
Das Dokument MDCG 2020-5 (Clinical Evaluation – Equivalence) erhöht die Anforderung an die Äquivalenz von Medizinprodukten, die Hersteller bei der klinischen Bewertung ihres Produkts heranziehen dürfen.
Ein klares Verständnis der regulatorischen Anforderungen hilft Ihnen,
- schnell zu entscheiden, ob Sie die Äquivalenzroute überhaupt anstreben oder gleich eigene klinische Daten und/oder Leistungsdaten nutzen, und
- die Äquivalenz anderer Produkte belastbar nachzuweisen, um Ihre klinische Bewertung zur Zufriedenheit Ihrer Benannten Stelle zu erstellen.
Damit vermeiden Sie unnötige Iterationen und Aufwände und schaffen die Voraussetzungen für eine schnelle Zulassung Ihrer Produkte.
Sie stehen noch am Anfang Ihrer klinischen Bewertung?
Unser Clinical Evaluation Jumpstart Kit hilft Ihnen, die ersten Schritte innerhalb der klinischen Bewertung Ihrer Medizinprodukte zu gehen und verschafft Orientierung, was konkret von Ihnen verlangt wird.
1. Regulatorische Anforderungen an die Äquivalenz
a) Die MDR bestimmt die Anforderungen an die Gleichartigkeit von Produkten
Die Aspekte der Gleichartigkeit (Äquivalenz)
Die MDR bestimmt im Annex XIV Part A (3) die Voraussetzungen, unter denen ein Produkt bei der klinischen Bewertung als Äquivalenzprodukt zum eigenen Produkt betrachtet werden darf:
Ein Produkt ist nur dann äquivalent, wenn es
- technisch,
- biologisch und
- klinisch
die gleichen Merkmale aufweist. Genau diese Gleichartigkeit (englisch: „equivalence“) muss der Hersteller bei der klinischen Bewertung nachweisen. Andernfalls darf er die klinischen Daten des anderen Produkts nicht heranziehen, um die Sicherheit, die Leistungsfähigkeit und den klinischen Nutzen des eigenen Produkts zu beweisen.
Diese Anforderung steckt bereits in der Definition des Begriffs „klinische Daten“:
„klinische Daten“ bezeichnet Angaben zur Sicherheit oder Leistung, die im Rahmen der Anwendung eines Produkts gewonnen werden und die aus den folgenden Quellen stammen: […] in nach dem Peer-Review-Verfahren überprüfter wissenschaftlicher Fachliteratur veröffentlichte Berichte über sonstige klinische Erfahrungen entweder mit dem betreffenden Produkt oder einem Produkt, dessen Gleichartigkeit mit dem betreffenden Produkt nachgewiesen werden kann,
Quelle: MDR Artikel 2
Das zweite Kapitel dieses Artikels stellt die Anforderungen der MDR an die technische, biologische und medizinische Gleichartigkeit (Äquivalenz) genauer vor.
Lesen Sie hier mehr zu den Themen klinische Daten und klinische Bewertungen.
Der Nachweis der Gleichartigkeit bedingt Zugriff auf Daten
Die Hersteller müssen über Daten verfügen, um in der klinischen Bewertung die Behauptungen (Claims) zu untermauern.
“It shall be clearly demonstrated that manufacturers have sufficient levels of access to the data relating to devices with which they are claiming equivalence in order to justify their claims of equivalence.”
