
Ist die STED (Summary Technical Documentation) und deren Nachfolger ToC (Table of Contents) die Lösung für Medizinproduktehersteller, die ihre Produkte international zulassen müssen?
Dieser Beitrag stellt die STED bzw. ToC und vor damit einen Vorschlag, wie Hersteller die technische Dokumentation strukturieren können.
1. STED: Eine kurze Einführung
a) Weshalb STED/ToC nützlich sein kann
Die Anforderungen der verschiedenen Rechtssysteme (z. B. Europa, USA/FDA, China, Japan, Kanada, Brasilien usw.) an die Zulassung von Medizinprodukten unterscheiden sich. Aber alle Zulassungsverfahren setzen eine technische Dokumentation voraus.
Leider unterscheiden sich sowohl die Inhalte als auch die Struktur dieser Akten, was dazu führt, dass international agierende Medizinproduktehersteller die technische Dokumentation mehrfach zusammenstellen müssen. Diese Arbeit ist wenig wertschöpfend.
b) Wie es zur STED und ToC kam
Auch deshalb haben sich Gremien aus Vertretern von Behörden und Herstellern wie die Global Harmonization Task Force (GHTF) gebildet, um Vorschläge für eine vereinheitlichte technische Dokumentation zu erarbeiten. Ein Ergebnis war die „Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices (STED)“ der GHTF (IMDRF/RPS WG/N9 FINAL:2018).
Inzwischen sind die Unterlagen nicht mehr auf den Seiten der GHTF, da die GHTF durch das International Medical Device Regulators Forum IMDRF abgelöst wurde. Diese bietet den STED unter dem Namen „Non-In Vitro Diagnostic Device
Regulatory Submission Table of Contents (nIVD ToC)“ zum Download an (inzwischen in der 4. Version von Juni 2024). Für In-vitro Diagnostika gibt es eine entsprechende Vorlage.
In den Veröffentlichungen spricht das IMDRF von „nIVD MA ToC“, also der Table of Contents für non IVDs. Für IVD gibt es eine analoge ToC.
c) Regulatorischer Hintergrund
Die GHTF bzw. das IMDRF setzt sich zwar aus Vertretern von staatlichen Behörden und Gesetzgebern zusammen; dennoch haben die Veröffentlichungen keine gesetzliche Kraft. Sie sind empfehlend, aber nicht bindend.
Die Hersteller sind jedoch verpflichtet, die regulatorischen Anforderungen an die Inhalte der Dokumentation zu erfüllen. Dazu zählen u. a.:
- Identifikation (z. B. UDI) des Produkts
- Beschreibung des Produkt, seiner Varianten, Konfigurationen und seines Zubehör
- Zweckbestimmung inklusive Charakterisierung der Patienten (Indikationen, Kontraindikationen), der Anwender und der Nutzungsumgebung
- Labeling, z. B. Gebrauchsanweisung, Verpackung
- Informationen zur Entwicklung und Herstellung
- Nachweis aller gesetzlicher Anforderungen (typischerweise durch Tests) an die Sicherheit, Sterilität, elektromagnetische Verträglichkeit, Biokompatibilität usw.
- Informationen über das Qualitätsmanagementsystem
2. Struktur der Dokumentation laut ToC
a) Übersicht
Das ToC-Format strukturiert die technische Dokumentation in über 150 Kapitel und Unterkapitel. Die erste Kapitelebene verschafft einen Überblick (s. Abb. 1) und macht klar, dass nicht nur das Produkt, sondern auch das Qualitätsmanagement zu dokumentieren ist (abhängig von Land und Zulassungsverfahren).
