
Die EN ISO 17664-1:2021 trägt den Titel „Aufbereitung von Produkten für die Gesundheitsfürsorge – Vom Medizinprodukt-Hersteller bereitzustellende Informationen für die Aufbereitung von Medizinprodukten“.
1. Einführung in die Welt der ISO 17664
a) Begriffsdefinitionen
Definition „Aufbereitung“
Im Kontext der ISO 17664-1 lässt sich der Begriff der Aufbereitung wie folgt definieren:
Vergleichbar ist die Definition der MDR:
Definition „Reinigung“
Die Norm definiert auch die Begriffe Reinigung, Desinfektion und Sterilisation:
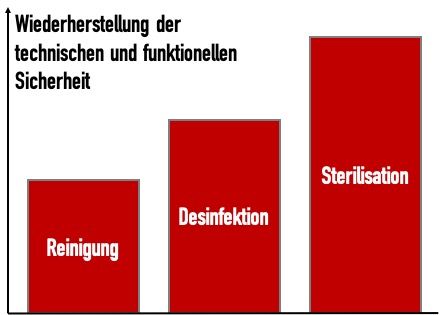
Die Norm merkt an, dass die Reinigung „das Entfernen von anhaftenden Verschmutzungen (z.B. Blut, Proteine und anderen Verunreinigungen) üblicherweise mit Reinigungsmittel und Wasser von den Oberflächen, Spalten, Rillen, Verbindungsstücken und Lumen eines Medizinprodukts durch ein manuelles oder maschinelles Verfahren umfasst, welches die Produkte für eine sichere Handhabung und/oder eine weitere Aufbereitung vorbereitet.“
Definition „Desinfektion“
Zu dieser Definition ergänzt die Norm keine Anmerkung.
Definition „Sterilisation“
Hier ergänzt die ISO 17664-1 die Definition mit einer Anmerkung: „Bei einem Sterilisationsverfahren verläuft die Inaktivierung von Mikroorganismen exponentiell. Deshalb kann das Überleben eines Mikroorganismus auf irgendeinem Einzelgegenstand als Wahrscheinlichkeit ausgedrückt werden. Obgleich diese Wahrscheinlichkeit auf eine sehr kleine Zahl verringert werden kann, kann sie niemals auf Null reduziert werden.“
Zusammenfassung
Damit wird deutlich, wie die verschiedenen Verfahren der Aufbereitung zu unterschiedlichen „Reinheitsgraden“ führen (Abb. 1).
Bei den Mikroorganismen unterscheidet man:
- Bakterien z.B. Coliforme, Staphylokokken, Pseudomonaden
- Pilze und Hefen
- Parasiten, z.B. Würmer, Amöben, Lamblien
- unbehüllte Viren (Hepatitis A, Rotaviren, Adenoviren, Norovirus) und behüllte Viren (HIV, Influenza, FSME, Corona)
Hinweis: Die MDR definiert den Begriff „Aufbereitung“ breiter als die ISO 17664-1. Sie kennt zudem die „Neuaufbereitung“, die aber nicht notwendigerweise ein Sonderfall einer Aufbereitung gemäß ISO 17664-1 sein muss.
b) Beispiele für Produkte, die aufbereitet werden müssen
Viele Einmalprodukte wie Kanülen, OP-Handschuhe und Pflaster müssen vor ihrer Verwendung aufbereitet, konkret: sterilisiert werden. Diese Aufbereitung führen meist die Hersteller oder deren Dienstleister durch.
Aber auch wiederverwendbare Medizinprodukte wie Bettpfannen, OP-Mäntel, chirurgische Instrumente (z.B. Skalpelle) und Medizingeräte (z.B. Beatmungsgeräte) müssen vor ihrer erneuten Verwendung gereinigt, ggf. desinfiziert oder sogar sterilisiert werden. Das gilt auch für Endoskope, Patientenlagerungen und Handstücke für Dentalinstrumente
Beachten Sie, dass viele in diesem Kapitel genannten Produkte nicht in den Anwendungsbereich der ISO 17664 fallen.
Diese (Wieder-)Aufbereitung erfolgt meist durch die Gesundheitsdienstleister (z.B. Krankenhäuser) oder deren Betreiber.

c) Anwendungsbereich der ISO 17664-1
Die ISO 176641- fühlt sich allerdings nicht für alle Medizinprodukte zuständig. Sie gilt nicht für:
- Nicht-kritische Medizinprodukte
- Textilien (z.B. OP-Bekleidung)
- Medizinprodukte zum Einmalgebrauch, die steril bereitgestellt werden (z.B. Kanülen)
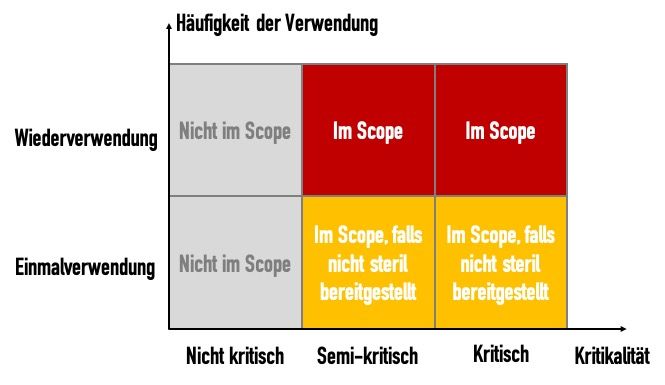
Im informativen Anhang C definiert die Norm die Kritikalitäten:
| Kritikalität der Produkte | Definition: Produkte… | Beispiel |
|---|---|---|
| Nicht kritisch | … kommen nur mit intakter Haut in Kontakt oder sind Produkte, die nicht für den direkten Patientenkontakt vorgesehen sind. | Blutdruckmanschetten, Bettpfannen, Krücken und Umgebungsoberflächen |
| Semikritisch | … kommen mit Schleimhäuten oder nichtintakter Haut in Kontakt. | Anästhesiesysteme, Beatmungsgeräte |
| Kritisch | … dringen für gewöhnlich in sterile Teile des menschlichen Körpers ein. | Chirurgische Instrumente, Implantate, invasive Medizinprodukte |
Für Einmalprodukte, die erst nach der Bereitstellung aufbereitet, d.h. gereinigt, desinfiziert oder gar sterilisiert werden, fühlt sich die Norm zuständig.