Quelle: MDR Annex XIV Part A (3
Besondere Anforderungen für bestimmte Produktklassen
Die MDR stellt bei bestimmten Produktklassen zusätzliche Anforderungen. Diese schränken den Kreis der Hersteller bzw. Produkte ein, die für die klinische Bewertung auf Basis der Äquivalenzroute (d.h. ohne klinische Prüfung) einbezogen werden dürfen:
|
Produktklasse |
Hersteller |
Erläuterung |
|
Klasse IIb implantierbar und Klasse III |
Gleich | Vorgängerprodukt mit Modifikationen, CE-gekennzeichnetes Vorgängerprodukt |
|
Klasse IIb implantierbar und Klasse III |
Verschieden | Vertraglich geregelter, vollständiger Zugriff auf technische Dokumentation |
|
Produkte ohne medizinische Zweckbestimmung |
Unabhängig | Begründung, wenn vorhandene klinische Daten von analogem Produkt verwendet werden |
|
Andere Produkte |
Unabhängig |
Zugang zu präklinischen und klinischen Daten |
b) MEDDEV 2.7/1 rev. 4 konkretisiert die Anforderungen an die Gleichartigkeit
Die Leitlinie MEDDEV 2.7/1 in der Revision 4 enthält einen über zweiseitigen Anhang A1, der genauer aufführt, unter welchen Voraussetzungen die klinische, technische und biologische Äquivalenz als gegeben angenommen werden darf. Auch diese genaueren Vorgaben stellt das zweite Kapitel vor.
Das MEDDEV-Dokument enthält darüber hinausgehende Anforderungen.
Lesen Sie hier mehr zum Thema MEDDEV 2.7/1 Revision 4.
Die Äquivalenz kann nur auf Basis eines einzelnen Medizinprodukts nachgewiesen werden
Hersteller könnten versucht sein, für jeden Aspekt der Äquivalenz (technisch, biologisch, klinisch) ein spezielles Vergleichsprodukt heranzuziehen (s. Abb. 1). Genau das schließt die MEDDEV 2.7/1 aber aus.
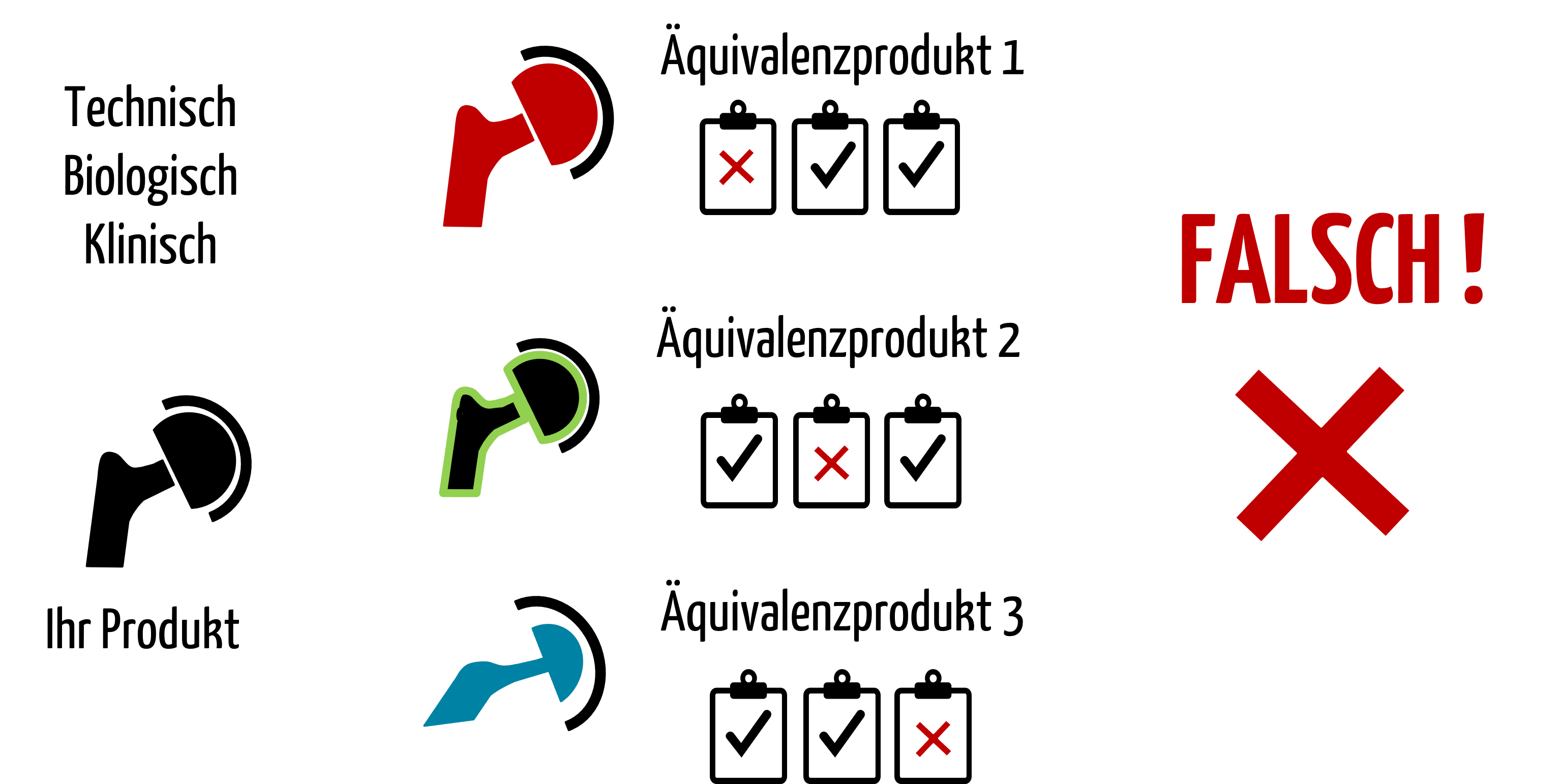
Abb. 1: Wenn ein Produkt nicht technisch oder nicht biologisch oder nicht klinisch äquivalent ist, darf es der Hersteller nicht als Äquivalenzprodukt heranziehen.
Die klinischen Daten eines Produkts dürfen Hersteller nur dann heranziehen, wenn dieses Produkt in allen drei Aspekten äquivalent ist (s. Abb. 2). Andernfalls zählt es nicht als Äquivalenzprodukt.

