
Wir haben diesen Artikel für Sie überarbeitet und die Verordnung (EU) 2024/1860 des Europäischen Parlaments und des Rates vom 13. Juni 2024 zur Änderung der Verordnungen (EU) 2017/745 und (EU) 2017/746 der Europäischen Kommission berücksichtigt.
Bereits im Dezember 2021 hat die EU die Übergangsfristen der Verordnung 2017/746 über In-vitro-Diagnostika (IVDR) verlängert (Verordnung 2022/112). Im März 2023 hat die Europäische Kommission den Wegfall der „Abverkaufsfrist“ für IVD, die konform mit der Richtlinie 98/79/EG (IVDD) sind, beschlossen (Verordnung 2023/607).
Im Januar 2024 hat die Europäische Kommission einen Vorschlag zur Änderung der IVDR veröffentlich, welcher die Übergangsfristen weiter verlängert. Als Grund hierfür wird vor allem der Mangel an Benannten Stellen vorangestellt. Das ändert zwar nichts am Geltungsbeginn der IVDR, dieser bleibt der 26. Mai 2022, allerdings verschaffen die verlängerten Fristen Herstellern und Benannten Stellen mehr Zeit, um IVD-Produkte durch das Konformitätsverfahren gemäß IVDR zu bringen; zudem werden sichere und leistungsfähige Produkte nicht unnötigerweise entsorgt. Dieser Vorschlag wurde als Verordnung (EU) 2024/1860 im Amtsblatt der Europäischen Union veröffentlicht und ist somit gültig.
Lesen Sie im Folgenden, welche Übergangsfristen bis 2030 bestehen und was das für Sie bedeutet.
1. Übersicht über die Übergangsfristen
a) Zweck der Änderungsverordnung der IVDR?
Die Änderungen gewähren insbesondere Herstellern von Produkten, die unter der IVDR eine Benannte Stelle benötigen, mehr Zeit. Solche Produkte dürfen auch noch nach dem Datum der Gültigkeit der IVDR am 26. Mai 2022 hergestellt und in Verkehr gebracht werden. Aber Achtung! Einige Anforderungen der IVDR müssen trotzdem erfüllt werden. Mehr dazu erfahren Sie weiter unten.
b) Welche Produkte betreffen die Änderungen?
Zunächst ist wichtig, dass die neuen Regelungen nur für Bestandsprodukte gelten. Das sind Produkte, für die vor dem 26. Mai 2022 die Konformität erklärt wurde UND die unter der IVDR in die Klassen D, C, B oder A (steril) fallen. Das bedeutet, dass alle Produkte von einer verlängerten Übergangsfrist profitieren, außer
- Klasse A-Produkte ohne Steril-Kennzeichnung und
- neue Produkte, für die erst nach dem 26. Mai 2022 eine Konformitätserklärung ausgestellt wird.
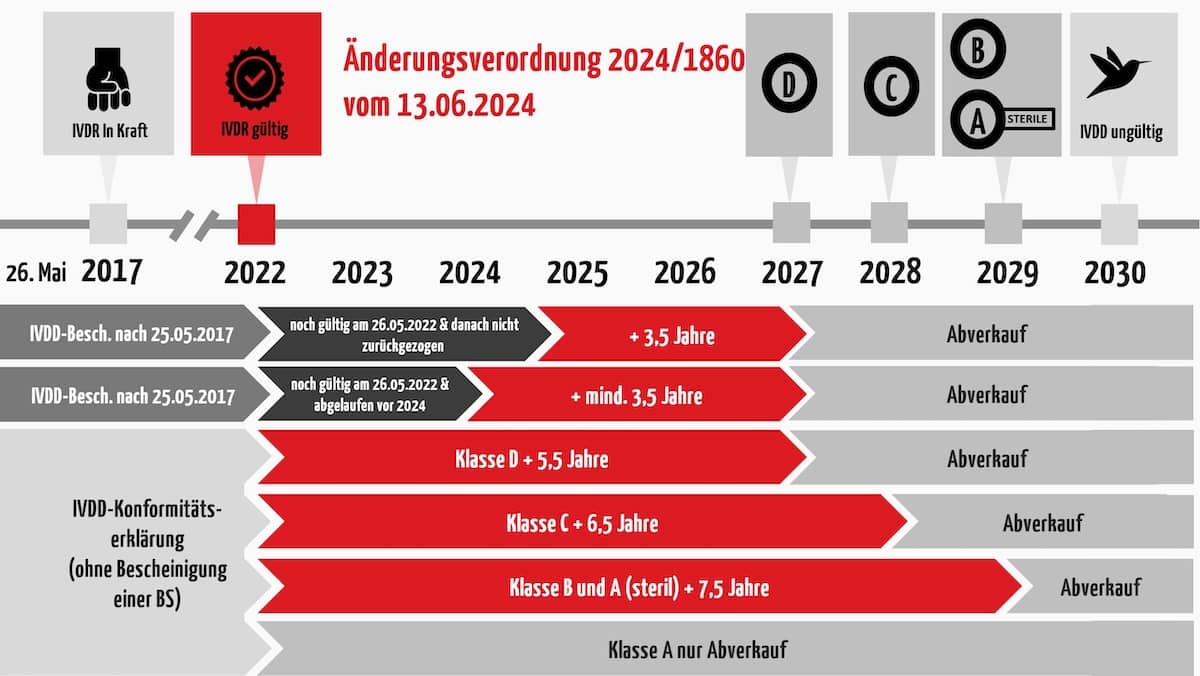
c) Wie lang sind die Übergangsfristen?