Die ISO 17664 war vor der Version aus dem Jahr 2017 auf den Sterilisationsprozess und re-sterilisierbare Medizinprodukte beschränkt. Jetzt umfasst sie auch Medizinprodukte, die gereinigt und desinfiziert werden.
d) Adressaten der Norm
Bereits der Titel der Norm („[…] Vom Medizinprodukt-Hersteller bereitzustellende Informationen […]“) macht die Adressaten klar: die Hersteller. Sie wendet sich somit nicht direkt an die Gesundheitseinrichtungen bzw. Anwender. Diese sind vielmehr Adressaten der Informationen der Hersteller.
e) Zielsetzung der ISO 17664 Reihe
Dass die ISO 17664 die Sicherheit der Patienten im Blick hat, überrascht nicht. Sie möchte diese Sicherheit aber nicht nur dadurch zu erreichen helfen, dass infektiöse Agenzien minimiert werden.
Vielmehr möchte sie auch andere nachteilige Auswirkungen durch die Aufbereitung auf das Medizinprodukt reduzieren. Beispiele dafür wären eine Reduzierung der Lebensdauer oder ein Verlust anderer grundlegender Sicherheits- und Leistungsanforderungen durch die Aufbereitung.
2. Regulatorische Anforderungen an die Aufbereitung
a) EU-Medizinprodukteverordnung MDR
Artikel 17 („Einmalprodukte und ihre Aufbereitung“)
Der Artikel 17 adressiert im Wesentlichen die „Aufbereitung und Weiterverwendung von Einmalprodukten“. Er stellt keine Anforderung an die initiale Aufbereitung von Einmalprodukten.
Der Artikel 17 definiert alle natürlichen und juristischen Personen, die Einmalprodukte für die weitere Verwendung aufbereiten, als Hersteller. Entsprechend gelten für diese Personen alle Herstellerpflichten.
Anhang I, Absatz 23.4 („Angaben in der Gebrauchsanweisung“)
Im Anhang I mit den grundlegenden Sicherheits- und Leistungsanforderungen finden sich die Anforderungen an die Gebrauchsanweisung. Diese muss folgende Angaben enthalten:
bei wiederverwendbaren Produkten Angaben über geeignete Aufbereitungsverfahren, z. B. zur Reinigung, Desinfektion, Verpackung und gegebenenfalls über das validierte Verfahren zur erneuten Sterilisation entsprechend dem/den Mitgliedstaat(en), in dem/denen das Produkt in Verkehr gebracht worden ist. Es ist deutlich zu machen, woran zu erkennen ist, dass das Produkt nicht mehr wiederverwendet werden sollte, z.B. Anzeichen von Materialabnutzung oder die Höchstzahl erlaubter Wiederverwendungen;
MDR, Anhang I, Kapitel III, 23.4 n)
Die MDR verpflichtet somit Hersteller, genau zu beschreiben, wie die Anwender
- die Produkte aufbereiten sollen und
- erkennen können, ob die Produkte für eine weitere Aufbereitung noch geeignet sind.
Damit verbunden ist auch die nächste Anforderung:
falls es sich um ein aufbereitetes Produkt zum Einmalgebrauch handelt, einen Hinweis auf diesen Sachverhalt, die Anzahl der bereits durchlaufenen Aufbereitungszyklen und mögliche Beschränkungen hinsichtlich der Anzahl der Aufbereitungszyklen;
MDR, Anhang I, Kapitel III, 23.4 o)
b) EU-Medizinprodukterichtlinie MDD
Die Anforderungen der MDD waren sehr vergleichbar. Demnach muss die Gebrauchsanweisung umfassen:
h) bei wiederzuverwendenden Produkten Angaben über geeignete Aufbereitungsverfahren, z. B. Reinigung, Desinfektion, Verpackung und gegebenenfalls Sterilisationsverfahren, wenn eine erneute Sterilisation erforderlich ist, sowie Angaben zu einer eventuellen zahlenmäßigen Beschränkung der Wiederverwendungen;
MDD, Anhang 1, Absatz 13.6
bei der Lieferung von Produkten, die vor der Anwendung zu sterilisieren sind, müssen die Angaben zur Reinigung und Sterilisation sicherstellen, daß das Produkt bei ihrer ordnungsgemäßen Befolgung die Anforderungen des Abschnitts I nach wie vor erfüllt;
i) Hinweise auf eine möglicherweise vor der Anwendung eines Produkts erforderliche besondere Behandlung oder zusätzliche Aufbereitung (z. B. Sterilisation, Montage usw.);
Der Anhang ZA bestätigt, dass die Konformität mit „13.6 h, nur erster und zweiter Abschnitt“ sowie mit 13.6 i durch Einhaltung der EN ISO 17664 vermutet werden darf.
