
Die EU-Medizinprodukteverordnung (MDR) reguliert nicht nur Medizinprodukte, sondern auch Produkte ohne medizinische Zweckbestimmung, z. B. Geräte zur Fettabsaugung, Brustimplantate oder farbige Kontaktlinsen.
Im Dezember 2022 – viereinhalb Jahre nach Erscheinen der MDR – hat die EU mit zwei Durchführungsverordnungen (2022/2346 und 2022/2347) notwendige Details geregelt.
Für Hersteller dieser Produkte sind besonders die folgenden Quellen relevant:
- MDR Anhang XVI
- Common Specifications: Durchführungsverordnung (EU) 2022/2346 im Amtsblatt (DE)
- Klassifizierung: Durchführungsverordnung (EU) 2022/2347 zur Reklassifizierung der Regeln 9 und 10 für Anhang-XVI-Produkte (DE)
Dieser Artikel verschafft Herstellern von Produkten ohne medizinische Zweckbestimmung (Anhang-XVI-Produkte) einen Überblick über die regulatorischen Anforderungen, die sie zu beachten haben, und gibt Tipps zur Umsetzung.
1. Produkte, die unter den Anhang XVI fallen
Die MDR listet im Anhang XVI sechs Produktgruppen.
| # | Produktgruppe | Beispiele |
| 1 | Kontaktlinsen oder andere zur Einführung in oder auf das Auge bestimmte Artikel | Farbige Kontaktlinsen |
| 2 | Produkte, die dazu bestimmt sind, durch chirurgisch-invasive Verfahren zum Zwecke der Modifizierung der Anatomie oder der Fixierung von Körperteilen vollständig oder teilweise in den menschlichen Körper eingeführt zu werden, mit Ausnahme von Tätowierungs- und Piercing-Produkten | Subdermale Implantate wie Hornimplantate, Brustimplantate |
| 3 | Stoffe, Kombinationen von Stoffen oder Artikel, die zur Verwendung als Gesichts- oder sonstige Haut- oder Schleimhautfüller durch subkutane, submuköse oder intrakutane Injektion oder andere Arten der Einführung bestimmt sind, mit Ausnahme derjenigen für Tätowierungen | Dermal Filler, z. B. Hyaluronspritzen |
| 4 | Geräte, die zur Reduzierung, Entfernung oder Zersetzung von Fettgewebe bestimmt sind, wie etwa Geräte zur Liposuktion, Lipolyse oder Lipoplastie | Bodyforming-Geräte, z. B. zur Fettabsaugung |
| 5 | Für die Anwendung am menschlichen Körper bestimmte Geräte, die hochintensive elektromagnetische Strahlung (Infrarotstrahlung, sichtbares Licht, ultraviolette Strahlung) abgeben, kohärente und nicht kohärente Lichtquellen sowie monochromatisches Licht und Licht im Breitbandspektrum eingeschlossen, etwa Laser und mit intensiv gepulstem Licht arbeitende Geräte zum Abtragen der oberen Hautschichten („skin resurfacing“), zur Tattoo- oder Haarentfernung oder zu anderen Formen der Hautbehandlung | IPL-Geräte zur Haarentfernung oder Hautverjüngung; gilt NICHT für Sonnenliegen |
| 6 | Geräte zur transkraniellen Stimulation des Gehirns durch elektrischen Strom oder magnetische oder elektromagnetische Felder zur Änderung der neuronalen Aktivität im Gehirn | Nicht-invasive Geräte für die magnetische oder elektrische Stimulation des Gehirns (ohne konkreten medizinischen Zweck) |
Die EU-Kommission darf diese Liste durch delegierte Rechtsakte (Durchführungsverordnungen) zukünftig erweitern und neue Produktgruppen hinzufügen. Voraussetzung ist allerdings, dass diese aufgrund des Risikoprofils oder aufgrund von Merkmalen bzw. deren Funktionalität entsprechenden Medizinprodukten ähnlich sind.
2. Anforderungen an Produkte ohne medizinische Zweckbestimmung
a) Es gelten fast die gleichen Anforderungen wie bei den Medizinprodukten
Die Kurzversion lautet: Die Anforderungen an Produkte ohne medizinische Zweckbestimmung sind (fast) die gleichen wie die Anforderungen an „normale“ Medizinprodukte.
Die MDR fasst Medizinprodukte, Zubehör und Produkte ohne medizinische Zweckbestimmung sogar unter dem Begriff „Produkte“ zusammen und unterscheidet diese (von wenigen Ausnahmen abgesehen) nicht.