Wie lang die neuen Fristen sind, hängt von der künftigen Risikoklasse ab und davon, ob eine Benannte Stelle bereits unter IVDD beteiligt war. Interessanterweise hat sich die EU entschieden, gemäß dem aktuellen Vorschlag das Stichpunktdatum vom 26. Mai auf den jeweiligen Jahreswechsel zu legen.
- Für Klasse-A-Produkte gibt es keine verlängerte Übergangsfrist (Ausnahme: Sterilprodukte. Diese erhalten eine Übergangsfrist für das Inverkehrbringen bis zum 31. Dezember 2029.)
- Für Klasse-B-Produkte gilt dieselbe Übergangsfrist bis zum 31. Dezember 2029.
- Für Klasse-C-Produkte endet diese Frist am 31. Dezember 2028 und
- für Klasse-D-Produkte am 31. Dezember 2027.
- Die sogenannte “Abverkaufsregelung” (Bereitstellung und Inbetriebnahme) entfällt.
Die folgende Abbildung fasst die Regelungen zusammen. Aber Achtung, die Regelungen sind an einige Bedingungen geknüpft. Lesen Sie im folgenden Abschnitt, wie das für die einzelnen Fälle im Detail aussieht.
2. Die neuen Fristen im Detail
a) IVD, für die unter IVDD eine Bescheinigung durch eine Benannte Stelle ausgestellt wurde und die auch unter IVDR eine Benannte Stelle benötigen
Für In-vitro-Diagnostika, für die unter der IVDD eine Bescheinigung durch eine Benannte Stelle ausgestellt wurde, endet die Frist spätestens am 31. Dezember 2027. Zu diesen IVD zählen Produkte, die dem Anhang II, Liste A und Liste B entsprechen, oder Produkte zur Eigenanwendung.
Das zugrundeliegende Konformitätsbewertungsverfahren ist hierbei zweitrangig. Es ist also nicht entscheidend, ob das betreffende Produkt über ein vollständiges Qualitätssicherungssystem, über die Baumusterprüfung oder über die Auslegungsprüfung bei Produkten zur Eigenanwendung eine Bescheinigung gemäß IVDD erhalten hat.
Beispiel: Sie haben für ein IVD der Liste B die Konformität mit Beteiligung einer Benannten Stelle gemäß Anhang IV der IVDD (vollständiges Qualitätssicherungssystem) erklärt. Die Ausstellung des Zertifikats erfolgte im Zeitraum vom 25. Mai 2017 bis 25. Mai 2022. In diesem Fall endet die Zertifikatsgültigkeit am 31. Dezember 2027. Die Verlängerung des Zertifikats ist direkt anwendbar, sodass Benannte Stellen das Datum auf den individuellen Zertifikaten nicht ändern müssen.
Eine weitere Bedingung ist, dass ein formaler Antrag bei der Benannten Stelle bis zum 26. Mai 2025 gestellt und bis zum 26. September 2025 ein Vertrag mit der Benannten Stelle geschlossen sein muss, damit das Zertifikat des IVDD-Produkts weiterhin seine Gültigkeit behält.