c) ISO 10993-1
Nicht nur die europäische Gesetzgebung, sondern auch Normen stehen im Bezug zur ISO 17664. So finden sich in der ISO 10993-1 mehrere entsprechende Stellen, die die Hersteller verpflichten, den Einfluss der Aufbereitung zu überprüfen:
- 7 „The biological safety of a medical device shall be evaluated by the manufacturer over the whole life-cycle of a medical device.“
- 4.5.1 „Typical changes that could alter the biological performance of a material or final medical device include, but are not limited to: processing e.g. sterilization, cleaning, surface treatment, welding, injection moulding, machining, primary packaging;“
- 8 „For re-usable medical devices, biological safety shall be evaluated for the maximum number of validated processing cycles by the manufacturer.“
Lesen Sie hier mehr zum Thema Biokompatibilität und ISO 10993.
3. Die Norm ISO 17664
a) Aufbau der Norm
Die DIN EN ISO 17664-1:2021 ist mit 34 Seiten relativ schlank. Das vierte und fünfte Kapitel umfassen zusammen nur eine einzige Seite. Das sechste Kapitel ist acht Seiten lang, das siebte nicht einmal eine Viertelseite.
Sehr hilfreich sind die informativen Anhänge, die 11 Seiten umfassen.
b) Anforderungen
Die ISO 17664-1 verpflichtet die Hersteller zu Folgendem:
- Validierung der Aufbereitung
Die Hersteller müssen alle Verfahren zur Aufbereitung des Medizinprodukts validieren. Darüber, wie die Hersteller diese Validierung durchführen sollen, lässt sich die Norm nicht aus. - Risikoanalyse der Aufbereitung konform ISO 14971
Die Hersteller müssen eine Risikoanalyse konform der ISO 14971 durchführen. Bei dieser Risikoanalyse müssen die Hersteller alle typischen Schritte einer Aufbereitung (s.u.) betrachten. Aus dieser Analyse müssen sie die bereitzustellenden Informationen bestimmen. - Bereitzustellende Informationen
Die bereitzustellenden Informationen müssen für alle Aufbereitungsschritte die im sechsten Kapitel jeweils vorgeschriebenen Elemente umfassen.
Eine typische Aufbereitung besteht aus mehreren Schritten (Abb. 5).

Die ISO 17664-1 fordert beispielsweise, bei der manuellen Reinigung unter anderem die folgenden Informationen zu liefern:
- Schritt-für-Schritt-Anweisung der Reinigung
- Zu verwendende Chemikalien und deren Konzentration
- Kontaktzeit der Chemikalien
- Notwendige Wasserqualität
- Art des Spülens
- Sonstige Prozessparameter
c) ISO 17664-2: Neuer zweiter Teil für nicht kritische Produkte
Ein zweiter Teil der Norm die ISO 17664-2:2021 trägt den Titel „Aufbereitung von Produkten für die Gesundheitsfürsorge – Vom Medizinprodukt-Hersteller bereitzustellende Informationen für die Aufbereitung von Medizinprodukten – Teil 2: Nicht kritische Medizinprodukte“.
Dieser Teil soll die Lücke mit den nicht-kritischen Medizinprodukten schließen (Abb. 3). Er befasst sich sowohl mit Medizinprodukten für die Einmalverwendung („single use“) als auch für die Wiederverwendung („reuse“).
Beispiele für solche Produkte sind Anästhesiegeräte, Teile vom Ultraschallgerät, User Interfaces, Blutdruckgeräte, EKG-Kabel, Handgriffe von OP-Leuchten.
Die ISO 17664-2 beinhaltet Informationen zur wirksamen Aufbereitung der nicht-kritischen Produkte ohne Sterilisation. Sie hilft mit einem Flowchart bei der Zuordnung, ob ein Produkt unter den Anwendungsbereich der ISO 17664-1 oder den der ISO 17664-2 fällt.
4. Acht Praxistipps zur Umsetzung der ISO 17664
Tipp 1: Aufbereitung bereits beim Produkt-Design bedenken
Bereits bei der Entwicklung sollten die Hersteller die Aufbereitung im Blick haben. Sie sollten insbesondere auf diese Aspekte achten:
- Wahl der Geometrie
Für die Reinigung, Desinfektion und Sterilisation müssen Bereiche vermieden werden, die nur schwer zu erreichen sind. Darunter fallen beispielsweise Hohlräume (Sacklumen, dünne Lumen), offene Mechaniken und Übergänge (Kanten, Nähte). - Anforderungen, die sich durch die Aufbereitung ergeben
Sind Geometrien nicht passiv für Reinigungs- und Desinfektionsmittel erreichbar, muss aktiv gespült werden (deshalb z.B. Berücksichtigung von Spülanschlüssen beim Design). - Wahl der Materialien
Die richtige Wahl der Materialien ist gleich im doppelten Sinn entscheidend: Zum einen müssen sie leicht aufzubereiten sein; zum anderen müssen sie in der Lage sein, viele Aufbereitungszyklen zu überstehen, ohne die mechanische oder biologische Sicherheit zu gefährden. Beispielsweise sind Materialien, die porös werden, schwer zu reinigen, zu desinfizieren oder zu sterilisieren. - Wahl des Aufbereitungsverfahrens
Viele Entwickler entwerfen das Produkt, um dessen Zweckbestimmung und damit Nutzen zu optimieren. Die Wahl des Aufbereitungsverfahrens rückt in den Hintergrund. Generell ist eine maschinelle Aufbereitung immer zu bevorzugen. Eine maschinelle Aufbereitung kann Anforderungen an das Produktdesign stellen.
