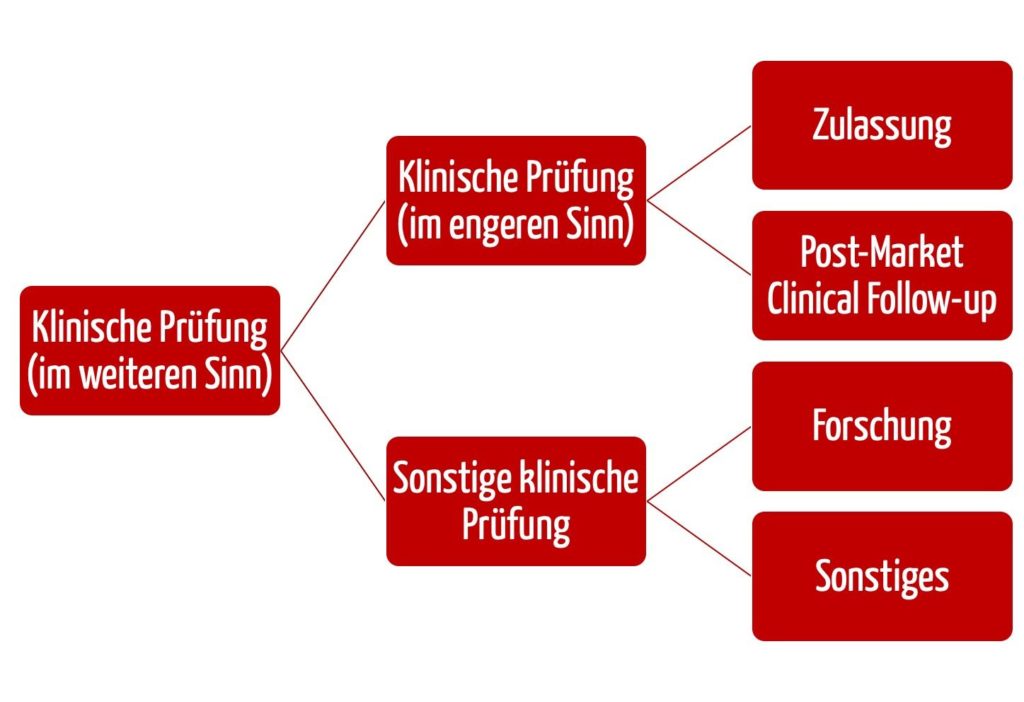
b) Abgrenzung von „klinischen Prüfungen” und „sonstigen klinischen Prüfungen”
Die EU-Medizinprodukteverordnung MDR hat den Begriff der „sonstigen klinischen Prüfungen“ eingeführt.
Klinische Prüfung
„Normale” klinische Prüfungen sind nach Art. 62 Abs. 1 MDR solche, die den folgenden Zwecken dienen:
- a) Die Eignung des Produkts für den bestimmten Zweck überprüfen
- b) Den klinischen Nutzen überprüfen
- c) Die klinische Sicherheit und Nebenwirkungen des Produkts prüfen
Diese Art der klinischen Prüfung können Hersteller im Konformitätsbewertungsverfahren nutzen. Sie müssen die Anforderungen aus Art. 62 bis 81 sowie des Anhangs XV der MDR erfüllen.
Klinische Prüfung nach dem Inverkehrbringen (PMCF-Studien)
Bei einer sogenannten klinischen Prüfung nach dem Inverkehrbringen (oder PMCF-Studie) gelten dagegen die Bestimmungen aus Art. 74 Abs. 1 MDR. Bei der klinischen Prüfung nach dem Inverkehrbringen geht es um eine weitergehende Bewertung von Produkten im Rahmen ihrer Zweckbestimmung, die bereits die CE- Kennzeichnung gemäß Artikel 20 Absatz 1 tragen.
Gilt die CE-Kennzeichnung für eine andere Zweckbestimmung, müssen Hersteller die klinische Prüfung nach den Regeln der „normalen” klinischen Prüfung (Art. 62 Abs. 1) durchführen.
Mehr zu Post-Market Clinical Follow-up (PMCF) erfahren Sie in unserem Beitrag zu Post-Market Surveillance
Sonstige klinische Prüfungen
Sonstige klinische Prüfungen sind nach Art. 82 MDR alle klinischen Prüfungen, die zu einem anderen Zweck als den in Art. 62 Abs. 1 MDR genannten Zwecken durchgeführt werden. Darunter fallen auch die Grundlagenforschung und Machbarkeitsstudien. Solche sonstigen klinischen Prüfungen müssen die Anforderungen aus Artikel 62 Absätze 2 und 3, Absatz 4 Buchstaben b, c, d, f, h und l und Absatz 6 MDR und ggf. weitere nationale Anforderungen erfüllen.
Für klinische Prüfungen und für sonstige klinische Prüfungen von Medizinprodukten gelten nicht die gleichen regulatorischen Anforderungen. Allerdings ist in beiden Fällen zu prüfen, ob das BfArM zu involvieren ist. In beiden Fällen mussten und müssen die Anforderungen der MDR beachtet und i.d.R eine Ethik-Kommission um Genehmigung gebeten werden.
Beispiel
| Sonstige klinische Prüfung | Klinische Prüfung | |
| Zweck | Sonstige Zwecke und NICHT im Rahmen des Konformitätsbewertungsverfahrens: Klinische Prüfungen, die nicht zu einem der in Artikel 62 Absatz 1 MDR genannten Zwecke durchgeführt werden.(Artikel 82 MDR) | a) zur Feststellung und Überprüfung, dass ein Produkt so ausgelegt, hergestellt und verpackt ist, dass es unter normalen Verwendungsbedingungen für einen oder mehrere der aufgelisteten spezifischen Zwecke geeignet ist und die von seinem Hersteller angegebene bezweckte Leistung erbringt;b) zur Feststellung und Überprüfung des von seinem Hersteller angegebenen klinischen Nutzens eines Produkts; c) zur Feststellung und Überprüfung der klinischen Sicherheit des Produkts und zur Bestimmung von bei normalen Verwendungsbedingungen gegebenenfalls auftretenden unerwünschten Nebenwirkungen des Produkts und zur Beurteilung, ob diese im Vergleich zu dem von dem Produkt erbrachten Nutzen vertretbare Risiken darstellen.(Artikel 62 MDR) |
| Situation | Ein Forscherteam nutzt ein (bereits zugelassenes) Kernspingerät, um MRT-Sequenzen zu erproben, zu kombinieren und zu verbessern, um so eine bestimmte Krebserkrankung zuverlässiger diagnostizieren zu können. Sie probieren diese Messsequenzen an Patienten aus, bei denen die Krebsdiagnose über eine Histologie bereits gesichert bzw. ausgeschlossen ist. | Anschließend baut ein Hersteller die vom Forscherteam entwickelten MRT-Sequenzen in die nächste Version seines Kernspingeräts ein. Weil die klinischen Daten des Forscherteams nicht ausreichend sind (z.B. weil nicht genügend Patienten untersucht wurden oder sich die Daten nicht 1:1 übertragen lassen), führt der Hersteller eine klinische Studie durch mit dem Gerät und den neu entwickelten Sequenzen, um Nutzen und Leistungsfähigkeit seines neuen Medizinprodukts zu beweisen. Dazu ist er im Rahmen der Konformitätsbewertung („Zulassung“) verpflichtet. |
| Bewertung | Dieses „Ausprobieren“ stellt eine „klinische Studie“ dar, d.h. eine „sonstige klinische Prüfung“, und bedingt die Zustimmung einer nach nationalem Recht eingesetzten Ethik-Kommission und ist somit genehmigungspflichtig ist. | Diese „Zulassungsstudie“, wurde bisher auch „MPG-Studie“ genannt, ist eine klinische Prüfung im Sinne des Medizinprodukterechts und wird in der MDR in Artikel 62 Absatz 1 ff reguliert. |
Fazit
Besonders bei Medizinprodukten, die auf neuen Verfahren beruhen, tun die Hersteller sich schwer, eine Abgrenzung vorzunehmen: Dient die klinische Prüfung dazu, das Verfahren auszuprobieren bzw. zu entwickeln? Oder dient sie dazu, bereits die klinischen Daten für die Zulassung des Medizinprodukts zu sammeln? Oder beides? Hersteller sollten dies eindeutig festlegen.