Somit gelten u. a. die Verpflichtungen gemäß Artikel 10 der MDR. Das heißt: Die Hersteller müssen:
- ein QM-System errichten und implementieren (dazu gehört auch ein Risikomanagement-System)
- eine klinische Bewertung durchführen
- eine technische Dokumentation nach Anhang II und III der MDR erstellen
- ein Konformitätsbewertungsverfahren durchlaufen und eine Konformitätserklärung ausstellen
- die UDI-Anforderungen erfüllen
- ein System zur „Überwachung nach dem Inverkehrbringen“ aufbauen (Post-Market Surveillance)
- die Meldepflichten (Vigilanz) zu erfüllen
- Vorkehrungen im Falle einer Haftung durch fehlerhafte Produkte treffen, z. B. einen angemessenen Versicherungsschutz einrichten
Die MDR fordert, dass die Produkte die für sie geltenden grundlegenden Sicherheits- und Leistungsanforderungen nach Anhang I erfüllen.
b) Es gibt jedoch auch Unterschiede
In einigen Punkten unterscheiden sich die Anforderungen an Medizinprodukte von den Anforderungen an die Anhang-XVI-Produkte.
Diese Unterschiede beschreiben die Common Specifications v. a. in den folgenden Anhängen:
| Anhang | Produkte | Kommentar |
| Anhang I | Alle Produkte | Der Anhang beschreibt v. a. die Anforderungen an das Risikomanagement und die „Sicherheitsinformationen“, z. B. die Gebrauchsanweisung und die Kennzeichnung. |
| Anhang II | Kontaktlinsen | Dieser Anhang beschreibt die für diese Produktklasse zusätzlichen spezifischen Anforderungen an das Risikomanagement und die Sicherheitsinformationen. |
| Anhang III | Produkte, die durch chirurgisch-invasive Verfahren in den Körper eingeführt werden, beispielsweise Hornimplantate und Brustimplantate | dito |
| Anhang IV | Gesichts- oder sonstige Haut- oder Schleimhautfüller, die durch subkutane, submuköse oder intrakutane Injektion oder andere Arten der Einführung bestimmt sind | dito |
| Anhang V | Geräte zur Reduzierung, Entfernung oder Zersetzung von Fettgewebe | dito |
| Anhang VI | Geräte, die für die Anwendung am menschlichen Körper bestimmt sind und hochintensive elektromagnetische Strahlung abgeben, wie Geräte zum Abtragen der oberen Hautschichten („skin resurfacing“), zur Tattoo- oder Haarentfernung oder zu anderen Formen der Hautbehandlung | dito Zusätzlich definiert dieser Anhang die Begriffe „Produkte für den beruflichen Gebrauch“ und „Produkte für den häuslichen Gebrauch“. |
| Anhang VII | Geräte zur transkraniellen Stimulation des Gehirns durch elektrischen Strom oder magnetische oder elektromagnetische Felder zur Änderung der neuronalen Aktivität im Gehirn | Dieser Anhang beschreibt die für diese Produktklasse zusätzlichen spezifischen Anforderungen an das Risikomanagement und die Sicherheitsinformationen. |
i) Klinische Bewertung ohne Nachweis des Nutzens
Es dürfte schwierig sein, wie für Medizinprodukte gefordert, den klinischen Nutzen nachzuweisen für Produkte ohne einen medizinischen Zweck. Aus diesem Grund muss die klinische Bewertung nach Artikel 61 „nur“ die Sicherheit und Leistung dieser Produkte nachweisen, aber nicht den klinischen Nutzen (siehe Artikel 61(9)).
Es kann Produkte geben, die sowohl als Medizinprodukt als auch als Anhang-XVI-Produkt gelten. Ein Beispiel sind Brustimplantate für medizinische und ästhetische Zwecke. In diesem Fall ist auch der klinische Nutzen nachzuweisen.
ii) Generelle Pflicht zu klinischen Prüfungen
Interessant ist die generelle Forderung nach einer klinischen Prüfung für Anhang-XVI-Produkte. Diese kann man nur umgehen, wenn zu einem „analogen“ Medizinprodukt ausreichend klinische Daten vorhanden sind und bewertet wurden.
Die MDR schreibt „analog“ und nicht „äquivalent“. Eine Definition des Begriffs „analog“ fehlt in der MDR. Diese liefern die Common Specifications:
„Unter einem analogen Produkt mit medizinischer Zweckbestimmung ist das gleiche Produkt mit medizinischer Zweckbestimmung oder ein Medizinprodukt zu verstehen, für das der Hersteller gemäß Anhang XIV Abschnitt 3 der Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates (1) die Gleichwertigkeit mit dem entsprechenden Produkt mit medizinischer Zweckbestimmung nachgewiesen hat.“
iii) Risikoakzeptanzkriterien, die nicht auf dem Nutzen basieren
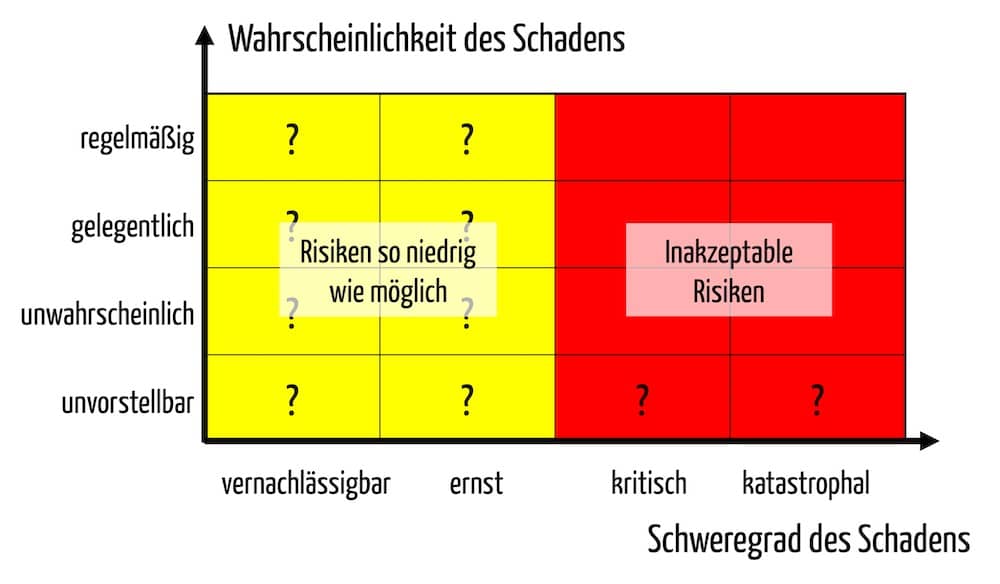
Ein weiterer Unterschied betrifft das in Anhang I geforderte Risiko-Nutzen-Verhältnis (siehe Anhang I, Kapitel I, 9.). Da kein medizinischer Nutzen vorliegt, müssen die Hersteller die Risikopolitik und die Akzeptanz auf einer anderen Basis festlegen.