Ist das Zertifikat jedoch bereits abgelaufen, bevor der neue Änderungsvorschlag angenommen ist, so ist die Verlängerung des Zertifikats daran geknüpft, dass der Hersteller entweder ein Vertrag mit einer Benannten Stelle geschlossen hat oder die nationale Zulassungsbehörde eine Ausnahme vom anwendbaren Konformitätsbewertungsverfahren gemäß Artikel 54 gewährt oder den Hersteller aufgefordert hat, das Konformitätsbewertungsverfahren gemäß Artikel 92 innerhalb eines bestimmten Zeitraums durchzuführen.
Falls Sie Ihr QM-System freiwillig nach ISO 13485 zertifizieren lassen, ist das keine Bescheinigung einer Benannten Stelle, sondern von einer Zertifizierstelle. Nur ein Zertifikat einer akkreditierten Benannten Stelle mit Bezug auf die IVD-Richtlinie 98/79/EG würde hier gelten.
b) “Sonstige IVD”, die ohne Beteiligung einer Benannten Stelle unter IVDD bereits in Verkehr gebracht wurden und unter IVDR eine Benannte Stelle benötigen
Für In-vitro-Diagnostika, für die Hersteller unter der IVDD die Konformitätserklärung selbst vor dem 26. Mai 2022 ausgestellt haben und die unter IVDR gemäß IVDR-Klassifizierungsregeln ein Konformitätsbewertungsverfahren mit einer Benannte Stelle durchlaufen müssen, gelten folgende neue Übergangsfristen:
- 31. Dezember 2027 für IVD der Klasse D
- 31. Dezember 2028 für IVD der Klasse C
- 31. Dezember 2029 für IVD der Klasse B
- 31. Dezember 2029 für IVD der Klasse A, die steril in Verkehr gebracht werden
Je nach Risikoklasse des IVD dürfen diese Bestandsprodukte somit auch nach dem Gültigkeitsdatum der IVDR am 26. Mai 2022 weiterhin in Verkehr gebracht werden, vorausgesetzt, die Produkte halten die Anforderungen der IVDD ein und es erfolgt keine signifikante Änderung am Design und der Zweckbestimmung.
Falls Sie diese Option nutzen wollen, achten Sie bitte auf die Details bei der Konformitätserklärung. Dieser Artikel hilft Ihnen dabei: EU-Konformitätserklärung – Declaration of Conformity
Beispiel: Für Ihr Produkt wurde unter der IVDD noch vor dem 26. Mai 2022 eine Konformitätserklärung gemäß Anhang III ohne Benannte Stelle ausgestellt. Dann stellt sich die Frage, unter welche Klasse das Produkt in Zukunft unter der IVDR fallen wird.
I) Fällt das Produkt künftig in Klasse D, dann dürfen Sie noch bis 31. Dezember 2027 diese Produkte herstellen und in Verkehr bringen, aber nicht mehr wesentlich ändern.
II) Fällt das Produkt künftig unter Klasse C, ist die Herstellung und Inverkehrbringung bis 31. Dezember 2028 erlaubt. Wesentliche Änderungen sind nicht gestattet.
III) Fällt das Produkt künftig unter Klasse B oder A (steril), ist die Herstellung und Inverkehrbringung bis 31. Dezember 2029 erlaubt, was somit die längste Übergangsfrist darstellt. Änderungen am Produkt sind auch hier nicht gestattet.
Auch diese Übergangsfristen sind an Bedingungen geknüpft! Hersteller müssen einen formalen Antrag bei der Benannten Stelle gestellt haben. Die Fristen hierfür sind die folgenden:
- Klasse D-Produkte: Antrag gestellt bis zum 26. Mai 2025
- Klasse C-Produkte: Antrag gestellt bis zum 26. Mai 2026
- Klasse B oder A (steril): Antrag gestellt bis zum 26. Mai 2027
Auf diese Fristen folgt sogleich die nächste Bedingung: Vier Monate nach der oben genannten Antragsstellung muss ein schriftlicher Vertrag mit der Benannten Stelle geschlossen sein, damit die IVDD-Produkte weiterhin in Verkehr gebracht werden dürfen.
Die MDCG-Leitlinie MDCG 2022-6 verschafft Ihnen Klarheit über das Konzept von „wesentlichen Veränderungen der Auslegung und Zweckbestimmung“ gemäß Artikel 110(3) der IVDR. Keine „wesentlichen Veränderungen“ umfassen insbesondere administrative Änderungen und organisatorische Änderungen – eng angelehnt an MDCG 2020-03. Im Weiteren werden die „wesentlichen Veränderungen der Auslegung und Zweckbestimmung“ definiert, wobei auf sechs Flussdiagramme verwiesen wird und ab Kapitel 4.3.2.1 konkrete Beispiele aufgeführt werden.