2. Kategorisierung von klinischen Prüfungen
a) Möglichkeiten der Kategorisierung
Klinische Prüfungen lassen sich nach verschiedenen Aspekten kategorisieren:
- Nach Zweck (z. B. Zulassung, Erforschung, etc. s. u.)
- Nach Phase (am Anfang der Entwicklung, vor der Zulassung, nach der Zulassung, etc. Die Einteilung in die Phasen 0 bis IV findet sich v. a. bei Medikamentenstudien und ist hier nicht Gegenstand der Betrachtung.)
- Nach Studiendesign (s. u.)
b) Kategorisierung nach Zweck
Die unterschiedlichen Arten von klinischen Prüfungen im Bereich Medizinprodukte dienen unterschiedlichen Zwecken. Je nach Zweck unterliegen sie verschiedenen Anforderungen.
| Typ | Ziel | Zeitpunkt bezogen auf Inverkehrbringung | Eingriff in Diagnose oder Therapie |
| Forschungsstudie (eine Form einer „sonstigen klinischen Prüfung“), Artikel 82 MDR und § 24 MPDG) | Erkenntnisse gewinnen, Machbarkeit feststellen | vorher | ja und nein |
| Zulassungsstudie (klinische Prüfung), Artikel 62 Absatz 1 MDR) | Sicherheit, Nutzen und Leistungsfähigkeit eines (ansonsten fertigen) Produkts nachweisen | vorher | ja |
| PMCF-Studie (klinischen Prüfung mit einem Produkt, das die CE-Kennzeichnung trägt, Artikel 74 MDR) | Sicherheit, Nutzen und Leistungsfähigkeit eines im Markt befindlichen Produkts nachweisen | nachher | ja |
| PMCF-Studie ohne zusätzliche belastende und/oder invasive Verfahren (entweder eine sonstige klinische Prüfung oder eine Form einer PMCF-Studie) | Entweder zu Forschungszwecke ohne Nachweis von Sicherheit, Nutzen und Leistungsfähigkeit oderZum Nachweis von Sicherheit, Nutzen und Leistungsfähigkeit eines im Markt befindlichen Produkts | nachher | nein |
Beispiele für „Maßnahmen“:
- Zusätzliche Ultraschall-Untersuchung
- Zusätzliche Blutabnahme
- Erweiterte körperliche Untersuchung
- Zuweisen (auch randomisiert) des Patienten zu einer Kontrollgruppe, die anders untersucht oder behandelt wird als eine andere Gruppe
Würde man hingegen medizinisches Personal nur bei seiner Arbeit beobachten oder Patienten befragen, würde man nicht von einem Eingriff bzw. einer Maßnahme sprechen. Es läge keine klinische Prüfung vor. Man spricht in diesem Fall von einer nicht-interventionellen klinischen Prüfung.
c) Kategorisierung nach Studiendesign
Diese Kategorisierung basiert auf den Attributen des Studiendesigns, beispielsweise:
- Anzahl der Patienten (< 100, > 1000 (= Kohorte))
- Anzahl der Studienzentren (z. B. multizentrisch)
- Zeitpunkt der Planung: retrospektiv versus prospektiv
- Verblindung: unverblindet („open label“), einfach verblindet („single blinded“), doppel verblindet („double blinded“)
- Randomisiert/nicht randomisiert: Einteilung der Patienten in Studienarme nach dem Zufallsprinzip
- Mit oder ohne den Einsatz von Placebos („placebo-kontrolliert“)
- Zeitraum: Longitudinale Studien verfolgen Patienten über einen längeren Zeitraum, teilweise über deren ganzes Leben.
- Interventionell, nicht interventionell
Abhängig von diesen Attributen definieren sich verschiedene Studiendesigns. Die folgende Tabelle nennt Beispiele.
| Studiendesign | Wissenschaftliche Aussagekraft | Anzahl Patienten | Anzahl Zentren | Zeitpunkt der Planung | Verblindung | Randomisiert | Zeitraum | Inter-ventionell |
| Randomisierte Studie | sehr hoch | mehrere bis viele | meist mehrere | prospektiv | ja/nein | ja | abh. vom Ziel | ja |
| Kohorten-Studie | hoch | viele | meist mehrere | prospektiv / retrospektiv | ja/nein | nein | Jahre | ja |
| Querschnittsstudie | mittel | viele | meist mehrere | prospektiv | nein | nein | Zeitpunkt, ggf. mehrere | ja |
| Fallserie | mittel bis hoch | mehrere bis viele | eins oder mehrere | prospektiv / retrospektiv | ja/nein | ja/nein | abh. vom Ziel | ja |
| Fallstudie | niedrig | einer | eines | prospektiv / retrospektiv | nein | nein | meist Zeitpunkt | ja/nein |
Die MDR fordert, dass die Hersteller das Evidenzniveau bestimmen. Je höher das Evidenzniveau ist, umso höher muss die wissenschaftliche Aussagekraft sein.
Das Ziel der klinischen Prüfung und der darauf abgestimmte primäre Endpunkt bestimmen das Design. Aus diesem ergeben sich die Evidenz und die wissenschaftliche Aussagekraft.
Eine randomisierte klinische Prüfung ist nicht immer möglich. Man kann auch eine Fallserie prospektiv und kontrolliert durchführen und dadurch Level 3. erreichen. Dieses Level allein bestimmt jedoch auch nicht die wissenschaftliche Aussagekraft.
3. Klinische Prüfungen in der Praxis
a) Phasen und Aktivitäten
Die klinischen Prüfungen von Medizinprodukten erfolgen meist in Phasen, die idealerweise sequenziell durchlaufen werden, in der Praxis manchmal auch iterativ.
- Ziele festlegen
In der ersten Phase ermitteln die Hersteller als Sponsor der klinischen Prüfung die regulatorischen Anforderungen, entscheiden über die Notwendigkeit einer klinischen Prüfung und legen deren Ziele im Groben fest. - Klinische Prüfung planen
Anschließend formulieren die Hersteller – meist mit Hilfe von Biostatistikern – die Hypothesen und Ziele genauer und legen das Studiendesign fest. Sie planen den Ablauf der Studie, wählen Personen aus und stellen Budgets bereit. - Klinische Prüfung vorbereiten
Die Hersteller wählen die Prüfzentren und klinischen Prüfer aus und holen das Votum von Ethik-Kommissionen bzw. Behörden ein. Sie bereiten die Prozesse (inkl. Dokumentation) und Werkzeuge (z. B. elektronische Datenerfassung) vor und schulen die Beteiligten. - Klinische Prüfung durchführen
Die Prüfärzte und das beteiligte medizinische Personal erheben die Daten. Die klinischen Monitore überprüfen die Daten fortlaufend auf Plausibilität und Vollständigkeit sowie die Durchführung am Prüfzentrum im Rahmen von Besuchen vor Ort. Die Datenmanager werten die Daten begleitend aus, um bei Problemen Behörden informieren bzw. die klinische Prüfung anzupassen oder abbrechen zu können. - Daten auswerten, Sicherheit und Leistungsfähigkeit des Produkts bewerten
Die Hersteller bzw. deren Dienstleister werten die Daten aus. Sie bewerten, ob die Hypothesen verifiziert und damit die Sicherheit und Leistungsfähigkeit des Produkts belegt werden konnten. Sie erstellen Berichte und die klinische Bewertung.
b) Wichtige Punkte bei klinischen Prüfungen
Worauf bei klinischen Prüfungen zu achten ist, legt die MDR in Anhang XV Kapitel I fest:
- Anerkannte ethische Grundsätze werden beachtet.
- Die Prüfung wird nach einem Prüfplan durchgeführt, der dem Stand der Wissenschaft und Technik entspricht und an das zu prüfende Produkt angepasst ist.
- Es wird unter voraussichtlichen Verwendungsbedingungen geprüft und unter Einbeziehung einer repräsentativen Zahl von Anwendern.
- Alle Merkmale des Produkts werden berücksichtigt und dokumentiert.