Tipp 2: Risiken für Biokompatibilität im doppelten Sinn beachten
Viele Hersteller analysieren „nur“ die Risiken, die sich dadurch ergeben, dass die Anwender einen Aufbereitungsschritt nicht spezifikationsgemäß durchführen und daher die Anforderungen an die Reinheit nicht erfüllt sind.
Die Hersteller sollten auch die Risiken beachten, die sich durch die spezifikationsgemäße Aufbereitung ergeben. So sollten sie Reinigungs- und Desinfektionsmittel auf kritische Inhaltsstoffe prüfen.
Spezielle Tests ermöglichen es, die Biokompatibilität von Reinigungs- und Desinfektionsmitteln ausreichend zu belegen.
Leider stehen nicht alle Inhaltsstoffe immer auf dem Sicherheitsdatenblatt ( Anhang D der DIN EN ISO 17664-1:2021).
Die Benannten Stellen fragen verstärkt, wie sichergestellt ist, dass keine Rückstände aus der Produktion (z.B. von Prozesschemikalien) auf dem Produkt vorhanden sind. Wären die Prozesschemikalien definierter und kontrollierter, wäre das Problem geringer oder nicht existent.
Tipp 3: Vollständigkeit des Design-Outputs prüfen
Als „Design-Output“ sollten u.a. vorliegen:
- Festlegung des Designs und der Geometrie, Konstruktionszeichnungen
- Spezifikation aller Materialien; ggf. Festlegung von Alternativen für die Einkaufsabteilung
- Genaue Spezifikation des Aufbereitungsverfahrens inklusive aller Arbeitsschritte, Prozessparameter und Reinigungsmaterialien
- Festlegung der Anzahl von Wiederaufbereitungen bzw. der Lebensdauer bzw. von Kriterien, mit denen sich die Fähigkeit des Produkts bestimmen lassen, erneut aufbereitet zu werden
- Risikomanagementakte
Tipp 4: Mit Produktfamilien statt Einzelprodukten arbeiten
Medizinproduktehersteller sollten von der Möglichkeit Gebrauch machen, nicht jedes einzelne Produkt, sondern die ganze Produktfamilie zu betrachten. Damit sparen sie redundante und damit unnötige Aufwände, z.B. beim Nachweis der Biokompatibilität.
Worst-Case-Betrachtungen haben sich bewährt. Diese beziehen sich z.B. auf:
- Risiken für Patienten durch mangelnde Aufbereitung
- Ungeeignete Prozessparameter bei der Aufbereitung
- Wahrscheinlichkeit und Art möglicher Nutzungsfehler
- Geometrie des Produkts
- Art (Keime), Ort und Grad der Verschmutzung des Produkts
Tipp 5: Geeignetes Prüflabor auswählen
Hersteller sind gut beraten, ein Prüflabor auszuwählen, das
- Erfahrungen speziell mit Medizinprodukten hat,
- risikobasiert arbeitet und nicht versucht, nur Umsätze zu maximieren,
- abschätzen kann, was für die gegebene Produktfamilie die zu prüfenden Worst-Case-Produkte sind,
- in der Lage ist, normenkonforme Prüfprotokolle zu erstellen,
- die Aufbereitung und(!) die Aufbereitungsanweisungen validieren kann und
- erfahren darin ist, die Auswirkungen einer unzureichenden Aufbereitung sowie der Reinigungs- und Desinfektionsmittel auf die Biokompatibilität zu bewerten.
Tipp 6: Typische Fehler bei der Validierung vermeiden
Die Experten des Johner Instituts beobachten bei der Validierung häufig diese Fehler:
- Ungeeignete Prüfanschmutzung
Das Prüflabor betrachtet nicht den wirklichen Worst-Case bezüglich Produkt (einer Produktfamilie), Auswahl der Keime, Ort der Anschmutzung (besonders bei kritischen Geometrien). - Unvollständige Risikoanalyse
Der Hersteller bzw. das Labor analysieren nicht die Risiken durch Reinigungs- und Desinfektionsmittel. Insbesondere sind deren Auswirkungen auf die Lebensdauer des Produkts nicht systematisch untersucht und dokumentiert. Das Gleiche gilt für Risiken durch Rückstände der Desinfektions- und Reinigungsmittel. - Fehlende Usability-Betrachtung
Die Prüflabore bewerten, ob die Aufbereitungsanweisung geeignet ist. Hingegen fehlt eine Bewertung deren Verständlichkeit (Usability) und möglicher Nutzungsfehler. - Fehlende Revalidierung
Nicht nur Änderungen im Produktdesign, sondern auch bei der Endreinigung, der Reinigung, der Desinfektion und Sterilisation erfordern eine Neubetrachtung der Biokompatibilität.