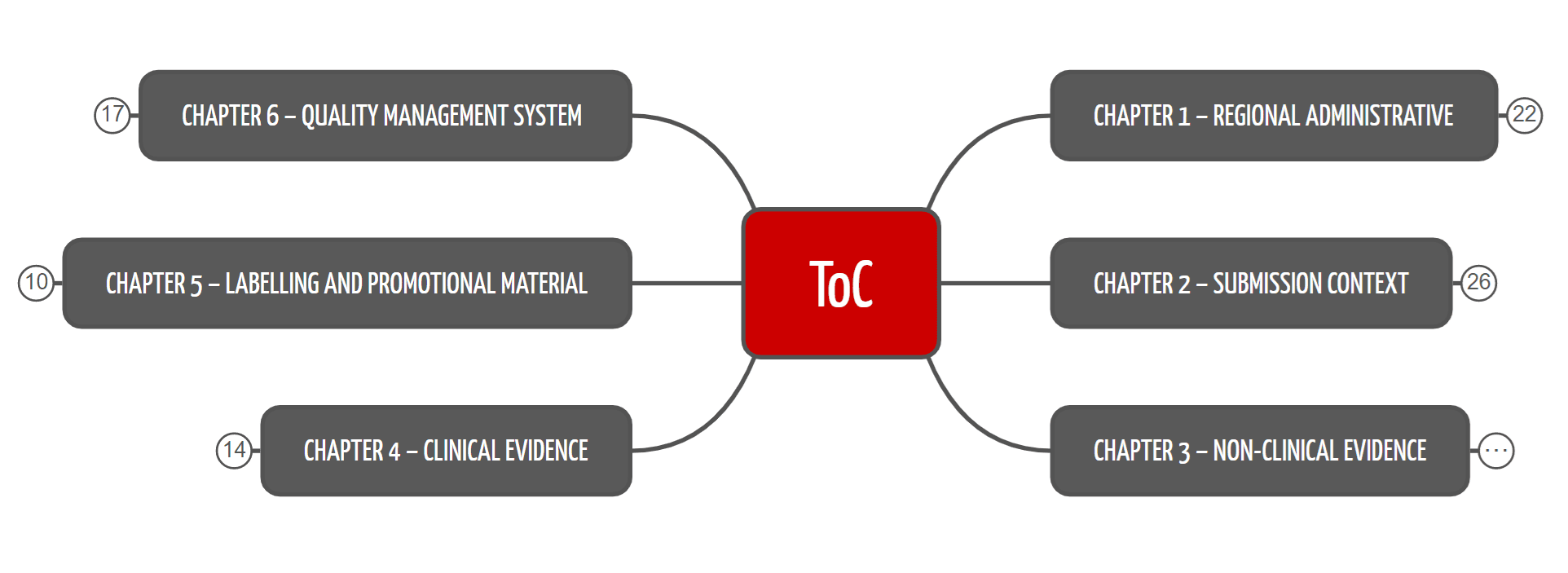
Die Kapitel lauten:
- Regulatorisches: Hier finden sich die marktspezifischen und regulatorischen Dokumente.
- Kontext der Einreichung: Hier findet sich die Beschreibung des Produkts.
- Nicht-klinische Nachweise, z. B. Labortests
- Klinische Nachweise
- Labeling
- QM-System: Verfahren, Methoden und produktspezifische Vorgaben
b) Kapitel 1: Regional Administative
Das erste Kapitel umfasst alle Inhalte, die spezifisch für den jeweiligen Markt bzw. das konkrete Zulassungsverfahren sind. Das reicht vom „Device Listing“ über Erklärungen bis hin zu den „User Fees“.
c) Kapitel 2: Submission Context
Das zweite Kapitel des ToC ist mit „Submission Context“ übertitelt. Es umfasst eine genaue Vorgabe, wie das Produkt mit seiner Zweckbestimmung, seinen Varianten, mit der Abgrenzung zu Vorgängerprodukten und ähnlichen Produkten zu beschreiben ist.