- Der Prüfer und seine Mitarbeiterinnen und Mitarbeiter erhalten Zugang zu den technischen und klinischen Daten des Produkts und werden angemessen in die Handhabung eingewiesen.
- Der Bericht über die klinische Prüfung enthält eine kritische Bewertung aller gesammelten Daten, inklusive aller negativen Ergebnisse.
c) Typische Fehler, die Hersteller vermeiden sollten
Das Johner Institut stößt bei Herstellern, die als Sponsoren klinische Prüfungen durchführen, regelmäßig auf folgende Fehlerquellen:
Unklarheit über Notwendigkeit einer klinischen Prüfung
Die Hersteller wissen nicht, ob eine klinische Prüfung überhaupt notwendig ist. Insbesondere ist unklar, ob die bisherigen Daten ausreichen, um die Sicherheit und Leistungsfähigkeit zu belegen. Eine unnötig durchgeführte Prüfung ist Verschwendung von Zeit und Geld. Wenn dagegen eine klinische Prüfung vorgeschrieben ist, aber nicht durchgeführt wurde, gefährdet das die rechtskonforme Vermarktung.
Unpräzise oder falsche Ziele
Die Hersteller müssen genau festlegen, welche sogenannten „Endpunkte“ die Studie mit klinischen Daten belegen muss. Andernfalls laufen sie Gefahr, dass die Studie zwar eine Hypothese beweist, dieser Beweis aber nicht geeignet ist, um die Sicherheit, die Leistungsfähigkeit und den klinischen Nutzen zu belegen.
Falsches Studiendesign
Die Bedeutung der Planung kann man kaum genug betonen: Der falsche Populationsumfang, eine ungeeignete Form der klinischen Prüfung oder ein unrealistischer Projektplan können zum Scheitern einer klinischen Prüfung beitragen. Auch ein agiles, iterativ inkrementelles Vorgehen ist im Kontext klinischer Prüfungen kein zielführendes Rezept.
Unzureichendes Monitoring und Datenmanagement
Fehler und Lücken in den Daten und eine Erfassung, die nicht dem klinischen Prüfplan entspricht, machen die Aussagekraft einer klinischen Prüfung zunichte. Ein engmaschiges Monitoring und ein fundiertes Datenmanagement sind für die klinische Prüfung daher unerlässlich.
Mangelnde Äquivalenz
Manche Hersteller nutzen die Erkenntnisse aus der klinischen Prüfung, um das Produkt weiter zu verbessern. Sie müssen dann die Äquivalenz der Produkte nachweisen, die bei der klinischen Prüfung eingesetzt werden, und der Produkte, die zugelassen werden sollen.
Unzureichende Teilnehmerzahl
Viele Hersteller tun sich schwer damit, ausreichend viele Studienteilnehmer zu rekrutieren, um die notwendige Evidenz zu erreichen. Das trifft besonders in diesen Fällen zu:
- Seltene Krankheit
- Der Vorteil (= Überlegenheit) des Produkts ist im Vergleich zu Alternativen gering oder sogar fragwürdig.
- Das Produkt erscheint den Patienten als fremd oder „riskant“.
- Es gibt nicht ausreichend Prüfärzte, die an der Studie interessiert sind.
Unklarheit über regulatorische Anforderungen
Die MDR und v. a. das MPDG haben die Anforderungen an „sonstige klinische Prüfungen“ nun eindeutig definiert und festgelegt. Das trifft auch auf PMCF-Studien zu. Dies sollte den Herstellern bewusst sein. Sie sollten auch nicht davon ausgehen, dass „Studien“ mit bereits zugelassenen Produkten genehmigungsfrei sind.
d) Aufgaben und Auswahl von Clinical Research Organizations (CROs)
Gegebenenfalls können Clinical Research Organizations (CROs) bei allen oben genannten Schritten helfen. Die meisten CROs haben sich allerdings auf die Erforschung von Arzneimitteln spezialisiert.
Es ist nicht immer nötig, eine (u. U. teure) CRO in Anspruch zu nehmen, denn nicht jedes Produkt braucht eine klinische Prüfung oder PMCF-Studie. Mehr dazu erfahren Sie in unseren Beiträgen zur MDCG 2020-6 sowie der klinischen Bewertung oder in unserem Seminar zur klinischen Bewertung.
e) Einsatz von Digital Health Tools
Der Einsatz von Digital Health Tools kann die Geschwindigkeit und Güte beispielsweise der Datenerhebung signifikant erhöhen. Dabei sind aber regulatorische Randbedingungen zu beachten. Die folgenden wissenschaftlichen Fachartikel verschaffen darüber eine Übersicht:
- Digital Tools—Regulatory Considerations for Application in Clinical Trials (springer.com)
- Digital Health Technology (DHT) in European Clinical Trials, How to Improve the Status-Quo of the Regulatory Landscape? (springer.com)
4. Regulatorische Anforderungen an klinische Prüfungen
a) Medizinprodukteverordnung MDR
Die MDR legt ihre Anforderungen an klinische Prüfungen in den Artikeln 62 bis 80 sowie dem Anhang XV fest. Insgesamt sind die Anforderungen der MDR umfangreich und spezifisch. So gibt es bei besonderen Personengruppen wie Kindern, Schwangeren und Stillenden etwa konkrete Anforderungen an die Aufklärung und an die Prüfungen.
Die MDR legt außerdem fest, wann und wie die Daten (künftig) in der EUDAMED zu hinterlegen sind, wie man bei Änderungen des Studiendesigns vorgehen muss und was die Anforderungen an klinische Prüfungen mit Produkten sind, die bereits ein CE-Zeichen tragen.
Der Anhang XV der MDR stellt weitere Anforderungen an Durchführung, Dokumentation und Sponsoren (Hersteller) von klinischen Prüfungen.
Zudem behält sich die MDR vor, über gemeinsame Spezifikationen (Common Specifications, CS) weitere Anforderungen zu ergänzen.
Eine erste Abhilfe schafft die Leitlinie MDCG 2021-6 „Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation“. Das Dokument richtet sich an Sponsoren von klinischen Prüfungen von Medizinprodukten, die im Geltungsbereich der Verordnung (EU) 2017/745 (MDR) durchgeführt werden.
b) Medizinprodukte-Durchführungsgesetz MPDG
Das Medizinprodukte-Durchführungsgesetz MPDG hat das MPG und die MPKPV abgelöst. Es stellt inzwischen fast die gleichen Anforderungen an „Forschungsstudien“ („sonstige klinische Prüfungen“) und an „klinische Prüfungen“. Auch bei den „sonstigen klinischen Prüfungen besteht das MPDG auf:
- Minimierung von Risiken und Belastungen sowie deren Vereinbarkeit mit dem erwartetem Nutzen
- Qualifikation der Prüfärzte
- Versicherungsschutz
- Einwilligung der Probanden (es gibt Sonderregelungen)
- Positives Votum der Ethik-Kommission (Ausnahmen ggf. bei CE-gekennzeichneten Produkten möglich)
- Anzeige bei der zuständigen Bundesoberbehörde (Ausnahmen ggf. bei CE-gekennzeichneten Produkten möglich)
Bei den „sonstigen klinischen Prüfungen“ verlangt das MPDG, dass „die sonstige klinische Prüfung der zuständigen Bundesoberbehörde nach § 53 Absatz 1 angezeigt wurde.“ Bei den „klinischen Prüfungen“ ist erforderlich, dass „die zuständige Bundesoberbehörde hierfür eine Genehmigung erteilt hat“.
PMCF-Studien gemäß Artikel 74 MDR werden im MPDG nicht explizit beschrieben und definiert. Hersteller müssen sich aber an die Anforderungen der MDR halten.