Tipp 7: Verschiedene Validierungen unterscheiden
Die Auditoren und Qualitätsmanager sollten sicherstellen und das folgende Verständnis überprüfen:
Eine validierte Endreinigung ersetzt NICHT die Notwendigkeit der Validierung der Aufbereitung. Die eingesetzten Reinigungsmittel zielen auf andere Rückstände. Rückstände aus der Produktion, die nach der Aufbereitung noch zu Problemen führen, sehen wir immer wieder bei Produkten.
Eine validierte Aufbereitung ist demzufolge kein Garant für ein „sauberes“ Medizinprodukt, das frei von Rückständen aus dem Produktionsprozess ist. Über die Lebensdauer des Produkts kann zudem eine Anreicherung von Reinigungsmitteln in schwer zugänglichen Bereichen des Produkts erfolgen.
Eine validierte Aufbereitung ersetzt NICHT die Überprüfung der Biokompatibilität. Aufgrund einer möglichen Anreicherung von Rückständen und einer möglichen Materialveränderung (Sprödigkeit, Korrosion usw.) ist es notwendig, ein „End of Life“ zu definieren. Dies geht nur unter Berücksichtigung der Biokompatibilität.
Tipp 8: Unnötige Tests vermeiden
Die Überprüfung der Biokompatibilität bedarf meist keiner Tierversuche. Bitte beachten Sie den Artikel zur Biokompatibilität, um unnötige Tests, regulatorische Probleme und Risiken für Patienten zu vermeiden.
5. Unterstützung
Das Johner Institut unterstützt Hersteller bei allen Aktivitäten, nicht nur im Kontext der ISO 17664:
- Bewertung des Produktdesigns (Geometrie, Materialien)
- Festlegung der Lebensdauer
- Spezifikation der Aufbereitung
- Formulierung und Gestaltung von Informationen zur Aufbereitung
- Formative und summative Bewertung der Gebrauchstauglichkeit
- Risikoanalyse
- Schreiben von Validierungsplänen
- Durchführung der Validierung
- Auswahl von Prüflaboren
6. Fazit
Weil die Aufbereitung nicht nur die Desinfektion und Sterilisation der Produkte umfasst, sondern auch deren Reinigung, müssen viele Hersteller die ISO 17664-Reihe und weitere Normen beachten.
Die ISO 17664-1 und -2 ist eine kompakte und gut verständliche Norm. Der Anwendungsbereich der ISO 17664-1 beschränkt sich auf semikritische und kritische Produkte. Sie schließt auch Einmalprodukte aus, die bereits steril bereitgestellt werden. Die ISO 17664-2 dehnt den „Scope“ auf nicht-kritische Medizinprodukte aus.
Um eine gesetzeskonforme Aufbereitung und damit die Sicherheit der Patienten zu gewährleisten, müssen die Hersteller den kompletten Lebenszyklus der Produkte beachten: von der Produktspezifikation über das Produkt-Design bis zur Festlegung und Feststellung des „End-of-Life“ eines Produkts.
Mit der Aufbereitung versuchen die Hersteller, Risiken durch mangelnde biologische Sicherheit zu minimieren. Gleichzeitig sollten sie die Risiken durch mangelnde Biokompatibilität im Blick behalten, die sich speziell durch die Aufbereitung ergeben.
Die kluge Wahl einer Validierungsstrategie hilft, unnötige Aufwände zu vermeiden, ohne die regulatorische Konformität und die Sicherheit von Patienten zu kompromittieren.
Änderungshistorie
- 2023-03-24: Aktualisierung der Norm: EN ISO 17664-1:2021 / ISO 17664-2:2021
- 2020-03-03: Artikel initial veröffentlicht



Sehr geehrter Herr Rudolf,
vielen Dank für die Aufarbeitung der ISO 17664. Ich habe eine Frage zum Anwendungsbereich der Norm. Wir diskutieren aktuell, in wie weit sich die Norm auch auf (semikritische) Einpatientenprodukte bezieht, die vor der Wiederverwendung am selben Patienten nicht sterilisiert werden müssen. Bei einigen dieser Produkte ist nur eine Reinigung vorgesehen (ohne Desinfektion). Dies ist für uns aus der Norm nicht wirklich ersichtlich. Was würden Sie sagen?
Vielen Dank und Grüße,
M. May
Liebe Frau May,
vielen Dank für Ihre Nachfrage. Sie sprechen hier einen wunden Punkt an, denn tatsächlich äußert sich die Norm zur Aufbereitung von Einpatientenprodukten nicht explizit.
Dies bedeutet für Sie zunächst, dass Sie, wenn Sie nicht ausschließen können in deren Geltungsbereich zu fallen, Sie die Norm voll anwenden müssen.
Zwar ist anzunehmen, dass das „Risiko der Übertragung infektiöser Agenzien“, wenn nur ein Patient Kontakt aufweist, überschaubar ist, allerdings gibt es auch hier problematische Produkte und Sonderfälle (z.B. Re-Infektion bei Trachealkanülen u.ä.).