Auch hier hilft ein Blick in die gemeinsamen Spezifikationen:
„Restrisiken können als vertretbar angesehen werden, wenn die unerwünschten Nebenwirkungen vorübergehender Natur sind und keinen medizinischen oder chirurgischen Eingriff erfordern, um eine lebensbedrohliche Erkrankung oder eine dauerhafte Beeinträchtigung einer Körperfunktion oder eine dauerhafte Schädigung einer Körperstruktur zu verhindern. Werden eine oder mehrere der in diesem Abschnitt festgelegten Bedingungen nicht erfüllt, muss der Hersteller eine Begründung vorlegen, warum die Risiken vertretbar sind.“
Durchführungsverordnung 2022/2346
Die Risikoakzeptanz muss somit anhand des Schweregrades und nicht der Wahrscheinlichkeit möglicher Schäden erfolgen. Dies ist eine unglückliche Forderung, weil in beliebig unwahrscheinlichen Fällen immer ein schwerer (z. B. irreversibler) Schaden entstehen kann.
iv) Produktspezifische Anforderungen
Der eigentliche Unterschied besteht in den Produktgruppen-spezifischen Anforderungen, welche die Common Specifications, die „gemeinsamen Spezifikationen“ in den Anhängen I bis VII der Durchführungsverordnung 2022/2346 fordern.
Diese basieren auf den grundlegenden Sicherheits- und Leistungsanforderungen aus Anhang I der MDR wie dem Risikomanagement oder der Kennzeichnung (Labeling), sind aber produktspezifisch.
Risikoanalyse
Anhang I des Rechtsakts gilt für sämtliche Anhang-XVI-Produkte und beschreibt u. a. den geforderten Risiko-Management-Prozess. Dieser ist sehr nah an der ISO 14971 ausgerichtet.
Er nennt produktspezifische Gefährdungen, welche Hersteller bewerten müssen.
Beispiel: Abschnitt 2 Produkte wie subdermale Implantate (Anhang III des Rechtsakts): Spezifische Gefährdungen, z. B. mikrobiologische Kontamination, Implantatversagen und -bewegung, Schrumpfen und Faltenbildung des Implantats, Missempfinden oder Schmerzen usw.
Bedauerlicherweise verwenden die Autoren die Begrifflichkeiten nicht präzise und abgestimmt:
- Im englischen Text heißt es „hazards“, im deutschen „Gefahren“. Die korrekte Übersetzung wäre „Gefährdungen“.
- Auch klassifizieren die Autoren Missempfinden und Schmerzen als Gefährdungen. Besser wäre es, von Schäden zu sprechen.
Risikobeherrschung
Die Common Specifications nennen nicht nur Gefährdungen, die die Hersteller betrachten müssen, sondern auch spezifische Risikokontrollmaßnahmen. So fordern sie etwa, dass Implantate steril und nicht pyrogen sein dürfen; dass Langzeitdaten erhoben werden müssen, um das Vorhandensein von nicht abbaubaren Stoffen zu bewerten; oder dass die Hersteller Schulungen zur Implantation und sicheren Verwendung des Produkts anbieten müssen.
Kennzeichnung
Mit der Kennzeichnung müssen die Hersteller auf den nichtmedizinischen Zweck klar hinweisen. Bei Implantaten ist auf der Kennzeichnung der Hinweis Pflicht, dass „die Produkte nicht bei Personen unter 18 Jahren verwendet werden dürfen“.