Das BfArM erläutert in seiner Anleitung für Sponsoren wie klinische Prüfungen und klinische Leistungsstudien im DIMDS zu erfassen sind.
c) MDCG und MEDDEV-Dokumente
Die EU-Kommission und die Benannten Stellen haben weitere Dokumente publiziert. Dazu zählen folgende Guidance-Dokumente der MDCG sowie einige MEDDEVs, die teilweise noch als Stand der Technik angesehen und daher zu Rate gezogen werden können:
- MDCG 2021-28: Substantial modification of clinical investigation under Medical Device Regulation
- MDCG 2021-8: Clinical investigation application/notification documents
- MDCG 2020-10/1 und MDCG 2020-10/2: Guidance on safety reporting in clinical investigations und Appendix: Clinical investigation summary safety report form
- MDCG 2020-6: Guidance on sufficient clinical evidence for legacy devices
- MDCG 2020-5: Guidance on clinical evaluation – Equivalence
- MEDDEV 2.12/2: Post Market Clinical Follow-up Studies
d) ISO 14155
Die genauesten Vorgaben an die Durchführung klinischer Prüfungen macht die DIN EN ISO 14155: 2021. Diese Norm trägt den Titel „Klinische Prüfung von Medizinprodukten an Menschen — Gute klinische Praxis“. Sie gibt beispielsweise vor,
- wie Prüfpläne zu erstellen sind,
- wie mit Änderungen umzugehen ist,
- welche Dokumente und Formulare mit welchen Inhalten gefordert sind und
- wer welche Verantwortung trägt.
e) Weitere regulatorische Anforderungen im Kontext von Medizinprodukten
Hersteller sollten diese Vorschriften und Best-Practices beachten:
- Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV)
- Verordnung über das datenbankgestützte Informationssystem über Medizinprodukte des Deutschen Instituts für Medizinische Dokumentation und Information (DIMDIV)
- IMDRF MDCE WG/N 65 FINAL:2021: Post Market Clinical Follow-up Studies
- FDA Guidances zur „Good Clinical Pratice“
f) Regulatorische Anforderungen für klinische Prüfungen im Rahmen der Forschung
Dient die klinische Prüfung NICHT dem Nachweis der Sicherheit und Leistungsfähigkeit im Rahmen der Zulassung eines Produkts, zählt sie zu den „sonstigen klinischen Prüfungen“ (Art. 82 MDR), weshalb die Anforderungen der MDR greifen.
Diese Daten können nicht für die Konformitätsbewertung herangezogen werden. Allerdings können Hersteller die Daten als prä-klinische Daten für die klinische Bewertung nutzen.
Außerdem sind die Regeln der „Good Clinical Practice“ beispielsweise auch bei der klinischen Forschung einzuhalten. Die Deklaration von Helsinki ist in jedem Fall zu beachten. Meist muss ein Votum der Ethik-Kommission eingeholt werden.
5. Fazit und Empfehlung
Dass sich viele Hersteller scheuen, ihre Medizinprodukte einer klinischen Prüfung zu unterziehen, ist sehr verständlich. Die Aufwände dafür und die regulatorischen Anforderungen daran sind immens. Zu leicht unterlaufen hier Fehler, die den Wert der klinischen Prüfung zunichte machen oder gar strafrechtliche Konsequenzen haben können.
Die MDR erhöht zum einen die Anforderungen an die klinischen Prüfungen und zum anderen die Anzahl der Fälle, in denen eine klinische Prüfung notwendig wird. Das liegt auch daran, dass die MDR und viele Benannte Stellen nur neue klinische Daten akzeptieren, da die Anforderungen an äquivalente Produkte gestiegen sind.
Daher empfiehlt das Johner Institut insbesondere bei innovativen Produkten, bei implantierbaren Produkten und solchen der Klasse III entweder die klinische Strategie schon früh im Entwicklungsprozess einzuplanen oder bei eigenen CE-gekennzeichneten Produkten im Rahmen von PMCF-Studien die noch fehlenden klinische Daten zu erheben, um Abweichungen und im schlimmsten Fall Zertifikatsentzüge zu vermeiden.
Das Johner Institut berät zu Dokumenten, Inhalten oder zur Durchführung von klinischen Prüfungen. Auch beraten wir Sie zu Strategien und möglichen Alternativen zur klinischen Prüfung.
Versionshistorie:
- 2024-11-08: Kapitel 3.e) mit Digital Health-Tools eingefügt, Nummerierung des Kapitels 3 korrigiert
- 2022-09-23: Leitlinie des BfArM für Sponsoren ergänzt
- 2022-09-07: Komplettes Update des Beitrags



Hallo Prof. Johner, vielen Dank für den wieder sehr aufschlussreichen Artikel. Ich habe durch ein MedTechEurope Dokument erfahren, dass die ISO 14155 voraussichtlich Mitte des Jahres 2018 als Draft International Standard (DIS) und damit auch als Entwurf DIN EN ISO 14155 erscheinen und ggf. Mitte 2019 veröffentlicht wird. Es macht sicher Sinn, dass bei der Überarbeitung der Prozesse im Unternehmen nicht nur die MDR, sondern auch gleich die kommenden Anforderungen der neuen ISO 14155 berücksichtigt werden, damit die demnächst erhobenen Daten auch gleich internationale Annerkennung finden. Leider ist es mir nicht gelungen jemanden aus dem Gremium ISO/TC 194/WG zu ermitteln, der mir Infos zu den „Hauptänderungen“ geben kann. Haben Sie Kontakte zur ISO/TC 194/WG und können Sie ggf. mal einen Artikel zu den kommenden Änderungen publizieren? Ich denke das wäre sehr interessant für Viele.
Danke für diese großartige Anregung, lieber Herr Matz!
Das kommt gleich in den Redaktionsplan!
Viele Grüße, Christian Johner
Ergänzend zu meinem Kommentar: ich habe gesehen, dass auch ein FDA Guidance referenziert wurde. Dazu vielleicht auch ein Update -> es gibt seit 02/2018 eine neue Richtlinie, die in den nächsten 12 Monaten gültig wird (new guidance ‚Acceptance of Clinical Data to Support Medical Device Applications and Submissions) https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM597273.pdf
Wer seine Produkte international, insbesondere in den Gebieten EU, USA und Japan zulässt und in den nächsten 2 Jahren Studien plant, sollte m.E. die MDR, die neue ISO 14155 und die neuen FDA Richtlinien berücksichtigen. Sonst besteht die Gefahr, dass „doppelte Arbeit“ geleistet werden muss.
Wie würden Sie Anwendungsbeobachtung in Englischer Sprache benennen?
Ich gebe mal ein paar Vorschläge:
– observational study
– surveillance study
– clinical application study
– clinical application observation?
Meist übersetzt man es mit „clinical observation study“.
Surveillance wird eher im Kontext der Post-Market Surveillance genutzt.
Die angekündigte ISO/DIS ist gerade erschienen:
https://www.beuth.de/de/norm-entwurf/din-en-iso-14155/289646651
Beste Grüße. T. Matz
Hallo Herr Johner, zwischenzeitlich ist auch die ISO/DIS 14971 erschienen:
https://www.beuth.de/de/norm-entwurf/din-en-iso-14971/290649384
Dies ist m.E. so interessant, da die ISO 14155 nur im Zusammenhang mit der ISO 14971 gültig ist und beide Drafts auch schon einen Z-Anhang mit Bezug zur MDR aufweisen. Wer seine Prozesse in Bezug auf die MDR gerade überarbeitet, sollte dies gleich mit aufnehmen! „Rund“ wird dies m.E. wenn man auch gleich das QM-System auf Basis MDR und 13485:2016 überarbeitet. Auch dazu gibt es schon eine Veröffentlichung (PD CEN TR 17223_2018 Guidance on the relationship between EN ISO 13485_2016). MfG T. Matz
Sehr geehrter Herr Professor Johner,
Ich bin Student der TU München und führe gerade ein Projektstudium zum Thema Digialisierung von klinischen Studien für Medizin Produkte durch.
Um den Markt besser zu verstehen stellt sich mir die Frage, welche Kategorie von Medizinproduktherstellern führen im Rahmen der klinischen Bewertung bei der Zulassung der Medizinprodukte und/ oder der Post Market Surveillance (PMS) eigene klinische Studien durch? Hängt es von der Risikoklasse oder Neuartigkeit der Technologie/ Verfahren ab?
Was sind weitere Faktoren dafür, dass klinische Daten (durch Post Market Clincal Follow Up) von den Medizin Produkt Herstellern erhoben werden müssen um die Audits zu bestehen? Und bei welchen Herstellern ist es ausreichend die klinische Bewertung/ Evidenz durch Literatur Daten oder äquivalente Konkurrenz Medizin Produkte erfolgreich zu beweisen? Entscheiden das Auditoren nach Vorgaben der MDR oder MEDDEV?