Zu nennen wäre hier auch ein Fieberthermometer mit möglicher oraler und rektaler Messung. Hier wäre eine Desinfektion natürlich wünschenswert, auch wenn es immer der gleiche Patient sein sollte.
Eine Pauschalaussage ist somit nicht möglich und die Tendenz sollte im Zweifelsfall immer in Richtung „höchste Patientensicherheit“ gehen.
Prinzipiell gibt aus meiner Sicht die Norm es aber her, bei bestimmten Produkten das Risiko der Übertragung nach Reinigung zu argumentieren und die Desinfektion wegzulassen (mir fällt leider aber gerade kein Beispiel ein),
andererseits kann bei bestimmten Geometrien und Anwendungen trotzdem eine aktive Keimreduktion sinnvoll/nötig sein (z.B. Nasendusche).
Da die Norm hier keine expliziten Ausnahmen nennt, sollten Sie zunächst davon ausgehen, dass eine Reinigung mit Desinfektion sinnvoll und nötig ist.
Im Zweifelsfall sollten Sie dies mit Ihrer benannten Stelle im Vorfeld abklären.
Ich hoffe sehr Ihnen weitergeholfen zu haben.
Dipl.-Ing (FH) Hendrik Rudolf
Sehr geehrter Herr Rudolf,
ich frage mich ob der Anwendungsbereich der Norm auf den professionellen Sektor begrenzt ist, also davon ausgegangen werden kann, dass Reinigung bzw. Sterilisation von ausgebildeten Personal durchgeführt wird, oder ob die Norm auch für Produkte aus dem Home Healthcare Bereich anzuwenden ist.
Vielen Dank und viele Grüße
A. Thoma
Sehr geehrter Herr Thoma,
in der ISO 17664-1 und -2 wird nicht zwischen Medizinprodukt für die Klinik und dem Home-Healthcare Bereich unterschieden.
In der Definition für „Aufbereiter“ wird auch auf keine professionelle Ausbildung verwiesen.
Es gibt daher diesbezüglich keine Forderung, dass Medizinprodukte nur von Fachpersonal aufbereitet werden dürfen.
Daraus kann geschlussfolgert werden, dass die Norm auch auf Produkte aus dem Home-Healthcare Bereich anzuwenden ist.
Meines Erachtens gibt es keinen Grund warum das nicht nicht so sein sollte.
Herzliche Grüße
Sarah Gruber
Sehr geehrter Herr Rudolf,
es geht um die Definition „wiederverwendbar“ für Medizinprodukte Klasse I, z.B. eine Kniebandage.
Diese Kniebandage wird nur von einem Patienten, dem sie verordnet wurde, verwendet. Wenn der Patient diese Bandage nun reinigt (=wäscht) und wieder an sich selbst verwendet, zählt sie dann zu den wieder verwendbaren Medizinprodukten i.S. der MDR? Oder bezieht sich das Wiederverwendbar darauf, dass das MP nach Reinigung an einem ANDEREN Patienten angewendet wird?
Ich bin bisher von letzterem ausgegangen und hoffe, dass ich damit richtig liege. In einer internen Diskussion sind wir zu keinem Ergebnis gekommen.
Vielen Dank im Voraus!
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
nach der Definition in der ISO 17664 ist ein wiederverwendbares Medizinprodukt, ein Medizinprodukt, das vom Hersteller als für die Aufbereitung und Wiederverwendung geeignet bestimmt ist.
Bezüglich „Einpatientenprodukte“ äußert sich die Norm leider nicht. Dieser Punkt spielt aber bei der Einschätzung des Risikos für den Patienten eine große Rolle.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber,
vielen Dank für die Antwort, die meine Annahme leider nicht bestätigt.
Das bedeutet also, dass alle Medizinprodukte, die für einen Patienten zur mehrmaligen Verwendung vorgesehen sind, somit wiederverwendbare Medizinprodukte sind?
Das würde weiterhin bedeuten, dass der Hersteller Informationen zur Aufbereitung, wozu wohl auch die Reinigung gehört, bereitstellen muss, richtig?
Wie detailliert müssen die Informationen sein?
Sind Pflegesymbole ausreichend?
Im Zusammenhang mit der Kennzeichnung von Medizinprodukten gemäß MDR bedeutet es auch, dass das Medizinprodukt- eine Kniebandage- mit der UDI (Anhang VI, Teil C, 4.10: Wiederverwendbare Produkte tragen den UDI-Träger auf dem Produkt selbst ) direkt gekennzeichnet sein muss?
Bei einer Bandage gäbe es nur die Möglichkeit, die UDI auf das Einnähetikett zu bringen. Zählt das zur direkten Kennzeichnung des Medizinprodukts?
Vielen Dank im Voraus für Ihre Unterstützung !
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
richtig. Die Reinigungsmethode muss ja so gewählt sein, damit z.B. der bestimmungsgemäße Gebrauch, die Lebensdauer als auch die Biokompatibilität nicht negativ beeinflusst werden.
Bezüglich der Frage zur UDI, möchte ich Sie gern auf unsere Micro-Consulting Angebot verweisen, damit ein Experte für dieses Themengebiet Sie bei Ihrer Fragestellung unterstützen kann.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber,
ich habe mich für Ihre letzte Antwort noch nicht bedankt. Das hole ich hiermit nach.