Es ist allerdings möglich, dass der Hersteller die Daten mehrerer Äquivalenzprodukte verwendet, wenn diese jeweils bezüglich aller drei Aspekten äquivalent sind.
Der Hersteller muss auch die Unterschiede zum Vergleichsprodukt offenlegen
Die MEDDEV verpflichtet die Hersteller, auch alle Unterschiede offenzulegen zwischen dem eigenen Produkt und dem Produkt, die sie als Äquivalenzprodukt heranziehen wollen. Die Leitlinie erwartet Erklärungen, weshalb diese Unterschiede die klinische Leistungsfähigkeit und die klinische Sicherheit nicht signifikant beeinflussen.
Die Vergleichsprodukte müssen eine CE-Kennzeichnung tragen
Zu viele Diskussionen haben zu der Anforderung geführt, dass nur die klinischen Daten CE-gekennzeichneter Medizinprodukte überhaupt bei der klinischen Bewertung verwendet werden dürfen.
Bei der Prüfung der technischen Dokumentation bestehen die Benannten Stellen meist nicht mehr auf dieser Forderung und akzeptieren beispielsweise auch Produkte als Äquivalenzprodukte, die in den USA zugelassen sind.
c) MDCG 2020-5 „Clinical Evaluation – Equivalence“
Das MDCG-Dokument wendet sich eigentlich an Benannte Stellen. Weil es aber die Anforderungen der MDR und der MEDDEV 2.7/1 erläutert, sollten auch Hersteller diese MDCG-Leitlinie studieren.
Die MDCG-Leitlinie diskutiert Unterschiede zwischen MDR und MEDDEV 2.7/1
Die Anforderungen der MDR und der MEDDEV sind nicht deckungsgleich. Teilweise gehen die Anforderungen der MEDDEV über die der MDR hinaus, teilweise enthält die MDR Anforderungen, auf die die MEDDEV nicht eingeht.

Beispielsweise fordert die MDR, bei der technischen Gleichartigkeit der Produkte die Software-Algorithmen zu berücksichtigen. Darauf geht das MEDDEV-Dokument nicht ein. Die Leitlinie MDCG-2020-5 betont nochmals die Relevanz dieser Software-Algorithmen, es sei denn, die Software verfolgt keinen medizinischen Zweck sondern steuert beispielsweise nur die Hardware an.