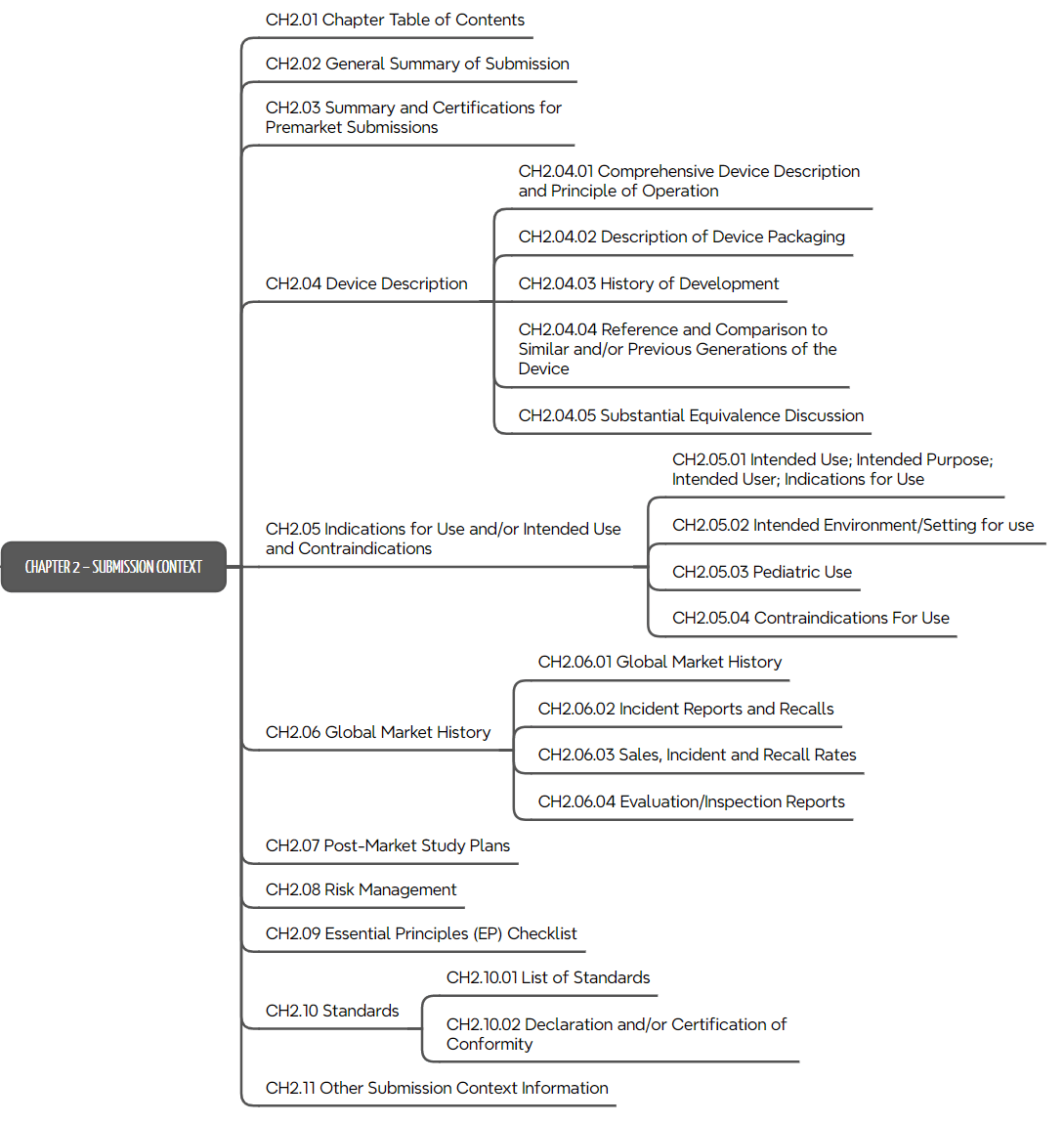
d) Kapitel 3: Non-clinical Evidence
Das dritte Kapitel des ToC ist das umfangreichste. Es spezifiziert die Nachweise der regulatorischen Anforderungen wie Labortests, Simulationen, Software-Tests, Tierversuche, Usability-Studien usw.