Bei mir hat sich eine weitere Frage aufgetan:
In der Norm steht am Ende des Kapitels 1 Anwendungsbereich, dass die Norm nicht anzuwenden ist auf „Unkritische Produkte“. Laut Definition sind es Produkte, die nur mit intakter Haut in Kontakt kommen.
Eine Kniebandage oder medizinische Kompressionsstrümpfe , z.B., kommen nur mit intakter Haut in Kontakt. Nach meinem Verständnis ist die Norm für Medizinprodukte dieser Art nicht anwendbar.
Wie ist Ihre Meinung dazu?
Vielen Dank für Ihre hilfreiche Antwort.
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
das ist richtig.
Für unkritische Produkte beachten Sie bitte die ISO 17664-2.
Diese wird auch im Blogartikel angesprochen.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber, sehr geehrter Herr Rudolf,
viele Dank für diesen verständlichen Artikel.
Ich hätte noch eine Frage zu den Reinigungsmitteln/Chemikalien die bei der Aufbereitung zum Einsatz kommen.
Muss der Hersteller für die Validierung bzw. Aufbereitung explizite Marken/Hersteller nennen oder reicht es aus, wenn man Hinweise auf Anforderungen eines Reinigungsmittels (ph-Wert o.ä.) gibt?
Vielen Dank für Ihre Auskunft!
Antje Neumann
Sehr geehrte Frau Neumann,
vielen Dank für Ihre Frage.
Sie müssen nach der ISO 17664 die Chemikalie benennen und die Konzentration angeben.
In der Regel gibt man in der Aufbereitungsanweisung eine spezifische Beschreibung der Chemikalie mit Angabe der Wirkstoffgruppe z.B. „… auf Alkohol-Basis“ an und nennt zusätzlich explizit die Chemikalie, welche für die Validierung verwendet wurde (Handelsname, Hersteller, Artikelnummer, Einzelwirkstoffe). So lassen Sie Spielraum für die Verwendung andere vergleichbare Chemikalien und stellen gleichzeitig die Nachvollziehbarkeit bezogen auf die Validierung sicher.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Damen und Herren,
leider erschließt sich mir Kapitel 7.2 nicht ganz.
„Bei Medizinprodukten, die keine beigefügten Aufbereitungsanweisungen erfordern, müssen andere Kommunikationsmittel verwendet werden[…]“
Wieso sollen hier andere Kommunikationsmittel verwendet werden, wo doch keine Aufbereitungsanweisung benötigt werden?
Sehr geehrter Herr Gerwers,
das von Ihnen angesprochene Kapitel 7.2 bezieht sich aus unsere Sicht auf die Produkte die aus dem Anwendungsbereich der ISO 17664 ausgeschlossen sind:
– Textilien zur Verwendung für OP-Bekleidung oder Patientenabdeckungssystemen
– Medizinprodukte für den Einmalgebrauch, welche gebrauchsfertig bereitgestellt werden
Andere Kommunikationsmittel wären dann z.B. Symbole wie z.B. Wasch-und Pflegesymbole für Textilien oder Hinweise auf der (Steril-)Verpackung.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber, Sehr geehrter Herr Rudolf,
Vor kurzem bin ich aus dem Pharma- in den IVDR-Bereich gewechselt und habe die Sterilisationsvalidierung eingeführt. Häufig sehe ich mich konfrontiert mit der Fragestellung ob die entsprechenden Normen überhaupt anwendbar sind und aus Kostengründen nicht geringere Prüfumfänge gewählt werden können. Da die IVDR sowie die ISO 13485 meines Wissens keine Vorgaben zur Sterilisation des Prozessequipments geben (bei unsterilen Endprodukten), konnte ich bislang dazu keine Stellung beziehen. Können Sie mir hier weiterhelfen?
Viele Grüße
Oliver Öder
Sehr geehrter Herr Öder,
vielen Dank für Ihren Kommentar. Generell ist immer ein risikobasierter Ansatz für den spezifischen Prozess, das zu sterilisierende Produkt und die Art der Sterilisation zu wählen und entsprechend zu begründen.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber,
Wie kann die Definition von Reinigung mit einer Definition der Sauberkeit / cleanliness von Produkte in Verbindung gebracht werden, die in der (EN) ISO 13485:2016 Kap 7.5.2 gefordert ist?
Wo kann eine Definition von Sauberkeit nachgelesen werden? Bislang kenne ich die ISO 19227:2018 – cleanliness of orthopedic implants, die genauere Vorgaben beschreibt, aber nicht unbedingt für andere Produkte übertragbar ist.
Vielen Dank für Ihre Hilfe!
M. Widmann
Sehr geehrte Frau Widmann,
die Reinigung hängt natürlich von der erforderlichen Sauberkeit ab.
Die benötigte Sauberkeit ist Produktspezifisch und hängt von der Produktanwendung ab und sollte in einer Risikobewertung adressiert werden.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber,
Besten Dank – das machen wir bereits. Gibt es irgendwo konkrete, quantitative Aussagen, bei welcher Anwendung welche Sauberkeit gefordert ist – so wie es die ISO 19227:2018 für die Implantate vorgibt?
Wie ist im allgemeinen Sauberkeit definiert?