3. Konformitätsbewertung von Produkten ohne medizinische Zweckbestimmung
a) Konformitätsbewertungsverfahren
Die MDR sieht für die Anhang-XVI-Produkte die gleichen Konformitätsbewertungsverfahren wie bei Medizinprodukten vor. Auch bei den Produkten ohne medizinische Zweckbestimmung bestimmt die Klasse des Produkts, welches Konformitätsbewertungsverfahren die Hersteller anwenden dürfen.
b) Klassifizierung
Diese Klassifizierung richtet sich auch bei den Anhang-XVI-Produkten nach den Regeln des Anhangs VIII der MDR. Allerdings lassen sich nicht alle Regeln anwenden. So gehen die Regeln 9 und 10 (das sind die Regeln für aktiv therapeutische und diagnostische Produkte) von einem medizinischen Zweck aus.
Daher wurde zeitgleich zu den gemeinsamen Spezifikationen eine weitere Durchführungsverordnung (2022/2347) verabschiedet zur Reklassifizierung entsprechender Produkte:
| Produktklasse | Beschrieben in Abschnitt | Klasse |
| Bodyforming-Geräte | 4 | IIb |
| Geräte zur Hautverjüngung, Haarentfernung etc. | 5 | Haarentfernung: Klasse IIa, andernfalls IIb |
| Geräte zur transkraniellen Stimulation des Gehirns | 6 | III |
4. Übergangsfristen für Anhang-XVI-Produkte
Die Durchführungsverordnungen sind erst viereinhalb Jahre nach der MDR erschienen. Möglicherweise haben diese Tatsache sowie die Erfahrungen mit den Übergangsfristen bei den Medizinprodukten zu teilweise großzügigen Übergangsfristen geführt.
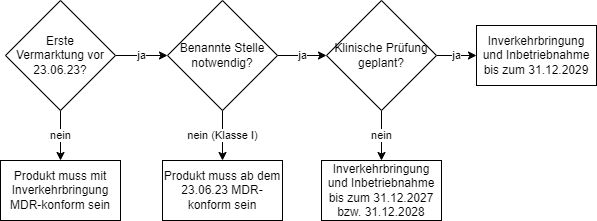
Welche Übergangsfristen gelten, hängt von der jeweiligen Konstellation ab:
| # | Konstellation | Übergangsfrist | Voraussetzungen |
| 1 | Klinische Prüfung in Durchführung oder geplant UND Einbeziehung einer Benannten Stelle notwendig (> Klasse I) | Inverkehrbringung und Inbetriebnahme bis zum 31. Dezember 2029 | – Erste rechtmäßige Vermarktung vor dem 22. Juni 2023 (Achtung: Fehler in der deutschen Übersetzung! Diese spricht von Inverkehrbringung.) – Vermarktung weiterhin gemäß den vor der MDR gelten Rechtsvorschriften für die jeweilige Produktgruppe – Keine wesentlichen Änderungen an der Auslegung oder Zweckbestimmung – Vollständiger Antrag samt Bestätigung der Behörde zur klinischen Prüfung bis zum 22. Juni 2024 – Start der klinischen Prüfung vor dem 23. Dezember 2024 – Unterzeichneter Vertrag zur Konformitätsbewertung mit Benannter Stelle vor dem 01.01.2028 |
| 2 | KEINE klinische Prüfung geplant UND Einbeziehung einer Benannten Stelle notwendig (> Klasse I) | Inverkehrbringung und Inbetriebnahme bis zum 31. Dezember 2028 | – Erste rechtmäßige Vermarktung vor dem 22. Juni 2023 (Achtung: Fehler in der deutschen Übersetzung! Diese spricht von Inverkehrbringung.) – Vermarktung weiterhin gemäß den vor der MDR geltenden Rechtsvorschriften für die jeweilige Produktgruppe – Keine wesentlichen Änderungen an der Auslegung oder Zweckbestimmung – Unterzeichneter Vertrag zur Konformitätsbewertung mit Benannter Stelle vor dem 01.01.2027 |
| 3 | MDD-Zertifikat vorhanden (abgelaufen zwischen dem 26. Mai 2021 und dem 20. März 2023) | Auch nach Ablauf des Zertifikats bis zum 31. Dezember 2027 (Klasse III und gewisse Klasse IIb implantierbare Produkte) oder 31. Dezember 2028 (sonstige) | – Kein unterzeichneter Vertrag zur MDR-Zertifizierung mit Benannter Stelle vor Zertifikatsablauf UND keine „Sonderzulassung“ gemäß Artikel 59 oder 97 der MDR – Vermarktung weiterhin gemäß den MDD-Anforderungen – Keine wesentlichen Änderungen an der Auslegung oder Zweckbestimmung – Kein unannehmbares Risiko – QMS gemäß Artikel 10(9) MDR spätestens zum 26.05.2024 – Antrag auf Konformitätsbewertung MDR spätestens zum 26.05.2024 – Unterzeichneter Vertrag zur Konformitätsbewertung MDR spätestens zum 26.09.2024 |
| 4 | Keine Einbeziehung einer Benannten Stelle notwendig (Klasse I) | Inverkehrbringung und Inbetriebnahme bis zum 22. Juni 2023 | – Konformitätserklärung nach MDR bis spätestens zum 22. Juni 2023 |
Die Übergangsfristen hängen somit ab von:
- Zeitpunkt der ersten Vermarktung
- Klasse des Produkts
- Notwendigkeit einer klinischen Prüfung
Unser Experte Luca Salvatore erläutert im Gespräch mit Professor Johner, um welche Produkte es geht, welche Anforderungen die Hersteller und deren Produkte erfüllen müssen, welche Übergangsfristen Sie dazu beachten sollten und wie die Hersteller herausfinden, ob sie eine Benannte Stelle einbeziehen müssen.