Laut MDCG 2020-5 bezieht sich die Forderung nach Äquivalenz der Software-Algorithmen nicht auf die Gleichartigkeit des Codes selbst. Es geht „nur“ um das „functional principle”, um die “clinical performance(s)“ und den „intended purpose(s)“ des Software-Algorithmus.
Allerdings besteht die Leitlinie darauf, dass der Code nach internationalen Standards wie der IEC 62304 entwickelt wurde. Ob sie den Code des eigenen Produkts oder des Äquivalenzprodukts meint, bleibt unklar. Beim eigenen wäre dies regulatorisch eh festgeschrieben.
MDCG 2020-5 warnt vor Verwechslung von „ähnlich“ mit „äquivalent“
Viele Hersteller verwechseln äquivalente und ähnliche Produkte, wenn sie die klinische Bewertung erstellen. Genau davor warnt die Leitlinie MDCG 2020-5. Als ähnliche Produkte sieht die MDCG:
“The term ‘similar devices’ may be understood as devices belonging to the same generic device group. The MDR defines this as a set of devices having the same or similar intended purposes or a commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics.”
Quelle: MDCG 2020-5, Kapitel 5
Da ähnliche Produkte nicht anhand von spezifischen Merkmalen verglichen werden, dürfen Hersteller sie nicht für die Äquivalenzbetrachtung heranziehen.
Jedoch sind die Daten von ähnlichen Produkten für eine Vielzahl anderer Zwecke nützlich. Das Kapitel „Tipps“ gibt dazu weitere Hinweise.
MDCG 2020-5 konkretisiert Anforderungen an den wissenschaftlichen Nachweis
Das MDCG 2020-5-Dokument wünscht sich zum einen, dass die Hersteller die Vergleiche der technischen, biologischen und klinischen Äquivalenz tabellarisch dokumentieren. Dazu stellt die Leitlinie Tabellen zur Verfügung, die die Vergleichsattribute enthalten (s. Abb. 4). Das zweite Kapitel stellt diese Attribute (Merkmale) vor.
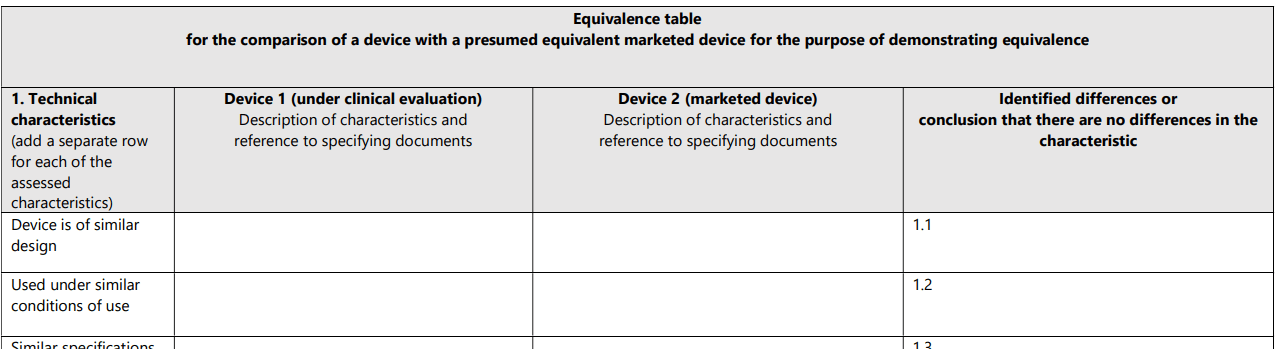
Für jedes Merkmal erwartet die MDCG 2020-05 einen Beleg. Dieser Beleg muss auf validen wissenschaftlichen Daten beruhen, z.B.:
- Klinische Daten aus der Literatur
- Publizierte wissenschaftliche Literatur von Tierstudien
- Präklinische Daten aus der technischen Dokumentation des Herstellers, z.B.
- Spezifikationen,
- Testergebnisse,
- chemisch/physikalisch/biologische Analysen,
- Daten aus präklinischen Untersuchungen etc.