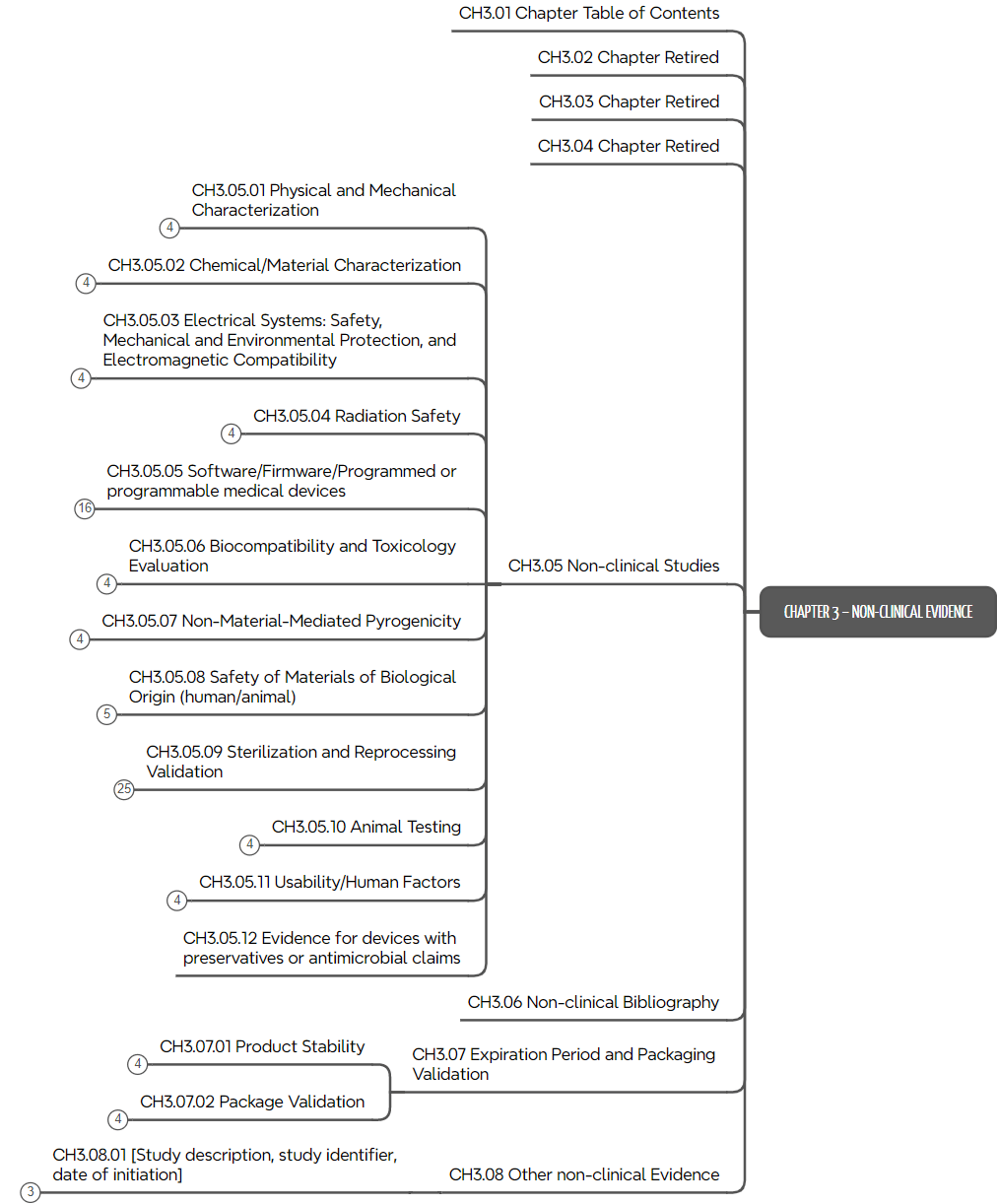
Das ToC liefert viel mehr als eine Kapitelstruktur. Es beschreibt auch typische Inhalte, wie in Abb. 4 am Beispiel der „Usability-Anforderungen“ zu sehen.

e) Kapitel 4: Clinical Evidence
Das vierte Kapitel gibt Vorgaben sowohl für die Dokumentation der klinischen Bewertung als auch der klinischen Prüfungen. Im Vergleich zur MEDDEV 2.7/1 und der ISO 14155 sind diese Vorgaben recht grob.
f) Kapitel 5: Labelling and Promotional Material
Der Titel des fünften Kapitel beschreibt treffend dessen Inhalt. Das Kapitel unterscheidet die verschiedenen Adressaten (Ärzte, Patienten, technische Anwender). Es unterscheidet beim Labeling die „Labels“ am Produkt, der Verpackung sowie die verschiedenen Anleitungen, Broschüren und Marketingmaterialien.
g) Kapitel 6: Quality Management System
Das sechste Kapitel stellt Anforderungen an die Dokumentation des QM-Systems. Dabei unterscheidet das ToC allgemeine und produktspezifische Vorgaben. Letztlich referenzieren die Kapitel einzelne Abschnitte der ISO 13485.

Möchten Sie sicherstellen, dass Ihre technische Dokumentation den Anforderungen der MDR/IVDR entspricht?
Unser E-Learning-Kurs zeigt Ihnen Schritt für Schritt, wie Sie eine konforme Struktur für Ihre Dokumentation erstellen können. Sie lernen, wie Sie Ihre Dokumente nicht nur auf Vollständigkeit, sondern auch auf Konsistenz und Konformität prüfen. Nach Abschluss des Kurses sind Sie in der Lage, Ihre Dokumentationen zu überprüfen und sicherzustellen, dass sie den Vorschriften entsprechen.