Diese und weitere Podcast-Episoden finden Sie auch hier.

5. Empfehlungen zum Vorgehen
Viele Hersteller von Produkten ohne medizinische Zweckbestimmung werden durch die Komplexität und Quantität der regulatorischen Anforderungen sehr gefordert, einige auch überfordert sein.
Abhängig vom Wissensstand sollten diese die folgende Schritte durchlaufen:
- Machen Sie sich mit den regulatorischen Anforderungen vertraut. Dazu zählt v. a., die zu Beginn des Artikels genannten Quellen zu studieren.
- Recherchieren Sie einschlägige Normen, mit denen Sie die Konformität mit diesen Anforderungen nachweisen können.
- Bestimmen Sie die Klasse des Produkts.
- Eruieren Sie, ob eine klinische Prüfung verpflichtend ist.
- Bestimmen Sie die Übergangsfristen.
- Suchen Sie sich für Produkte der Klasse IIa und höher eine Benannte Stelle.
- Etablieren Sie ein Qualitätsmanagementsystem und lassen Sie dieses ggf. zertifizieren.
- Tragen Sie die Konformitätsnachweise zusammen.
- Planen Sie ggf. die klinische Prüfung und führen Sie diese durch.
- Erklären Sie die Konformität Ihrer Produkte und vermarkten Sie diese.
Das Johner Institut unterstützt Hersteller von Anhang-XVI-Produkten bei allen Schritten. Nehmen Sie gerne Kontakt auf!
6. Fazit und Zusammenfassung
Der Gesetzgeber hat die Anforderungen an Produkte ohne medizinische Zweckbestimmungen gemäß Anhang XVI der MDR sehr hoch gehängt. Das führt mit hoher Wahrscheinlichkeit zu einer massiven „Marktbereinigung“. Vermutlich war genau dies das Ziel des Gesetzgebers.
Es ist bedauerlich, dass der Gesetzgeber weit über vier Jahre benötigt hat, um Durchführungsverordnungen zu erstellen, die einem Anspruch auf Präzision nicht vollumfänglich gerecht werden.
Die Einhaltung dieser neuen Anforderungen obliegt nun den Behörden. Diese stehen vor der Aufgabe, den Markt zu überwachen, indem sie etwa Internet-Shops durchforsten und einschlägige Messen besuchen. Ohne dieses „Enforcement“ wird das Ziel der Gesetzgebung nicht erreicht.
Änderungshistorie
- 2023-06-22: Artikel angepasst an die neuen Übergangsfristen der Durchführungsverordnung 2023/1194 vom 20.06.2023



Sehr geehrter Herr Salvatore,
vielen Dank für diesen ausführlichen und hilfreichen Artikel zu Produkten nach Anhang XVI.
Als Hersteller eines Medizinprodukts zur transkraniellen Magnetstimulation MIT medizinischer Zweckbestimmung haben wir mit ein wenig Sorge die Durchführungsverordnung 2022/2347 zur Kenntnis genommen. Die darin beschriebenen Produkte zur transkraniellen Magnetstimulation OHNE medizinische Zweckbestimmung werden der Klasse III zugeordnet. Die Produkte MIT medizinischer Zweckbestimmung dagegen in Klasse IIa. Wie bewerten Sie diesen Unterschied, bzw. was für Folgen könnte dies für Hersteller der Produkte MIT medizinischer Zweckbestimmung haben? Ist eine Neuklassifizierung zu fürchten oder könnte das vorrangige „Ziel des Gesetzgebers“ eine „Marktbereinigung“ sein, wie Sie es auch schon angemerkt hatten?
Vielen Dank im Voraus für Ihre Einschätzung.
Freundliche Grüße
Peter Schenk
Sehr geehrter Herr Schenk,
Ihre Sorge kann ich gut verstehen. Die Durchführungsverordnung 2022/2347 rechtfertigt die Höherklassifizierung in den Erwägungsgründen. Dort nennt sie die möglichen Neben- bzw. Langzeitwirkungen. Diese sind vermutlich bei ähnlichen Geräte mit medizinischer Zweckbestimmung ebenso nicht auszuschließen. Nun gilt 2022/2347 allerdings nur für solche Produkte OHNE medizinische Zweckbestimmung. D.h. in Ihrem Fall wäre diese nur anwendbar, wenn Sie Ihre Produkte auch für nicht-medizinische Zwecke vermarkten. Leider haben wir aktuell keine Kenntnis darüber, ob eine ähnliche Reklassifizierung auch für die genannten Produkte mit medizinischer Zweckbestimmung zukünftig angedacht ist. Dies würde allerdings die Klassifizierungsregeln gemäß Anhang VIII generell in Frage stellen.