Beachten Sie hierzu auch die Artikel Design Changes und Software Changes.
Unabhängig von der Risikoklassifizierung nach IVDR können Sie bzw. der Distributor das Produkt auch nach Ende der Übergangsfrist weiterhin bereitstellen und inbetriebnehmen, da durch die Verordnung 2023/607 nun die „Abverkaufsregel“ entfallen ist.
Da die Begriffe “in Verkehr bringen”, “Bereitstellung” und “Inbetriebnahme” immer wieder Verständnisprobleme verursachen, empfehlen wir Ihnen diesen Artikel: https://www.johner-institut.de/blog/regulatory-affairs/inverkehrbringung/
c) Welche Anforderungen der IVDR müssen Hersteller trotz Übergangsfrist erfüllen?
Hersteller sollten jedoch beachten, dass auch für Produkte, für die die vorgeschlagenen Übergangsfristen anwendbar wären, die Anforderungen der IVDR an die Überwachung nach dem Inverkehrbringen (PMS), die Marktüberwachung, die Vigilanz und die Registrierung von Wirtschaftsakteuren und Produkten in EUDAMED einzuhalten sind. Der Vorschlag vom Januar 2024, welcher auch die Übergangsfristen der IVDR neu regelt, verpflichtet die Wirtschaftsakteure, jedes der sechs EUDAMED Module spätestens sechs Monate nach der Veröffentlichung zu nutzen.
Des Weiteren muss beachtet werden, dass Hersteller ein Qualitätsmanagementsystem gemäß Artikel 10(8) der IVDR bis zum 26. Mai 2025 einführen müssen – unabhängig davon, ob es sich um ein IVDD-Produkt mit oder ohne Beteiligung einer Benannten Stelle handelt.
d) Keine Verlängerung für (nicht-sterile) Klasse-A-Produkte und „neue“ IVD
Die von der Kommission vorgeschlagenen Verlängerungen sollen für zwei Produktgruppen allerdings NICHT gelten:
- (Nicht-sterile) Klasse-A-Produkte
- „Neue“ IVD
Fällt das Produkt künftig unter Klasse A und ist nicht steril, müssen Hersteller in jedem Fall seit dem 26. Mai 2022 einen Konformitätsnachweis gemäß IVDR erbringen und alle anwendbaren Anforderungen der IVDR erfüllen. Sie können damit keine verlängerte Übergangsfrist nutzen. Da keine benannte Stelle erforderlich ist, liegt es also einzig an den Ressourcen und der Zeitplanung des Herstellers, dieses Ziel zu erreichen.
Ebenso wenig gelten die verlängerten Fristen für „neue“ In-vitro-Diagnostika. Als solche sieht die Kommission jene IVD an, deren Konformität nicht unter der IVDD erklärt wurde. Auch für diese Produkte müssen Hersteller in jedem Fall seit dem 26. Mai 2022 einen Konformitätsnachweis gemäß IVDR erbringen.
e) Inhouse-IVD (auch Laboratory Developed Tests, LDT, genannt)
Auch Produkte, die von medizinischen Einrichtungen selbst produziert und verwendet werden, profitieren von verlängerten Übergangsfristen.
Doch Vorsicht: Ausnahme bilden die grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I, IVDR inkl. der Leistungsbewertung (IVDR, Artikel 5 (5) Satz 1, Satz 2 Buchstabe a) und Sätze 3 bis 5). Diese müssen ab dem 26. Mai 2022 in der EU erfüllt werden.
Eine Verlängerung der Frist bis zum 26. Mai 2024 gilt für die Implementierung der Anforderungen gemäß Artikel 5 (5), Unterpunkte b) und c) sowie e) bis i).