3. STED und ToC in der Praxis
a) Eine umfangreiche Zusammenfassung
Die Struktur der STED (Summary Technical Documentation) bzw. ToC ist mit über 150 Kapiteln und Unterkapiteln sehr umfangreich, so dass der Begriff „Summary“ fast ein Euphemismus ist.
Allerdings stellen diese Kapitel die Übermenge dessen dar, was ein Hersteller für ein Produkt benötigt. Viele Kapitel sind auf das konkrete Produkt nicht anwendbar: Eine Software bedarf keiner Diskussion der Biokompatibilität, und für viele Produkte sind keine Tierversuche notwendig.
b) Detailgrad
Das mit über 160 Seiten umfangreiche Dokument verschafft den Herstellern mehr als nur eine Übersicht dessen, was eine technische Dokumentation enthalten sollte. Der STED und auch das ToC sind und wollen aber nicht so granular sein, dass ihre Vorgaben Normen überflüssig machen.
c) Anerkennung durch Behörden
Die FDA arbeitet aktiv mit an der Weiterentwicklung des STED bzw. ToCs. Allerdings forciert sie mit ihrem eSTAR-Format einen alternativen Weg zur digitalen und datengetriebenen Zulassungsakte. Dennoch bietet sie in der eSTAR-Vorlage ein Mapping zum ToC an.
Die kanadischen Behörden finden Gefallen am STED bzw. dessen Nachfolger ToC. Auf dessen Struktur basierend haben sie ihre eigene Vorstellung von der Struktur der technischen Dokumentation publiziert. Allerdings arbeitet Health Canada inzwischen eng mit der FDA zusammen und nutzt das eSTAR-Format im Rahmen eines Pilotprogramms.
Die Chinesische Behörde NMPA setzt ebenfalls auf das ToC-Format bzw. macht dieses verpflichtend.
Die Japanische Behörde PMDA setzt weiterhin auf das alte STED-Format.
d) Fazit
Die Summary Technical Documentation (STED) und deren Nachfolger Table-of-Content-Format (ToC) sind ein wertvolles Hilfsmittel, um die technische Dokumentation zu strukturieren. Es gibt auch alternative Strukturen, die sich eignen.
Darüber, ob das STED bzw. ToC auch die Struktur der QM-Unterlagen festlegen sollte, lässt sich streiten.
Eine technische Dokumentation im STED- oder ToC-Format ist keine Garantie für eine erfolgreiche Zulassung. Denn dafür bedarf es nicht nur einer übersichtlichen Struktur, sondern vollständiger und korrekter Inhalte.
Es ist bedauerlich, dass sich die Behörden weltweit noch nicht auf ein Format einigen konnten. Die STED und ToC ist diesem Ziel aber bereits ziemlich nahe gekommen.
Wir vermuten allerdings, dass zukünftig immer mehr Länder das eSTAR-Format der FDA für ihre Zulassungsverfahren einsetzen werden.
Bitte beachten Sie den ausführlichen Beitrag zur technischen Dokumentation
Änderungshistorie:
- 2024-10-16: Aktualisierung basierend auf der aktuellen Vorlage des ToCs



Hallo Herr Johner,
zusätzlich würde ich noch folgende Guidance zum ASEAN CSDT empfehlen:
Guidance on Preparation of a Product Registration
Submission for General Medical Devices using the
ASEAN Common Submission Dossier Template (CSDT)
http://www.asean.org/archive/SnC/Guidance%20to%20ASEAN%20CSDT_Final_21%20Oct%202010.pdf
Viele Grüsse,
Timur Resch
#
Anmerkung von CJ vom März 2019: Neuer Link ist hier:https://www.asean.org/storage/images/archive/SnC/Guidance%20to%20ASEAN%20CSDT_Final_21%20Oct%202010.pdf
Bitte auch Artikel zur technischen Dokumentation beachten, der auf ASEAN Dokument eingeht.
Super, danke, lieber Herr Resch!
In den Beitrag werde ich Ihren Tipp mehr als gerne mitaufnehmen.