Herzliche Grüße
Luca Salvatore
Vielen Dank für Ihre Einschätzung Herr Salvatore.
Der Entwurf der Durchführungsverordnung wurde auch von anderen „indirekt Betroffenen“ entsprechend kommentiert, jedoch führte es letztendlich nicht zu einer Anpassung der Verordnung.
Das für uns Entscheidende ist, wie Sie auch beschrieben haben, das ausschließliche Vorliegen einer medizinischen Zweckbestimmung, die uns die Klassifizierungsregeln gemäß Anhang VIII anwenden lassen.
Freundliche Grüße
Peter Schenk
Sehr geehrter Herr Salvatore,
könnte auch Software, beispielsweise ChatGPT ein Produkt nach Anhang XVI MDR darstellen?
Liest man die MDCG 2019-11 heißt es dort: „Wenn das Produkt ein MDR Anhang XVI Produkt oder ein Zubehör für ein Medizinprodukt ist oder Software die die Verwendung eines Medizinprodukts steuert oder beeinflusst, muss es in seinem Regulierungsprozess als Teil dieses Produkts betrachtet werden oder unabhängig, wenn es sich um ein Zubehör handelt.
Das hört sich ja so an als könnte Software die ein Produkt nach Anhang XVI darstellt nur in Verbindung mit einem anderen Produkt existieren und nur bei einer Qualifizierung als Zubehör eigenständig zu regulieren sein.
Sehr geehrter Herr Keiber,
ich kann mir keine Software vorstellen, die für sich alleine genommen, als Anhang XVI-Produkt qualifiziert. Sicherlich werden Geräte (aktive Produkte) in den Gruppen 4-6 in Anhang XVI Software enthalten. Diese Software wird die Geräte steuern, aber nicht für sich genommen als MDSW qualifizieren.
Freundliche Grüße
Luca Salvatore
Sehr geehrtes Johner Team
Wo genau finde ich denn die Vorgaben für die Kennzeichnung von Flyern (promotional material) für Produkte ohne medizinische Zweckbestimmung (LIposuktion Kanüle für den Einmalgebrauch)?
Die ISO 15223-1 ist ja schon mal ausgeschlossen.
Vielen Dank und Viele Grüsse
Natascha Ohr
Liebe Frau Ohr,
es gibt in der MDR nur eine konkrete Anforderung an Werbematerialien und zwar im Artikel 20 bezüglich der CE-Kennzeichnung. Dort heißt es: “ Diese Kennnummer ist auch auf jeglichem Werbematerial anzugeben, in dem darauf hingewiesen wird, dass das Produkt die Anforderungen für die CE-Kennzeichnung erfüllt.“
Generell empfiehlt es sich aber, auch Werbematerialen als gelenktes Dokument im QM-System zu führen. Denn spezifische Angaben wie Claims oder Indikationen auf Werbematerialien können als Zweckbestimmung des Produkts verstanden werden (siehe hierzu die Definition von Zweckbestimmung in Artikel 2 12.). D.h. diese Angaben sollten nicht von denen im Konformitätsbewertungsverfahren geprüften abweichen.
Herzliche Grüße
Luca Salvatore
Guten Tag,
wir haben ein Produkt, welches sowohl eine medizinische als auch eine nichtmedizinische Zweckbestimmung aufweist und auch damit vermarktet wird. Gemäß MDR müssen in diesem Falle die Anforderungen beider Rechtsvorschriften eingehalten werden. Leider ergeben sich dadurch an einigen Stellen Konflikte im Hinblick auf die IFU, bspw. bei der Angabe der Klassifizierung (IIa gemäß MDR vs. IIb gemäß Durchführungsverordnung (EU) 2022/2346). Auch die Kennzeichnung „Medical Device (MD)“ vs. „nichtmedizinische Zweckbestimmung“ steht nach meiner aktuellen Auffassung in einem Widerspruch zueinander. Zudem schreibt die Durchführungsverordnung die Integration von vorformulierten Warnhinweisen vor, welche zum Teil nicht gänzlich korrekt sind, wenn wir die medizinische Zweckbestimmung mit betrachten.
Haben Sie eine Empfehlung, wie Kennzeichnung und IFU in einem solchen Fall rechtskonform umzusetzen ist?
Beste Grüße,
Chris
Lieber Chris,
ich bin nicht ganz sicher, ob ich die Frage verstehe. Geht es um ein Produkt, das einmal mit und einmal ohne medizinische Zweckbestimmung verkauft wird, oder um ein Produkt, das sowohl eine medizinische Zweckbestimmung als auch eine nicht-medizinische hat? Der letztere Fall tritt häufig auf, ohne das Konflikte auftreten:
Daher sehe ich keinen Konflikt. Aber möglicherweise habe ich die Frage noch nicht verstanden gehabt.
Viele Grüße
Christian Johner
Hallo Herr Johner,
vielen Dank für Ihre rasche Antwort. Verstehe ich es also richtig, dass bei gleichzeitigem Vorliegen einer medizinischen UND nicht-medizinischen Zweckbestimmung lediglich die Anforderungen aus der MDR umzusetzen sind und die Durchführungsverordnung (EU) 2022/2346 und 2022/2347 keine Anwendung finden?