Diese beinhalten:
- Qualitätsmanagementsystem
Buchstabe b) bezieht sich auf die Forderung nach einem geeigneten Qualitätsmanagementsystem, das mindestens die Entwicklung und Herstellung von IVD aus der ISO 13485 berücksichtigen müsste, um dem Stand der Technik gerecht zu werden. - Qualitätssicherung im Labor
Buchstabe c) fordert die Einhaltung der Vorgaben der ISO 15189 für die Qualitätssicherung im Labor und die Erfüllung nationaler Akkreditierungs-Vorschriften wie in Deutschland die RiLiBÄK (Richtlinie der Bundesärztekammer für die Qualitätssicherung in der Laboratoriumsmedizin). - Bereitstellung von Information
Buchstabe e) Bereitstellung von Information für Behörden über Herstellung, Modifikation und Verwendung - Öffentliche Erklärung
Buchstabe f) Erklärung über die Einhaltung des Anhang I der IVDR - Dokumentation
Buchstabe g) Dokumentation zu Fertigung, Konstruktion und Leistung, also sozusagen die “Produktakte” - Nachweis Herstellung nach Vorgaben (Batch record)
Buchstabe h) Nachweis der Herstellung gemäß den Spezifikationen in der “Produktakte” - Schwerwiegende Vorkommnisse und Korrekturmaßnahmen
Buchstabe i) Sammeln und Bewerten von Erfahrungen in der klinischen Anwendung sowie Ableiten von Korrekturmaßnahmen
Für die Erfüllung des Artikel 5 (5, d) wird mit dem neuen Vorschlag der EU Kommission eine Übergangsfrist bis zum 31. Dezember 2030 gewährt. Dieser besagt:
“die Gesundheitseinrichtung liefert in ihrer Dokumentation eine Begründung dafür, dass die spezifischen Erfordernisse der Patientenzielgruppe nicht bzw. nicht auf dem angezeigten Leistungsniveau durch ein gleichartiges auf dem Markt befindliches Produkt befriedigt werden können;”
Diese Forderung sollte erreichen, dass zwischen Inhouse-IVD und kommerziellen Produkten am Markt kein Wettbewerb herrscht, da sie unterschiedlich reguliert werden. Nun hat die EU diesen Punkt ganz ans Ende der Übergangsfrist gesetzt mit der Begründung, dass Labore erst dann die Produkte am Markt mit dem eigenen Produkt vergleichen können, wenn auch alle IVD-Produkte am Markt unter IVDR zugelassen und in EUDAMED registriert sind.
Lesen Sie hier, wie die EU die medizinischen Labore reguliert.
f) EUDAMED
Die EUDAMED sollte ursprünglich erst nach ihrer Fertigstellung verpflichtend für alle Wirtschaftsakteure werden. Mit der Verordnung EU 2024/1860 vom Sommer 2024 soll jedes der sechs Module spätestens sechs Monate nach der Bekanntmachung, dass die Module den funktionellen Spezifikationen entsprechen, von den Akteuren genutzt werden.
Die verpflichtende Nutzung der ersten Module bezüglich der Wirtschaftsakteure, der UDI, der Benannten Stellen und der Zertifikate könnte somit im vierten Quartal 2025 starten. Die gesamte Funktionalität der EUDAMED kann laut dem Vorschlag vom Januar 2024 nicht vor dem vierten Quartal 2027 erwartet werden. Als finale Übergangsfrist für die Nutzung der EUDAMED wird das zweiten Quartal 2029 genannt.
Mehr Informationen finden Sie im Beitrag zu den Übergangsfristen von EUDAMED.
3. Was sich nicht ändert
Die IVDR gilt seit 26. Mai 2022. Damit gelten auch die Überwachungspflichten der Produkte im Markt gemäß Artikel 78 bis 81 sowie zur Vigilanz (Artikel 82 bis 87) und der Überwachung durch zuständige Behörden, ungeachtet der Übergangsfristen. Somit ist das gesamte Kapitel VII der IVDR anzuwenden, auch wenn die Produkte noch unter IVDD in Verkehr gebracht werden.
Außerdem müssen sich Wirtschaftsakteure in der EUDAMED registrieren.
However , the requirements of this Regulation relating to post-market surveillance, market surveillance, vigilance, registration of economic operators and of devices shall apply to devices referred to […] instead of the corresponding requirements in Directive 98/79/EC.
Vorschlag der EU Kommission vom 14. 10.2021 zur Anpassung der IVDR (EU) 2017/746
Die Benannte Stelle, die unter der IVD-Richtlinie Zertifikate ausgestellt hat, ist auch in der Übergangsfrist weiterhin zuständig für die Überwachung der Konformität. Wichtig ist zudem, dass keine wesentlichen Änderungen an den Produkten in der Übergangsfrist durchgeführt werden; ist dies der Fall, müssen Hersteller unmittelbar alle Anforderungen der IVDR erfüllen.