Beste Grüße
Christian Johner
Hallo Herr Prof. Johner und Lesende,
im Anhang II der MDR/ VO (EU) 2017/745 Abschnitt 4 Unterpunkt b wird von einer Zusammenfassung der technischen Dokumentation gesprochen. Leider habe ich nur an dieser einzigen Stelle die Erwähnung dieses Dokumentes gefunden. Allerdings wird im Vorspann der Verordnung Abschnitt (5) von GHTF und IMDRF gesprochen. Kann es sich unter Berücksichtigung dieser Abschnitte tatsächlich um eine STED nach GHTF handeln? Wenn ja, wann wird diese gebraucht?
Sehr geehrte Frau Szymkiewicz,
danke für die spannenden Fragen! Derzeit gibt es keine verbindliche Antworten darauf. Dennoch hier erste Gedanken:
Die Struktur nach STED ist sicher hilfreich und bietet eine gute Möglichkeit, die Akte zu strukturieren. Der MDR geht es aber v.a. um die Übersichtlichkeit. Wenn es zu viele Dokumenten sind, wünscht sie sich eine Zusammenfassung. Harte Kriterien für die Notwendigkeit gibt es nicht. Aber die Verordnung gibt damit den benannten Stellen die Möglichkeit, so eine Zusammenfassung anzufordern.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
Die neue Struktur des IMDRF wird in vielen Veröffentlichungen „nIVD MA ToC“ genannt (z.B. in dem IMDRF Dokument selbst), teilweise wird sie auch als STED Nachfolger bezeichnet. Ich würde Ihnen empfehlen zumindest den neuen Begriff „nIVD MA ToC“ einmal in Ihrem Artikel zu erwähnen.
Wertvoller Hinweis! Danke, Herr Malm!
Ihren Tipp habe ich sofort berücksichtig!
Nochmals besten Dank!
Viele Grüße, Christian Johner
Der Link aus (1) scheint sich geändert zu haben. Hier die heutige Version:
https://www.asean.org/storage/images/archive/SnC/Guidance%20to%20ASEAN%20CSDT_Final_21%20Oct%202010.pdf
Vielen herzlichen Dank, lieber Herr Brunner!
Ich tausche den Link gleich aus.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
bzgl. des Links in 1b): „[…] die nun den STED unter dem Namen „Non in Vitro Diagnostic Device Market Authorization Table of Contents (nlVDMAToC)“ zum Download (inzwischen in der 2. Version vom März 2019) anbietet.“ gibt es offenbar ein Update.
Seit Juni 2024 ist der nIVD ToC (neue Benennung: „Non-In Vitro Diagnostic Device Regulatory Submission Table of Contents“) nun in der Version 4 verfügbar. Der Link zum Dokument hat sich wie folgt gerändert:
https://www.imdrf.org/documents/non-vitro-diagnostic-device-regulatory-submission-table-contents-nivd-toc
Best Grüße
Lars Meinecke
Sehr geehrter Herr Meinecke,
vielen Dank für Ihren wichtigen Hinweis. Da in Version 4 auch größere strukturelle Änderungen vorgenommen wurden, werden wir den Artikel baldmöglichst überarbeiten.
Beste Grüße
Luca Salvatore
Guten Tag,
Welche Länder akzeptieren die STED? Beim MDSAP teilnehmenden Länder klar. Zur STED konnte ich aber bislang keine vollständige Liste finden.
Vielen Dank im Voraus
Guten Tag,
eine veröffentlichte Übersicht über die Länder, welche STED oder ToC akzeptieren bzw. verlangen, ist uns leider nicht bekannt. Einige Länder wie Japan orientieren sich beispielsweise noch am „alten“ STED-Format. Andere Länder wie Kanada und China referenzieren bzw. verlangen das ToC-Format. Die FDA nutzt inzwischen eSTAR, bietet dort aber ein Mapping zum ToC.
Die an der Erstellung des ToC-Formats beteiligten Behörden finden Sie direkt im Dokument beschrieben. Diese sind:
– EU Kommission
– Anvisa / Brasilien
– Health Canada
– PMDA / Japan
– Health Sciences Authority / Singapur
– Ministry of Food and Drug Safety / Korea
– MHRA / UK
– NMPA / China
– TGA / Australien
– FDA / USA
– Russland
Allerdings bedeutet dies nicht, dass die genannten Länder auf das ToC-Format setzen. Vermutlich werden einige Länder zukünftig eher das FDA eSTAR-Format implementieren (siehe z.B. Pilot-Projekt mit Health Canada).
Freundliche Grüße
Luca Salvatore