Besten Dank
Margit Widmann
Sehr geehrte Frau Widmann,
Für das Festlegen der Grenzwerte zur quantitativen Überprüfung der Reinigungswirksamkeit Im Zuge der Aufbereitungsvalidierung können Sie die ANSI/AAMI ST 98:2022 und die DIN EN ISO 15883-5:2021 heranziehen.
Die erforderliche Sauberkeit eines Medizinprodukts kann meines Erachtens nicht pauschal definiert werden, da diese von der Produktanwendung und den Gefährdungspotentialen abhängt.
Für eine weitere Produktspezifische Diskussion bezüglich Anforderungen an die Sauberkeit/Endreinigung nutzen Sie gerne unser Kontaktformular. Danke Ihnen und herzliche Grüße
Sehr geehrte Frau Gruber,
Vielen Dank für diese beiden Tipps!
M. Widmann
Sehr geehrte Frau Gruber,
wir entwickeln, produzieren und vertreiben ophthalmologische OP-Geräte der Risikoklasse 2b (aktive Geräte). Die Geräte selbst sind nach der o.g. Definition nicht kritisch, denn sie haben weder Patientenkontakt noch kommen sie mit intakter/nicht intakter Haut in Kontakt. Dies trifft nur für das Zubehör zu, welches separat zugelassen ist und die 17664 erfüllt.
Fällt die Wischdesinfektion solcher Geräte im OP unter die 17664-2?
Vielen Dank für Ihre Antwort.
Lieber Herr Müller-Albinus,
vielen Dank für Ihre spannende Frage.
Unkritische Medizinprodukte, welche nicht für eine Sterilisation vorgesehen sind aber aufbereitet werden, sind im Scope der ISO 17664-2. Beispiele für unkritische Medizinprodukte sind z.B. Benutzeroberflächen und Überwachungsgeräte. Aufgrund von wahrscheinlichen Kontaktpunkte Ihres Produktes mit dem Anwender ist eine Kreuzkontamination nicht auszuschließen was eine Aufbereitung notwendig macht und damit auch eine Validierung dieser Methode für Ihr Produkt. Ich hoffe ich konnte damit Ihre Frage beantworten.
Herzliche Grüße
Sarah Gruber
Sehr geehrte Frau Gruber,
vielen Dank für Ihre Ausführung. Nun werden bei dem Gerät Bedienflächen mit einer sterilen single-use Display-Folie abgeklebt, so dass der sterile Bereich, das sterile Bedienpersonal oder das sterile Zubehör nicht in Kontakt mit dem Gerät kommt und so m.E. keine Kreuzkontamination in keine Richtung möglich ist.
Vielen Dank.
Sehr geehrte Frau Gruber,
muss die ISO 17664 auch für IVD Laborgeräte ohne Patientenkontakt für den Gebrauch durch einen professionellen Nutzer angewendet werden? Oder dibt es für IVD Produkte weitere Standards die relevant sind? Primär geht es hier um Reinigung und Disinfektion da das Produkt keinen sterilen Status hat.
Herzlichen Dank und viele Grüße
Maria Pinkerneil
Liebe Frau Pinkerneil, die ISO 17664-2 ist bei einer Aufbereitung von Medizinprodukte mit unkritischen oder ohne direkten Patientenkontakt anwendbar. Der Fokus einer Wiederaufbereitung liegt meist auf die Vermeidung von Kreuzkontamination durch die Entfernung von Schmutz und Mikroben aus der vorangegangen klinischen Anwendung. Für den Nachweis der Effektivität einer solchen Aufbereitung ist eine Validierung notwendig. Herzliche Grüße Sarah Gruber
Sehr geehrte Frau Gruber,
als Hersteller von Edelstahlband-Zahnmatrizen, Matrizenzange und Pinzetten die wiederverwendbar sind nach Klasse I (nicht ir oder is), verkaufen wir diese Produkte an Händler und Zahnärzte. Die Zahnärzte selbst sterilsieren unsere Produkte. Reicht es aus, dass wir sagen 4 Minuten bei 134 Grad im Dampfsterilisator? Natürlich schreiben wir dazu, „muss zuvor nach Benutzung der Instrumente gereinigt werden in Reinigungsflüssigkeit, abgebürstet und dann in Steri. Gibt es eine Anleitung (verschiedene Sprachen) die man verwenden kann für den Zahnarzt. Hersteller müssen ja Aufbereitungshinweise bereit halten. Aber jeder Zahnarzt verwendet ja einen anderen Sterilisator und andere Flüssigkeiten unterschiedlicher Hersteller. Was muss ich tun, um hier auf der sicheren Seite zu sein auch bei evtl. Prüfungen der Behörde.
Vielen Dank für Ihre Hilfe. Viele Grüße Claudia
Liebe Claudia,
Als Hersteller sind Sie nach MDR verpflichtet mindestens eine validierte Aufbereitungsanweisung Ihrer wiederverwendbaren Medizinprodukte (inkl. Angabe der konkreten Prozesschemie) den Anwendern bereitzustellen.
Die Anforderungen sind in der ISO 17664-1/2 konkretisiert und die Norm gibt Ihnen Hilfestellung für die bereitzustellende Information.
Um auf der sicheren Seite zu sein, unterstützen wir Sie sehr gerne innerhalb unserer Beratungsleistung. Melden Sie sich gerne konkret mit Ihrer Anfrage.
Ich freue mich von Ihnen zu hören.
Herzliche Grüße Sarah Gruber