Die präklinischen sowie klinischen Daten des Äquivalenzprodukts müssen sich auf eine bestimmte Generation oder Version des Produkts beziehen.
2. Nachweis der Äquivalenz konform MDR, MEDDEV 2.7/1 und MDCG 2020-5
a) Klinische Äquivalenz
Die MDR legt Kriterien für die klinische Äquivalenz fest:
“The device is used for the same clinical condition or purpose, including similar severity and stage of disease, at the same site in the body, in a similar population, including as regards age, anatomy and physiology; has the same kind of user; has similar relevant critical performance in view of the expected clinical effect for a specific intended purpose.”
Quelle: MDR Annex XIV Part A (3)
Das heißt, dass Ihr eigenes Produkt und das Äquivalenzprodukt
- unter den gleichen klinischen Bedingungen zum gleichen medizinischen Zweck,
- für die gleichen Patienten (inklusive Stadium der Erkrankung, Anatomie und Physiologie) und
- von den gleichen Anwendern benutzt werden müssen.
Andernfalls dürfen Sie die „Äquivalenzroute“ für die klinische Bewertung nicht verwenden.
Es empfiehlt sich, diese Attribute tabellarisch zu vergleichen:
|
Klinische Eigenschaften |
Eigenes Produkt |
Äquivalenzprodukt |
Bewertung |
|
Klinische Bedingung oder Zweck |
|
|
Gleich |
|
Patientenpopulation: Alter |
Ab 18 |
Erwachsene |
Gleich |
|
Patientenpopulation: Erkrankung (Schweregrad, Stadium) |
[..] |
[..] |
[..] |
|
Anwender |
Medizinisches Fachpersonal |
Ärzte, Intensivpflegekräfte |
Gleich |
|
Leistung im Hinblick auf die erwartete klinische Wirkung |
[..] |
[..] |
Ähnlich |
Gleichartigkeit der Anwender
Laut MDCG-5-Dokument ist unter einem Anwender jeder Angehörige eines Gesundheitsberufs oder Laie zu verstehen, der ein Produkt verwendet.
Ein Laie kann auch eine Person sein, die nicht über eine formale Ausbildung in einem relevanten Bereich des Gesundheitswesens oder einer medizinischen Disziplin verfügt.
Sie müssen daher berücksichtigen, ob die beabsichtigte Kompetenz oder das Wissen des Anwenders eine Auswirkung auf die Sicherheit, die klinische Leistung und das gewünschte klinische Ergebnis haben könnte.
Klinische Bedingungen, klinischer Zweck, Patientenpopulation
Das Äquivalenzprodukt soll unter den gleichen klinischen Bedingungen oder dem gleichen klinischen Zweck einschließlich ähnlichem Schweregrad und ähnlichem Krankheitsstadium verwendet werden.
Die MDR verlangt nicht explizit die gleiche Patientenpopulation. Sie brauchen aber eine wissenschaftliche Begründung, warum relevante Aspekte keine signifikante Rolle für die klinische Leistung, die Sicherheit und den Nutzen des Produkts spielen.
Hier sollten Sie folgende Aspekte in der klinischen Bewertung betrachten:
- Alter
- Geschlecht
- Physiologie
- Anatomie
- Andere relevante Aspekte
Jedoch sollte die Anwendung an der gleichen Körperstelle erfolgen.
Zusätzlich muss das Äquivalenzprodukt eine ähnliche, maßgebliche und entscheidende Leistung im Hinblick auf die erwartete klinische Wirkung für eine spezielle Zweckbestimmung haben.
b) Biologische Äquivalenz
Die MDR legt die Kriterien für die biologische Äquivalenz fest:
“The device uses the same materials or substances in contact with the same human tissues or body fluids for a similar kind and duration of contact and similar release characteristics of substances, including degradation products and leachables;”
Quelle: MDR Annex XIV Part A (3)
Das bedeutet, dass die Äquivalenzroute nur möglich ist, falls Ihr Produkt und das Äquivalenzprodukt aus den gleichen Materialien oder Stoffen bestehen für den Kontakt mit dem gleichen menschlichen Gewebe oder der gleichen Körperflüssigkeit. Andernfalls ist spätestens hier Schluss.