Können Sie mir dazu Produkt Beispiele oder Kategorien?
Vielen Dank für ihre Hilfe im Voraus.
K. Amm
Es gibt keine generelle Kategorien. Diejenigen, die noch nicht ausreichend klinische Daten haben, müssen klinische Prüfungen durchführen. Das kann mit der MDR auch Hersteller von alten Produkten treffen.
Bei neuartigen Verfahren, bei denen es keine Vergleichsprodukte gibt, ist die klinische Prüfung hingegen meist notwendig.
Die Anforderungen, wann die MDR eine klinische Prüfung verlangt (insbesondere bei Klasse III und bei Implantaten der Klasse IIb), beschreibt die MDR ganz gut. Außer in den dort genannten Ausnahmen immer.
Die Audits und Zulassungen sollten nicht verwechselt werden.
Die klinische Bewertung muss immer auf Basis klinischer Daten erfolgen. Wenn die vorhandenen Daten nicht ausreichend sind, um die Sicherheit, die Leistungsfähigkeit und den klinischen Nutzen zu beweisen, ist immer eine klinische Prüfung notwendig.
Sehr geehrter Herr Prof. Dr. Johner,
Ich habe mit Interesse ihren Beitrag zum Thema Klinische Prüfungen von Medizinprodukten gelesen. Da mich das Thema „generelle Forderungen nach GCP Compliance in klinischen Studien“ interessiert hab ich folgende Fragen an Sie:
1) Aufgrund welcher Rechts- bzw. Normenbasis kommen Sie zum Schluss dass die Regeln der „Good Clinical Practice“ auch bei der klinischen Forschung einzuhalten sind?
2) Gibt es konkrete regulatorische Anforderungen (Gesetze/Normen) an nicht-interventionelle Studien (z.B. Anwendungsbeobachtung), wenn die daraus gewonnenen Daten für die Argumentation in der Klinischen Datenakte eines MPs verwendet werden, z.B. Anforderungen an den Prozess der Datengewinnung und -auswertung/ Statistische Analyse/ Berichterstellung durch den MP Hersteller?
Viele Grüße,
Karin Pickl
Sehr geehrte Frau Pickl,
die Anforderungen an die GCP stellt u.a. die FDA im zitierten Dokument, Gesetze und Verordnungen wie die MPKPV sowie Normen wie die ISO 14155. Das gleiche gilt für die MDR. Ebenso ist die Deklaration von Helsinki zu nennen.
Die zweite Frage insbesondere die darin genannte Zielsetzung verstehe ich nicht. Die o.g. Regeln gelten jedenfalls auch für Anwendungsbeobachtungen. Leichte Unterschiede gibt es abhängig davon, ob die Studie der Forschung oder dem Nachweis von Leistung, Sicherheit und Risiko von Medizinprodukten dient.
Beste Grüße, Christian Johner.
Hallo Herr Prof. Dr. Johner,
das Konzept von agiler Produktentwicklung sieht ja vor, frühzeitig und kontinuierlich Kundenfeedback in die Produktentwicklung einfließen zu lassen. Bei SaMD ist es dabei leicht, das neue Produkt direkt beim Kunden als nicht-klinischer Prototyp verfügbar zu machen, um Feedback vom Kunden zu erhalten.
Nun kann laut MDR ein Inverkehrbringen allerdings nur über entweder CE-Markierung oder klinische Prüfung stattfinden. Und darüber hinaus gibt es nun die angesprochene Definition von „sonstiger klinischer Prüfung“.
Würden Sie sagen, eine solche Entwicklungsbegleitung durch Kunden durch kontinuierliches Feedback jeweils auf aktuellsten Prototypen ist ein Fall von „sonstiger klinischer Prüfung“, basierend auf der Tatsache, dass aus der Definition von „sonstiger klinischer Prüfung“ Punkt c) „andere Fragestellungen“ erfüllt ist? Oder würden Sie die Definition so anwenden, dass alle Unterpunkte a-d mit einem „und“ verknüpft sind und deswegen schon a) „nicht Teil eines … Prozesses zur Produktentwicklung“ zum Ausschluss führt? Falls letzteres, kann man dann davon ausgehen, dass gar keine Inverkehrbringung stattfindet, da eben keiner der beiden definierten Zwecke zutrifft, und da es sich bei einem Prototypen nicht um ein Produkt handelt?
Beste Grüße
Thomas Frank
Sehr geehrter Herr Frank,
Danke für Ihre wichtige Fragen!
Ich würde empfehlen, das Einsammeln des produktbegleitenden Kundenfeedbacks keinesfalls als klinische oder sonstige klinische Prüfung zu erheben. Vielmehr könnte man im Rahmen z.B. der formativen Bewertung dieses Kunden- — genauer gesagt — Nutzerfeedback einholen. Es muss darauf geachtet werden, dass keine Patienten dabei sind bzw. dass keine Intervention stattfindet.
Fazit: Wenn man zwischen Kunden bzw. Anwendern und Patienten entscheiden kann, dann ist man aus dem Schneider.
Viele Grüße, Christian Johner
Hallo Prof. Johner,
besteht nur Registrierungspflicht und Publikationspflicht für klinische Studien mit Medizinprodukten?
Herzliche Grüße
Dorothea Waser
Sehr geehrte Frau Waser,
eine Publikationspflicht für klinische Prüfungen existiert nicht. Dieser Aspekt wird im jeweiligen klinischen Prüfplan reguliert: In Anhang XV, Kapitel II Satz 3.17 der MDR findet sich dies als Anforderung: Vorgehensweise bei der Erstellung des klinischen Prüfberichts und der Veröffentlichung von Ergebnissen im Einklang mit den rechtlichen Anforderungen und den ethischen Grundsätzen gemäß Kapitel I Abschnitt 1.
Hallo Herr Prof. Johner,
vielen Dank für die sehr ausführliche Darstellung des Themenkomplexes. Eine Frage die ich mir gestellt habe ist: Wie würden Sie das aktive und nicht fall- bzw. fehlerbezogene Einsammeln und Auswerten von Leistungsdaten aktiver Medizinprodukte direkt nach dem Inverkehrbringen (auch z.B. eines SW Updates) im Kontext der klinischen Prüfung einordnen?
Vielen Dank & Viele Grüße
Philipp Holzfuß
Hallo Herr Prof. Johner,
wir würden gern ein Produkt von außerhalb des EAA einführen. Bevor wir uns vollständig vertraglich mit dem Hersteller festlegen, wollen wir sehr gern ausgewählten Stammkunden ein Testprodukt zur Verfügung stellen, um das Anwendungsverfahren und die Patientenzufriedenheit zu testen. Demnach geht es nur um einen Anwendungstest, aber nicht um die Bestimmung der Leistung oder Sicherheit des Produktes. Das Produkt ist bereits CE-zertifiziert.
Gibt es die Möglichkeit, einen solchen Anwendungstest innerhalb der Zweckbestimmung durchzuführen, ohne vorweg Zeitraum X auf die Registrierungsbestätigung des DIMDI warten zu müssen?
Allerbeste Grüße und vielen Dank im Voraus!
Sehr geehrte Frau Wagener,
vielen Dank für Ihre interessante Frage. Ich hoffe, ich habe Sie richtig verstanden. Sie schreiben, dass Ihr Produkt bereits CE-zertifiziert ist. Somit sollten Leistung und Sicherheit sowie der klinische Nutzen bereits über die klinische Bewertung nachgewiesen worden sein. Wenn Sie Ihr Produkt nun im Rahmen einer Anwendungsbeobachtung verwenden möchten, könnte ich mir vorstellen, dass dafür eine einfache PMCF-Studie, oder sogar eine Usability-Studie in einem Labor in Frage kommen. Das kommt auf die genaue Fragestellung an. Dafür braucht es meist keine BfArM-Genehmigung.
Bezüglich der Registrierung bleibt zu sagen, dass Hersteller das Inverkehrbringen von Medizinprodukten über das Medizinprodukte-Informationssystem des DIMDI/BfArM anzeigen müssen. Das sollte für Ihr CE-gekennzeichnetes Produkt bereits geschehen sein. Klinische Studien hingegen müssen beim Deutschen Register Klinischer Studien (DRKS, Teil des DIMDI) registriert werden.