Falls das Produkt einmal als „medizinische Variante“ und einmal als „nicht-medizinische Variante“ verkauft wird, müssten die jeweiligen Rechtsvorschriften gesondert auf die beiden Varianten angewendet werden (inkl. höherer Klassifizierung, bedingt durch Durchführungsverordnung (EU) 2022/2347)?
Hallo Chris,
meine Kollegin hat Ihnen per Email geantwortet.
Die Anforderungen an die sogenannten „Dual-Purpose“ Produkte finden Sie im MDCG 2023-5, u.a. in Kapitel 3.3. Demnach sind die GS zusätzlich zu den Anforderungen der MDR zu beachten. Im Falle der Klassifizierung gilt die höhere Klasse (Kapitel 4.1 Absatz 2 im MDCG-Dokument).
Herzliche Grüße
Luca Salvatore
Hallo Herr Salvatore,
vielen Dank für die tollen Ausführungen! Mich beschäftigt eine Frage:
Ist es zulässig, dass für einen Haarentfernugs-Laser zwar eine Konformitätsbescheinigung einer benannten Stelle nach alter MDD ausgestellt wurde, am Gerät jedoch ein CE-Kennzeichen ohne vierstellige Kennnummer angebracht ist? Vielen Dank im Voraus und beste Grüße
Ivan
Guten Tag Ivan,
wenn das Gerät unter dieser MDD-Konformitätsbescheinigung der Benannten Stelle in Verkehr gebracht wurde, muss die Kennnummer der Benannten Stelle angegeben sein. Eventuell hilft ein Blick auf die zugehörige Konformitätserklärung des Herstellers. Denkbar wäre, dass dieser das Produkt in zwei unterschiedlichen Varianten in den Verkehr bringt. Einmal mit medizinischem Zweck (=Medizinprodukt) und einmal ohne. Im letzteren Fall wäre eine CE-Kennzeichnung gemäß Niederspannungsrichtlinie ohne Kennnummer möglich.
Freundliche Grüße
Luca Salvatore
Vielen Dank für die rasche Antwort!
Freundliche Grüße
Ivan
Guten Tag Herr Salvatore,
ich bin ein Privatperson, kein Hersteller. Ich möchte nur nachfragen, ob eine Hautcreme, welche ich ohne Rezept in der Apotheke oder Online kaufen kann, ein Medizinprodukt ist. Konkret meine ich Bepanthen Wundcreme, Narbencreme.
Nach meiner OP hat mein Arzt mir empfohlen, eine Narbencreme zu verwenden, die nur aus Silikonöl, und Panthenol (Provitamin B5) besteht, und sagte, das ist ein Medizinprodukt. Selbst der Hersteller schreibt auf seinem Beipackzettel: „Das ist ein Medizinprodukt.“ Ich habe bei dem Hersteller nachgefragt, die Antwort war:
„Die Narbencreme ist keine Kosmetika, sondern ein Medizinprodukt, weil sie von Behörden wie dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) sowie durch spezielle Prüfstellen kontrolliert wird. Zwischen Kosmetika und Medizinprodukt macht die spezielle Formulierung und die medizinische Wirksamkeit den Unterschied. Die Einstufung als Medizinprodukt bedeutet, dass Bepanthen nicht nur pflegt, sondern aktiv zur Regeneration der Haut beiträgt. “
Fakt ist: viele Kosmetika auf dem Markt enthalten schon eine hohe Anteil von Provitamin B5, weilche zur Regeneration der Haut beitragen. Selbst der Wirkstoff ist online erreichbar, und sogar Menschen, die zu Hause Naturkosmetik rühren, können ihn kaufen :-).
Also die Antwort finde ich sehr witzig, weil ich selbst Kosmetikwissenschaft und Cosmetic Chemistry studiert habe. Meine Frage ist, ob man eine Hautcreme ohne Rezeptpflicht -nur mit kosmetischen Wirkstoffen (z.B Provitamin B5) – tatsächlich als Medizinprodukt bezeichnen kann.
Vielen Dank, mit freundlichen Grüßen
Anett Harnisch
Guten Tag Frau Harnisch,
mit Unterstützung meiner Kollegin Frau Dr. Reinhold, Expertin für stoffliche Produkte, versuche ich gerne Ihre Frage zu beantworten.
Es kommt zunächst primär auf die Zweckbestimmung des Produkts an. Eine Hautcreme kann grundsätzlich ein Kosmetisches Mittel, auch ein (stoffliches) Medizinprodukt oder ein Arzneimittel sein.
Kosmetische Mittel sind laut Begriffsdefinition der Kosmetikverordnung Stoffe oder Gemische, die dazu bestimmt sind Teile des menschlichen Körpers zu reinigen, zu parfümieren, ihr Aussehen zu verändern, sie zu schützen, sie in gutem Zustand zu halten oder den Körpergeruch zu beeinflussen. Kommt ein konkreter medizinischer Zweck hinzu, handelt es sich entweder um ein Medizinprodukt oder Arzneimittel – je nach Wirkungsweise. Die Inhaltsstoffe entscheiden hier also zunächst grundsätzlich nicht, ob es sich um ein Kosmetikum oder ein Medizinprodukt handelt.