Auch hier empfehlen sich Tabellen, um die Attribute zu vergleichen:
|
Biologische Eigenschaften |
Eigenes Produkt |
Äquivalenzprodukt |
Bewertung |
|
Materialien und Stoffe |
A, B und C |
B, C und D |
Nicht gleich (Ende) |
|
Kontakt mit menschlichem Gewebe oder Körperflüssigkeiten |
A und B, nicht C |
C |
Nicht gleich |
|
Art und Dauer des Kontakts |
2h, direkter Hautkontakt |
Undefiniert, direkter Hautkontakt |
Ähnlich |
|
Abgabeverhalten der Stoffe, Abbauprodukte, Leachables |
[…] |
[…] |
[…] |
Freisetzungeigenschaften
Die Unterscheidung zwischen gleichen Materialien oder Stoffen und ähnlichen Freisetzungseigenschaften von Stoffen wird nur gemacht, weil die Verarbeitung, das Design und die Nutzungsumgebung Einfluss auf die Freisetzungseigenschaften des Materials haben können, selbst wenn die Rohstoffe gleich sind.
Die Verarbeitung kann Materialien auch anfälliger für Abbau machen. Zum Beispiel können kleine Änderungen des pH-Werts oder oxidativer Stress die Freisetzung erhöhen oder verringern. Aus diesem Grund sollte immer das Endprodukt getestet werden.
Stoffzusammensetzungen
Noch anspruchsvoller wird der Äquivalenzvergleich bei Produkten, die aus Stoffen oder Stoffzusammensetzungen bestehen oder sogar Arzneistoffe enthalten.
Mit „Stoffen“ und „Stoffzusammensetzungen“ sind auch alle damit verbundenen Hilfsstoffe und Beschichtungen gemeint. Hier müssen Hersteller weitere Merkmale für die klinische Bewertung überprüfen, z.B.:
- Absorption im Körper
- Verteilung im Körper
- Metabolismus
- Ausscheidung
- Lokale Toleranz
- Toxizität
- Interaktion mit anderen Geräten, Arzneimitteln oder anderen Substanzen und das Potenzial für unerwünschte Reaktionen
- Nützlichkeit der Substanz
Die MDCG-Leitlinie verweist auf die Normen der ISO-10993-Serie.
Lesen Sie hier mehr zum Thema Biokompatibilität und ISO 10993.
Die MEDDEV 2.7/1 erlaubt den Hersteller Ausnahmen in dem Fall, dass das eigene und das Vergleichsprodukt nicht die gleichen Stoffe bzw. Materialien verwenden. Die Leitlinie MDCG 2020-05 weist darauf hin, dass diese Ausnahmen unter der MDR nicht mehr zulässig sind.
c) Technische Äquivalenz
Laut MDR muss die technische Gleichartigkeit nur ähnlich sein, aber hier gibt es Einschränkungen.
“The device is of similar design; is used under similar conditions of use; has similar specifications and properties including physicochemical properties such as intensity of energy, tensile strength, viscosity, surface characteristics, wavelength and software algorithms; uses similar deployment methods, where relevant; has similar principles of operation and critical performance requirements;”
Quelle: MDR Annex XIV Part A (3)
Eine Vergleichstabelle sollte für die beiden Produkte beispielsweise die folgenden Attribute gegenüberstellen:
|
Technische Eigenschaften |
Eigenes Produkt |
Äquivalenzprodukt |
Bewertung |
|
Bauart |
|
|
|
|
Anwendungsbedingungen |
|
|
|
|
Spezifikation und physikalisch-chemische Eigenschaften |
|
|
|
|
Entwicklungsmethoden |
|
|
|
|
Funktionsprinzipien |
|
|
|
Anwendungsbedingungen
Die Anwendungsbedingungen müssen in dem Maße ähnlich sein, dass es keine klinisch signifikanten Unterschiede in der Sicherheit und der klinischen Leistung zwischen Ihrem und dem Äquivalenzprodukt gibt. Zu den Anwendungsbedingungen zählt u.a. die physikalische Umgebung (z.B. Helligkeit, Feuchtigkeit, Erschütterungen, Verschmutzungen, Temperatur und Druck).