Ich hoffe ich konnte Ihre Frage zufriedenstellend beantworten. Gerne können Sie für weitere Fragen auch unser kostenloses Microconsulting nutzen (https://www.johner-institut.de/micro-consulting/).
Freundliche Grüße
Julia Renz
Sehr geehrter Herr Johner,
zunächst vielen Dank für den interessanten Artikel. Eine Frage die ich mir gestellt habe:
Müssen Medizinprodukte die ausschließlich für sonstige klinische Prüfungen im Rahmen der Forschung Verwendung finden ein CE-Konformitätsverfahren durchlaufen?
Wenn ich die Definition in der MDR2017/745 richtig interpretiere fallen auch Produkte die lediglich im Rahmen von Forschungsprojekten eingesetzt werden, unter die Definition Medizinprodukt (z.B. 3. Spiegelstrich „Untersuchung […] der Anatomie oder eines physiologischen […] Vorgangs oder Zustands“) und müssten doch dementsprechend ein Konformitätsbewertungsverfahren durchlaufen oder gibt es eine Ausnahmeregelung für „research-only devices“?
Beste Grüße und vielen Dank im Voraus!
Marius Berthel
Guten Tag!
Danke für Ihren informativen Artikel. Ich hätte eine eher grundsätzliche Frage: gibt es rechtliche/ Vorgaben, die die Abgabe von Prototypen (ohne CE) an Kliniken für (klinik)-eigene Forschungszwecke (IIT) regeln?
Darf ein Hersteller so etwas? Wenn ja, unter welchen Bedingungen?
Danke für Ihre Einschätzung!
Guten Tag,
bitte haben Sie noch ein wenig Geduld. Wir werden uns zeitnah bei Ihnen melden!
Herzliche Grüße
Anja Segschneider | Redaktion
Liebe Frau Baruth,
Frau Schulze hat die folgende Antwort auf Ihre Frage gegeben:
Hier wäre einmal der Artikel 52, Abschnitt 13 der MDR zu nennen: (13) Für Prüfprodukte gelten die Anforderungen gemäß Artikel 62 bis 81.
„Prüfprodukt“ bezeichnet ein Produkt, das im Rahmen einer klinischen Prüfung bewertet wird;
Allerdings wäre eine (klinik)-eigene Forschung keine klinische Prüfung.
Weiterhin regelt Artikel 5, Abschnitt 5 der MDR:
(5) Mit Ausnahme der einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I gelten die Anforderungen dieser Verordnung nicht für Produkte, die ausschließlich innerhalb von in der Union ansässigen Gesundheitseinrichtungen hergestellt und verwendet werden, sofern alle folgenden Bedingungen erfüllt sind: …
Hier geht es allerdings nicht um Produkte eines externen Herstellers, sondern um Produkte, welche die Klinik selbst herstellt, weil sie am Markt nicht verfügbar sind.
Zusammenfassend ist mir keine rechtliche Vorgabe bekannt welche die Abgabe von Prototypen (ohne CE) an Kliniken für (klinik)-eigene Forschungszwecke direkt regelt. Aus den o.g. Regeln entnehme ich jedoch, dass eine Klinik die Verantwortung für so ein Produkt übernimmt, also quasi zum Hersteller wird und dass dieses Produkt die Grundlegenden Sicherheit- und Leistungsanforderungen erfüllen muss (Anhang I der MDR).
Es ist also weniger die Frage, ob der Hersteller das darf, denn er ist hier einfach nur Lieferant eines Prototypen ohne CE, worauf er hinweisen muss.
Es ist mehr die Frage, ob die Klinik das darf und die Bedingungen dafür sind im Artikel 5 Abschnitt 5 geregelt.
Mit besten Grüßen
Astrid Schulze
Sehr geehrtes Johner Institut,
vielen Dank für Ihren Artikel.
Mir ist noch nicht wirklich klar, ob Machbarkeitsstudien (Feasibility Studies) bzw. Pilotstudien wirklich unter „sonstige klinische Prüfung“ fallen oder nicht doch unter reguläre „klinische Prüfung“. Sie ordnen es an mehreren Stellen so ein, dass Machbarkeitsstudien genau wie Grundlagenforschung und „Ausprobieren“ unter Artikel 82 fallen (im Text und u.a. in den Tabellen). Unter Artikel 62 fallen demnach Studien zum Nachweis von Konformität, Leistung, Sicherheit oder Zulassungsstudien. Aber auf anderen Webseiten, welche die MDR interpretieren, heißt es, dass Feasibility Studies schon als Teil von Artikel 62 (klinische Prüfung) gelten.
Nach meiner Lesart (als absoluter juristischer Laie) klingt die Definition der „sonstigen klinischen Studie“ eher so, als wenn Machbarkeitsstudien nicht mehr darunter fallen, da davon gesprochen wird, dass die Studie nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung sein darf.
„a) nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung oder der Produktbeobachtung eines gegenwärtigen oder künftigen Herstellers ist“
Die Machbarkeitsstudie ist aber für alle Produkthersteller/innen ja ein Teil des geplanten Prozesses, weil sich auf deren Grundlage überhaupt erst entscheidet, ob die Produktentwicklung Sinn macht und verfolgt werden soll, auch wenn hier heraus noch keinerlei Schlüsse über Sicherheit, Performance und Konformität, geschweige denn einer Zulassung gezogen werden können. In dem Sinne ist es eine „Grundlage“. Aber eben auch eine, die Teil des systematischen Produktentwicklungsprozesses ist. In der Regel wird hier ja ein Proof of Concept geprüft.
Die Feasibility Study wird auch in Annex XIV (Clinical Evaluation), S. 164 des MDR, im Rahmen des Clinical Development Plans genannt:
„a clinical development plan indicating progression from exploratory investigations, such as first-in-man
studies, >>feasibility and pilot studies<<, to confirmatory investigations, such as pivotal clinical investigations,
and a PMCF as referred to in Part B of this Annex with an indication of milestones and a description of
potential acceptance criteria;"
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745&from=EN
Können Sie mir vielleicht aufzeigen, wo ggf. mein Verständnisfehler liegt oder etwas zu den widersprüchlichen Interpretationen anderer Medizinseiten sagen?
Vielen Dank und beste Grüße
A. Lambert
Liebe Frau Lambert, vielen Dank für Ihre interessante Frage, die uns Gelegenheit gibt, tiefer in das Thema einzusteigen.
Inzwischen gibt es ein MDCG Dokument 2021-6. Dort werden Pilotstudien grundsätzlich unter den Artikel 62 gestellt, es kann jedoch auch Ausnahmen geben. Der Anhang I des MDCG 2021-6 Dokuments gibt hier Hinweise. Machbarkeitsstudien werden im Kapitel 5 dieses MDCG Dokuments den Pilotstudien zugeordnet und es wird auf die ISO 14155 verwiesen, die diese Studientypen genauer beschreibt. Das in unserem Artikel erwähnte Medizinproduktedurchführungsgesetz (MDPG) nennt 4 Kriterien, wann eine sonstige klinische Prüfung vorliegt:
a) wenn die klinische Prüfung nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung oder der Produktbeobachtung eines gegenwärtigen oder künftigen Herstellers ist,
-> Hier können die Hersteller beispielsweise steuern, indem sie die Machbarkeitsphase (welche von der ISO 13485 ja nicht gefordert ist) nicht Teil Ihres im Qualitätsmanagementprozess definierten Entwicklungsprozesses werden lassen.
b) wenn die klinische Prüfung nicht mit dem Ziel durchgeführt wird, die Konformität eines Produktes mit den Anforderungen der Verordnung (EU) 2017/745 nachzuweisen,
-> z.B. wissenschaftliche Forschung oder auch klinische Prüfungen, die sich nicht auf den Konformitätsbewertungsprozess eines bestimmten Produkts beziehen. Auch hier können Hersteller etwas steuern, in dem sie die Forschungsfragen entsprechend allgemein formulieren und außerhalb der systematischen Produktentwicklung und Konformitätsbewertung eines konkreten Produkt bzw. einer konkreten Produktfamilie ansiedeln.
c) wenn die klinische Prüfung der Beantwortung wissenschaftlicher oder anderer Fragestellungen dient und
d) wenn die klinische Prüfung außerhalb eines klinischen Entwicklungsplans nach Anhang XIV Teil A Ziffer 1 Buchstabe a der Verordnung (EU) 2017/745 erfolgt;
-> Hier muss man aufpassen, da Punkt a) immer noch zutreffen kann. Wenn d) erfüllt ist, kann es also sein, dass es sich doch um eine klinische Prüfung nach Artikel 62 handelt, weil Punkt a) erfüllt ist.