Wund- und Narbencremes, die generell dazu bestimmt sind, die Wundheilung zu fördern und damit helfen, die Haut zu regenerieren, erfüllen einen medizinischen Zweck (z.B. Behandlung von Verletzungen). Das geht also über die Pflege und den Schutz der Haut und damit über ein reines Pflegeprodukt hinaus. Daher können Hautcremes auch Medizinprodukte sein. Der Antwort, die Sie vom Hersteller erhalten haben, wäre dahingehend also zuzustimmen. Die medizinische Wirksamkeit macht den Unterschied.
Uns ist unklar, was genau mit „kosmetischer Wirkstoff“ gemeint ist. Bei Panthenol handelt es sich unseres Wissens nach sogar um einen biologisch aktiven Wirkstoff. Es scheint die Zellproliferation der Haut zu stimulieren und die Differenzierung von Hautzellen zu fördern. Es stellt damit die natürliche Barriere der Haut wieder her und verhindert den Feuchtigkeitsverlust. Es hat damit also eine pharmakologische bzw. metabolische Wirkungsweise. Hier muss der Hersteller dann die Wirkungsweise genaustens darstellen, um damit die Qualifizierung zu rechtfertigen. Bei dieser Abgrenzung kommt es sicher auch auf die Konzentrationen und die damit verbundenen Wirkmechanismen an. Erfüllt die Creme insgesamt also einen medizinischen Zweck, ist eher die Frage zu klären, ob es sich bei der Creme um ein (stoffl.) Medizinprodukt oder ein Arzneimittel handelt.
Wir vermuten, dass die von Ihnen beschriebene Narben-Creme aus Silikonöl und Panthenol ein stoffl. Medizinprodukt ist, da diese primär über eine Filmbildung auf der Haut bzw. auf der Narbe durch das Silikon wirkt (physikalisch) und das Dexpanthenol unterstützend wirkt. Die Bepanthen Wund- und Heilsalbe hingegen kennen wir eigentlich als Arzneimittel (laut Beipackzettel), da diese ausschließlich Dexpanthenol als aktiven Wirkstoff enthält.
Fazit: Eine Hautcreme, die Panthenol enthält, kann auch ein (stoffl.) Medizinprodukt sein, wenn sie einen medizinischen Zweck erfüllt. Die Förderung der Wundheilung durch Regenerierung der Haut wäre unserer Auffassung nach ein medizinischer Zweck, der über die reine Pflege hinaus geht. Der klinische Nutzen muss in diesem Fall allerdings durch den Hersteller nachgewiesen werden. Da Panthenol aber so vielseitige Wirkungen hat, befinden wir uns hier in einem Spannungsfeld was die rechtliche Einordnung solcher Produkte betrifft. Da hier auch die Konzentrationen der Inhaltsstoffe eine wichtige Rolle spielen, ist es nicht immer gleich ersichtlich, ob es sich um ein Kosmetikum, ein Medizinprodukt oder nicht doch ein Arzneimittel handelt.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
wir betreiben zwei ärztlich geführte Institute zur Tattoo-Entfernung planen weitere Standorte zu eröffnen.
Können Sie mir sagen, welche Zertifikate von den Laserherstellern vorgelegt werden müssen, damit wir die Geräte betreiben dürfen? Ich bin kein Jurist und blicke bei den Übergangsfristen zum Betrieb der Geräte nicht durch. Wir haben auch das Problem, dass ein Mitbewerber mit einem günstigen Gerät aus China behandelt und wir nicht sicher sagen können, ob der Betrieb schon jetzt nicht erlaubt ist.
Ich freue mich auf Ihre Antwort.
Mit freundlichen Grüßen
Claus Hasenkamp
Sehr geehrter Herr Hasenkamp,
die Übergangsfristen im Bereich der Anhang XVI-Produkte sind in der Tat schwer verständlich und hängen von verschiedenen Faktoren ab, wie im Artikel beschrieben. Daher müssen die Anforderungen im Einzelfall bzw. dem konkreten Produkte betrachtet werden. Generell sollte das Produkt eine CE-Kennzeichnung tragen. Falls das Produkt bereits vor den Übergangsfristen als Medizinprodukt unter der alten Richtlinie (MDD) deklariert war, dann sollte der Hersteller über eine entsprechende EU-Konformitätserklärung verfügen (declaration of conformity). Ich empfehle, den rechtlichen Status direkt beim Hersteller anzufragen (Medizinprodukt ja/nein vor der MDR, Nutzung der Anhang XVI-Übergangsbestimmungen, eventuell bereits konform zur MDR mit entsprechender EU-Konformitätserklärung).
Denken Sie auch an die Einhaltung der Medizinprodukte-Betreiberverordnung (https://www.johner-institut.de/blog/regulatory-affairs/mpbetreibv-medizinprodukte-betreiberverordnung/).
Herzliche Grüße
Luca Salvatore