Spezifikationen und physikalisch-chemischen Eigenschaften
Bei den Spezifikationen und physikalisch-chemischen Eigenschaften würden die Hersteller u.a. die folgenden Punkte in Betracht ziehen:
- Art und Intensität von Energie
- Zugfestigkeit
- Viskosität
- Oberflächeneigenschaften
- Wellenlänge
- Oberflächentextur
- Porosität
- Partikelgröße
- Nanotechnologie
- Spezifische Masse
- Atomare Einschlüsse wie Nitrocarburieren, Oxidierbarkeit
Zu den Funktionsprinzipien zählt die Software. Darauf geht bereits das Kapitel 1.c ein.
Die technische Äquivalenz muss in vielen Merkmalen nur ähnlich sein. Sie müssen jedoch diese Unterschiede wissenschaftlich begründen.
3. Tipps
a) Fehlende Daten ergänzen
Hersteller können nur selten auf die technische Dokumentation der Konkurrenzprodukte zugreifen. Falls Daten fehlen, um die Äquivalenz bezüglich einiger Attribute nachzuweisen, kann man diese über vergleichende Tests generieren.
Falls diese Tests ebenfalls nicht zu ausreichender Evidenz bzw. Konformität führen, wird eine klinische Prüfung wahrscheinlicher. Hersteller sollten dann genau festlegen, welche Nachweise sie damit führen wollen. Entsprechend sollten sie die Endpunkte der klinischen Prüfungen bestimmen und die Studienprotokolle gestalten.
b) Daten von ähnlichen Produkten dennoch nutzen
Hersteller dürfen die (klinischen) Daten von Produkten, die nur ähnlich, aber nicht äquivalent sind, nicht zum Nachweis der Sicherheit, der Leistungsfähigkeit und der klinischen Wirksamkeit nutzen. Dennoch können diese Daten für eine Vielzahl anderer Zwecke nützlich sein:
- Sicherstellen, dass das Risikomanagementsystem umfassend ist
- Den Stand der Technik in der klinischen Bewertung verstehen
- Als Hilfe dienen bei der Definition des Scopes der klinischen Bewertung durch Identifizierung von Designmerkmalen in ähnlichen Produkten, die besondere Bedenken aufwerfen
- Als Input für die Planung dienen von
- klinischen Prüfungen,
- der klinischen Nachbeobachtung (Post-Market Clinical Follow-Up) und
- des Überwachungssystems (Post-Market Surveillance)
4. Zusammenfassung
a) Vorteile
Die Leitlinie MDCG 2020-5 wendet sich an Benannte Stellen. Medizinproduktehersteller sind gut beraten, dieses Dokument auch zu studieren:
- Es hilft, sich auf die Argumentationslinien der Auditoren vorzubereiten.
- Die Tabellen helfen bei dem systematischen Nachweis von technischer, biologischer und klinischer Äquivalenz.
- Das Dokument interpretiert die Unterschiede zwischen der MDR und der Leitlinie MEDDEV 2.7/1.
- Es warnt vor der häufig vorkommenden Verwechslung von „ähnlichen Produkten“ mit „äquivalenten Produkten“.
b) Qualität und Konsistenz der regulatorischen Vorgaben
Dennoch stimmt es nachdenklich, dass es eines Dokuments wie der MDCG 2020-5 bedarf:
- Die gesetzlichen Anforderungen sind offensichtlich nicht ausreichend vollständig oder/und verständlich.
- Zwei von der EU herausgegebene Dokumente sind nicht ausreichend aufeinander abgestimmt, sodass es weiterer Erläuterungen bedarf.
- Eine systematische und regelmäßige Pflege z.B. der MEDDEV 2.7/1 findet nicht statt, weshalb Inkonsistenzen und Widersprüche nicht beseitigt werden.
- Hersteller müssen nun mindestens drei Dokumente lesen.
- Zwei dieser drei Dokumente haben keinen gesetzlichen Charakter. Über deren Verbindlichkeit gibt es unterschiedliche Ansichten.