Zusammenfassend aus dem MDPG Dokument kann man also sagen, dass eine sonstige Prüfung dann vorliegt, wenn sie nicht im Rahmen der (vom Hersteller definierten!) Produktentwicklung, des Konformitätsbewertungsverfahrens und des klinischen Entwicklungsplans erfolgt. Sie dient dann also wissenschaftlichen oder sonstigen Fragestellungen.
Das oben erwähnte MDCG Dokument 2021-6 stellt die Machbarkeitsstudien unter den Oberbegriff der Pilotstudien (Kapitel 5). Das heißt natürlich nicht, das jede Machbarkeitsstudie auch eine Pilotstudie ist, die dem Artikel 62 unterliegt. Vielmehr betrifft es die Machbarkeitsstudien, die in der ISO 14155 näher spezifiziert werden. Wenn Sie Ihre Machbarkeitsstudie außerhalb der ISO 14155 und der o.g. Kriterien aus dem MDPG Dokument ansiedeln können, dann ist es keine Pilotstudie im Sinne des MDCG Dokuments und fällt somit auch nicht unter Artikel 62.
Immer dann wenn es nicht um ein konkretes Produkt geht,
Herzliche Grüße
Astrid Schulze
DiGA / DiPA Circle
Sehr geehrte Mitarbeiter*innen des Johner Institutes,
sollte eine Studie für die CE Zertifizierung eines Medzinproduktes in mehreren europäischen Ländern durchgeführt werden, oder reicht dafür ein Land aus, wenn genügend Daten vorhanden sind?
Bei Vertrieb in Deutschland ohne deutsche Studienbeteiligung: Wird das BfArM involviert? Würden deutsche Krankenkassen überhaupt die Kosten für Medizinprodukt übernehmen, welches in einem anderen europäischen Land getestet wurde?
Vielen Dank und mit freundlichen Grüßen
Dr. Adriane Napp
Liebe Frau Dr. Napp,
Klinische Studien sind eine mögliche Quelle für klinische Daten, die für die klinische Bewertung gefordert werden. Grundsätzlich müssen die vorhandenen klinischen Daten weltweit recherchiert und ausgewertet werden, um eine klinische Bewertung durchzuführen. Sollten nicht genügend klinische Daten für das eigene oder ein äquivalentes Produkt vorhanden sein und eine klinische Bewertung auf der Basis von Leistungsdaten nicht möglich sein, dann muss eine klinische Studie durchgeführt werden. Wo diese Studie durchgeführt werden kann, hängt von den Forschungsfragen ab, die Sie untersuchen wollen. Wenn es zum Beispiel bei einer Digitalen Gesundheitsanwendung um den Nachweis positiver Versorgungseffekte für deutsche Patienten geht, dann müssen Sie die Versorgungsrealität in Deutschland erfassen und die Studie hier durchführen oder in einem Land mit vergleichbarer Versorgungssituation. Sie müssen die Studie grundsätzlich dort anmelden, wo Sie sie durchführen. Das BfArM ist nur in Deutschland zuständig. Ansonsten giltgilt die MDR, die Europäische Verordnung für Medizinprodukte. Alle Produkte, die der MDR entsprechen, können auch in ganz Europa vertrieben wurden. Es können zusätzliche Anforderungen erhoben werden, wenn das Produkt in den Heil- und Hilfsmittelkatalog aufgenommen oder als DiGA gelistet werden soll.
Ich hoffe, ich konnte Ihnen etwas weiterhelfen.
Mit besten Grüßen
Astrid Schulze
Seniorberaterin QM & RA
Hallo Herr Prof. Dr. Johner,
vielen Dank zuerst für die wertvolle Erklärung.
Ich habe über das Thema „Klinische Prüfung“ eine Frage:
Muss in einer klinischen Prüfung Patienten teilnehmen?
Unsere Firma entwickelt einen Roboterarm, der im Krankenwagen ein Behandlungsprozess beschleunigt. Unseres Produkt ist neu und deshalb haben wir keine klinischen Daten oder äquivalente Produkte auf dem Markt und sind verpflichtet, eine klinische Bewertung durchzuführen laut MDR-Anforderungen. Für die klinische Bewertung brauchen wir keine Patienten, wir müssen technische Daten über den Roboterarm überprüfen und mit einem herkömmlichen Verfahren einer Behandlung vergleichen.
Gilt diese Überprüfung (Studie) als klinische Studie, obwohl keine Patienten teilnehmen?
Vielen Dank
Jakob Muaz
Lieber Herr Muaz,
vielen Dank für Ihre interessante Frage! Die Definition für klinische Prüfungen steht in der MDR Artikel 2, 45: „klinische Prüfung“ bezeichnet eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird;
Möglicherweise können Sie ihre klinische Bewertung auf der Grundlage einer Leistungsbewertung durchführen, nach MDR Artikel 61 (10). Ob diese Daten ausreichen können Sie z.B. mit Hilfe Ihrer Risikoanalyse begründen und sollten Ihren Weg im Plan zur klinischen Bewertung gut begründen. So wie Sie Ihr Produkt beschreiben, könnte das der beste Weg für Sie sein.
Mit besten Grüßen
Astrid Schulze
Seniorberaterin QM & RA
Liebes Team vom Johner Institut,
auch von mir ein großes Dankeschön für diesen tollen und sehr hilfreichen Artikel!
Frage habe ich zu folgendem Abschnitt:
Im Artikel steht: „Die MDR fordert, dass die Hersteller das Evidenzniveau bestimmen. Je höher das Evidenzniveau ist, umso höher muss die wissenschaftliche Aussagekraft sein.“
1. Was sind die Kriterien nach denen ich das Evidenzniveau festlegen muss? Oder anders: Wann darf ich ein Studiendesign wählen, dessen wissenschaftliche Aussagekraft unterhalb des Optimums von „sehr hoch“ liegt?
2. (Wie) spielt hier die Risikoklasse mit hinein? (Wir arbeiten an Klasse I).
3. Macht die MDR hierzu Aussagen (ich konnte nichts finden), oder woraus leitet sich das ab?
Vielen herzlichen Dank!
Inka Benthin
Liebe Frau Benthin, vielen Dank für Ihre spannende Frage und bitte entschuldigen Sie die verspätete Antwort. Das Evidenzniveau der wissenschaftlichen Artikel wird mit Hilfe der Kriterien bestimmt, die im Guidancedokument Meddev 2.7/1 rev. 4, z.B. Kapitel 9.3.1 festgelegt sind. Je nach Risikoklasse und Produktart kann und muss nicht immer das höchste Evidenzniveau erreicht werden. So ist zum Beispiel eine einfache oder doppelte Verblindung nicht bei allen Produkten möglich bzw. sinnvoll. Eine Kontrollgruppe kann in bestimmtem Fällen ethisch nicht vertretbar sein. Für ein Medizinprodukt der Klasse I empfehle ich zunächst zu prüfen, ob klinische Daten für äquivalente Produkte verfügbar sind und falls dies der Fall ist, die Evidenz der gefundenen klinischen Daten zu prüfen und zu dokumentieren. Dafür können die oben genannten Kriterien genutzt werden. Wenn Sie eine klinische Prüfung durchführen wollen oder müssen, empfehle ich die Planung durch eine Clinical Research Organisation (CRO). Dort liegen entsprechende Erfahrungen bezüglich des Studiendesigns und der Fallzahlplanung vor.