- Welchen Qualitätssicherungsprozessen die MDCG-Dokumente unterworfen sind, bleibt intransparent. Jedes Gesetz muss Anhörungsverfahren durchlaufen und eine Abstimmung in einem demokratischen Prozess bestehen; die MDCG-Leitlinien offensichtlich nicht.
c) Fazit
Dass die Anforderungen an die Äquivalenz von Produkten und damit an die klinische Bewertung insgesamt hoch sein müssen, leuchtet ein. Schließlich müssen Hersteller stichhaltig beweisen, dass ihre Produkte sicher, leistungsfähig und für die Patienten wirklich nützlich sind.
Aber unverhältnismäßig hohe Anforderungen verhindern auch nützliche Produkte und bedeuten für Hersteller vermeidbare Aufwände. Diese Problematik hat die MDCG mit dem Dokument 2020-5 eher verschärft.
Das Team der klinischen Bewerterinnen und Bewerter am Johner Institut hat zahllose klinische Bewertungen konform mit den Anforderungen der MDR, MEDDEV 2.7/1 und MDCG 2020-5 erstellt und sicher durch die Prüfungen durch Benannte Stellen begleitet.
Nehmen Sie gerne per E-Mail oder Formular Kontakt auf, um Ihre klinische Bewertung zu prüfen, zu erstellen oder zu verbessern.
Möchten Sie mehr zur klinischen Bewertung erfahren? Dann melden Sie sich gleich hier zum Bootcamp an!



Hallo Fr. Martin,
Viele Medizinprodukte werden hauptsächlich weiter entwickelt. Funktionalität bleibt teilweise unverändert bestehen..
Kann für die, unveränderte Funktionalität das Äquivalenz-Argument herangezogen werden?
z.B. Das neue Gerät verhält sich bei der Erfassung der Daten wie in der Vorgängerversion – Auch das ist Äquivalenz?
Viele Grüße
Sehr geehrter Herr Helbrecht,
vielen Dank für ihre spannende Frage.
Solange die klinische Gleichartigkeit gegeben ist wie beispielsweise Zweckbestimmung, Indikation, Kontraindikation, etc. sehe ich hier kein Problem eine Äquivalenz in Betracht zu ziehen.
Ich gehe davon aus, dass Sie hier von einem Gerät ohne einen direkten Körperkontakt mit dem Patienten oder Anwender sprechen. Des Weiteren sind Unterschiede zum Vorgängerprodukt offen zu legen und wissenschaftlich zu begründen. Die klinische Leistung und Sicherheit sollte nicht signifikant unterscheiden.
Sehr geehrte Frau Dr. Martin,
vielen Dank für Ihren Artikel zur Äquivalenzbetrachtung bei der klinischen Bewertung.
Ich habe eine Frage zur Technischen Äquivalenz. Müssen die Wesentlichen Leistungsmerkmale (DIN EN 60601-1:2022-11) zwischen eigenem Produkt und Äquivalenzprodukt auch gleich sein?
Ich freue mich auf Ihre Rückmeldung und verbleibe mit freundlichen Grüße,
Andreas Denk
Lieber Herr Denk,
in technischen Aspekten müssen die Produkte „nur“ ähnlich sein. Damit ist gemeint: Sie haben ähnliche Spezifikationen und Eigenschaften einschließlich physikalisch-chemischer Eigenschaften wie Energieintensität, Zugfestigkeit, Viskosität, Oberflächenbeschaffenheit, Wellenlänge und Software-Algorithmen. Sie können aber immer eine eine wissenschaftliche Begründung liefern, warum es keinen klinisch signifikanten Unterschied in der Sicherheit und der klinischen Leistung des Produkts geben würde, ODER eine Beschreibung der Auswirkungen auf die Sicherheit und/oder die klinische Leistung, falls die Produkte sich hier unterscheiden.
Sehr geehrte Frau Dr. Martin,
vielen herzlichen Dank für Ihre Rückmeldung.
Mit freundlichen Grüßen,
Andreas Denk