Mit besten Grüßen
Astrid Schulze
Sehr geehrter Herr Prof. Johner,
ich habe eine Verständnisfrage zum Thema MPDG „sonstige klinische Prüfung“. Im Prinzip ist doch jede Studie, die ein Hersteller durchführen lässt, eine „klinische Prüfung im engeren Sinne“, oder? Entweder man benötigt die Daten für das Konformitätsverfahren vor der Zulassung, oder man sammelt Daten für das Konformitätsverfahren nach der Zulassung (für eine Erweiterung/Änderung des Scopes), oder man sammelt PMCF-Daten. Eine „sonstige klinische Prüfung“ würde für einen Hersteller m.E. eher in die Richtung gehen, wie man interessierten Ärzten Geld zukommen lassen könnte, oder irre ich mich da? Eine „sonstige klinische Prüfung“ können doch eigentlich nur Ärzte aus eigenen Stücken und unabhängig von Herstellern durchführen, um unter „not inteded use“ bzw. „offlabel use“ Bedingungen für sie interessante (wissenschaftliche) Fragestellungen zu bearbeiten, oder irre ich mich da?
Liebe Grüße
Dr. Kuerschner
Sehr geehrter Dr. Kuerschner,
der Begriff der sonstigen klinischen Prüfung stammt v.a. aus dem MPDG §3.
Demnach wäre beispielsweise eine wissenschaftliche Studie mit einem zugelassenen MRI eine solche sonstige klinische Prüfungen, bei der man herausfinden will, wie verschiedene Therapieoptionen den Verlauf einer Krebserkrankung beeinflussen.
Eine Gleichsetzung mit ökonomischen Incentivierungen sehe ich nicht. Ebenso ist es für diese Definition nicht primär relevant, ob ein off-label-use stattfindet. Für die Beurteilung durch die Ethikkommission hingegen schon.
Ebenso ist es nicht relevant, von wem die Studie initiiert wurde. Hingegen ist die Zielsetzung dieser Studie entscheidend.
Beste Grüße, Christian Johner
Guten Morgen,
gibt es allgemein einen Grund oder eine Vorschrift, weshalb Usability Prüfungen im Unternehmen mit noch nicht zugelassenen Medizinprodukten der Klasse IIa (Einwilligung der Mitarbeiter vorausgesetzt) nicht für die Zulassung in Europa verwendet werden dürfen?
Viele Grüße
Fabian
Lieber Fabian,
klinische Prüfungen sind „eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit, Leistung oder Nutzen eines Produkts durchgeführt wird“. Hingegen dient die Usability Prüfung dazu, ob spezifizierte Nutzer im spezifizierten Nutzungskontext die spezifizierten Nutzungsziele (Zweckbestimmung) effektiv und effizient erreichen können. Hersteller müssen die Anforderungen der MDR an die Gebrauchstauglichkeit (Usability) für ausnahmslos alle Medizinprodukte nachweisen.
Die Ergebnisse der Usability Prüfung können für die Zulassung des Medizinproduktes verwendet werden, sind jedoch nicht zum alleinigen Nachweis der Leistungs- oder Sicherheitsaspekte möglich.
Viele Grüße,
N. Jurrmann.
Liebes Team vom Johner Institut,
mir ist leider noch nicht klar, welche Anforderungen für Studien der letzten Kategorie der Tabelle in Kapitel 2b) gelten: PMCF-Studien, deren Zweck der Nachweis von Sicherheit, Nutzen und Leistungsfähigkeit eines im Markt befindlichen Produktes ist.
PMCF hat zum Ziel die klinische Bewertung zu Aktualisierung, und zwar mit Hilfe von klinischen Daten, die sich aus der Verwendung eines CE-konformen Produktes ergeben. Es kann sich dabei demnach nur um Daten handeln, die auf irgendeine Weise Sicherheit, Nutzen und Leistungsfähigkeit des Produktes belegen, denn das ist ja das Ziel der klinischen Bewertung und damit auch der Aktualisierung derselbigen.
Neben den „klinisch relevanten Angaben“ die sich aus PMS und PMCF selbst ergeben, bleiben nur klinische Prüfungen oder „in nach dem Peer-Review-Verfahren überprüfter wissenschaftlicher Fachliteratur veröffentlichte Berichte über sonstige klinische Erfahrungen“.
Artikel 74 gilt nur dann, wenn „im Rahmen dieser Prüfung Prüfungsteilnehmer zusätzlichen Verfahren“ unterzogen werden und diese „invasiv oder belastend“ sind.
Artikel 82 ist in diesem Fall auch nicht anwendbar.
Demnach bleibt nur Artikel 62, was für mich aber merkwürdig erscheint, da hier mehr Anforderungen zu erfüllen sind, als für Studien nach Artikel 74, die ja aber per Definition, eine Belastung über die normale Anwendung hinaus enthalten „müssen“.
Mit freundlichen Grüßen
J. Hüttel
Sehr geehrter Herr Hüttel,
der Artikel 82 MDR eingeführte Begriff der „sonstigen klinischen Prüfung“ umfasst klinische Prüfungen, die zur Beantwortung rein wissenschaftlicher Fragestellungen durchgeführt werden. PMCF-Studien, die im Rahmen der festgelegten Zweckbestimmung und ohne zusätzliche invasive oder belastende Verfahren ablaufen, fallen auch unter sonstige klinische Prüfung nach MDR, Artikel 82. Siehe hierzu ebenfalls die Definition § 3 Nr. 4 des MPDG in Deutschland.
Durch § 47 Abschnitt 3 des MPDG werden die Vorgaben aus den Abschnitten 1 und 2 des gleichen Paragraphen aufgehoben. Hierzu gehört auch die Notwendigkeit einer zustimmenden Bewertung durch die Ethik-Kommission und die Anzeigepflicht bei der Bundesoberbehörde. Unangetastet bleiben jedoch Pflichten, die sich aus der Berufsordnung für Ärzte ergeben und ggf. sind weitere Bestimmungen bei der Durchführung der Datensammlung zu berücksichtigen, die sich aus anderen gesetzlichen Vorgaben ergeben.
Viele Grüße,
N. Jurrmann.
Liebes Team vom Johner Institut,
herzlichen Dank für den schönen Übesichtsartikel!
Sie schreiben darin u.a., dass nicht-interventionelle Studien (Studien ohne Eingriffe/Maßnahmen, NIS) keine klinischen Prüfungen gemäß MDR darstellen.
Das Konzept der Interventionalität/nicht-Interventionalität ist mir aus Arzneimittelstudien geläufig und ist dort auch klar definiert (Clinical Trials Regulation).
Die Definition einer KP in Art. 2 MDR gibt diese klare Unterscheidung m.E. allerdings (leider) nicht her. (Die Interventionalität müsste sich ja hinter den Worten “systematische Untersuchung” verbergen). Auch ist mir keine Guidance oder Stellungnahme hierzu bekannt.
Könnten Sie mir bitte eine offizielle Quelle nennen, in der diese (aus meiner Sicht sehr sinnvolle) Einteilung in nicht-interventionelle und interventionelle Studien und die Ansicht, dass NIS keine klinischen Prüfungen sind, auch für Medizinprodukte festgelegt/angeregt wird?
Vielen Dank & herzliche Grüße
Berit Bachmann
Sehr geehrte Frau Bachmann,
da haben Sie vollkommen recht, die Definition in Art. 2 der MDR gibt dies nicht her. Aber die ISO 14155 gibt in Kapitel I.6.3 eine Definition vor.
Als nicht-interventionelle Studien können im Medizinproduktberereich Studien angesehen werden, die unter anderem folgende Kriterien erfüllen:
– Enthalten studienbedingt keine belastenden Maßnahmen
– Die Verwendeten Medizinprodukte haben ein CE
– Die Verwendung der Medizinprodukte erfolgt im Rahmen ihrer Zweckbestimmung
– Die Studienteilnehmer werden nicht auf die Anwendung eines Medizinproduktes randomisiert
In der ISO 14155 finden sich auch weitere Hinweise welche Prüfungen als „nicht interventionell“ angesehen werden können.
Eine Studie die alle oben genannten Punkte erfüllt, findet sich in keiner der Definitionen der MDR wieder und wird in Deutschland under dem MPDG und der Berufsordnung für Ärzte (BoÄ) durchgeführt und kann somit nicht als klinische Prüfung nach MDR betrachtet werden.
Viele Grüße,
Susanne Golombek