4. Was die Änderungen für Hersteller bedeuten
Hersteller sollten unbedingt weiter an ihrem Umsetzungsplan festhalten, damit ihre IVD rechtzeitig IVDR-konform werden. Gerade aufgrund der knappen Ressourcen sollten Hersteller nicht unnötig Zeit verstreichen lassen.
Zudem sollten sich Hersteller über die Risikoklasse ihrer IVD klar werden. Hiervon hängt nicht nur das Konformitätsbewertungsverfahren ab, sondern auch die Übergangsfrist.
Ein IVDR-konformes Qualitätsmanagementsystem ist bereits bis Mai 2025 zu etablieren.
5. Fazit
Die IVDR gilt seit dem 26. Mai 2022. Daran ändern auch die Änderungsverordnungen nichts. Allerdings erhält nun ein Großteil der IVD-Hersteller mehr Zeit, um die nötigen Konformitätsnachweise für ihre Produkte zu erbringen. Angesichts der wenigen Benannten Stellen, die es bislang für die IVDR gibt, dürfte dies für viele eine große Erleichterung sein – und wichtige IVD-Produkte vor dem Verschwinden vom Markt bewahren. Zudem wird die unnötige Entsorgung von sicheren Produkten verhindert.
Auch für medizinische Labore gelten nun einige Anforderungen erst mit Verzögerung. Dennoch müssen auch sie für Inhouse-IVD die Grundlegenden Sicherheits- und Leistungsanforderungen erfüllen.
Die Expertinnen und Experten des Johner Instituts unterstützen Sie gern bei Fragen zur Zulassungsstrategie von IVD oder speziell zum Thema Lab Developed Tests. Kontaktieren Sie uns einfach über das Formular oder schreiben Sie uns eine E-Mail.
Versionshistorie:
- 2024-10-08: Hinweise und neue Übergangsfristen gemäß der Änderungsverordnung 2024/1860 vom 13.06.2024 ergänzt.
- 2024-02-05: Hinweise und neue Übergangsfristen aus der Regulierung (EU) 2024/0021 ergänzt.
- 2023-04-11: Hinweise zur Regulierung (EU) 2023/607 ergänzt.
- 2023-01-09: Hinweis zum Kommissionsvorschlag ergänzt. Korrekturen, Anpassungen von Verlinkungen und redaktionelle Änderungen im gesamten Artikel.
- 2022-05-06: Hinweise zu MDCG 2022-6 ergänzt.
- 2022-03-22: Unter 2e) Hinweise zur Registrierungsfrist in EUDAMED eingefügt.



Sehr geehrter Herr Dr. Grömminger,
vielen Dank für Ihre Zusammenfassung!
Wenn wir als Hersteller die Übergangsfrist nutzen möchten, müssen wir aber trotzdem mindestens folgende Dokumente aufsetzen:
Post market surveillance Plan
Leistungsbewertung
Risikomanagement
Sehe ich das richtig?
Vielen Dank und mit freundlichen Grüßen,
Martina Mitterhuber
Liebe Frau Mitterhuber,
gerne geschehen. Danke für Ihre spanende Frage.
Als erstes sollten Sie die Risiko-Klasse A, B C oder D für Ihr IVD ermitteln gemäß IVDR Anhang VIII. daraus ergibt sich Ihre Übergangsfrist.
Ja, dann benötigen Sie für die Übergangsfrist einen Post-Market surveillance (PMS)-Plan gemäß Art. 79.
Gemäß Ihres Plans dann aber auch in zeitlichem Versatz einen PMS-Report (Art. 80) bzw. je nach Klasse des Produktes nach einem Jahr in der Übergangsfrist einen Periodic Safety Update Report, bei Klasse C und D (Art. 81)
Da solche Reports immer ein Ergebnis eines Prozesses sind, benötigen Sie auch noch entsprechende Verfahrensanweisungen:
– Eine PMS-Verfahrensanweisung
– eine für den Vigilanz-Prozess (Art. 82) sowie
– eine für das Trend Reporting (Art. 83)
Einen Leistungsbewertungsprozess und und eine Leistungsbewertung sollten Sie ja bereits für das bestehende Produkt haben. Hier sollten Sie die Übergangsfrist nutzen, die Leistungsbewertung auf die Anforderungen der IVDR anzupassen. Hiermit sind erhebliche Aufwände verbunden.
Das Risikomanagement ist erfahrungsgemäß die größte Schwachstelle bei Produkten, die bisher nicht in der Überwachung durch eine Benannte Stelle sind. Daher meine dringende Empfehlung auch diesen Prozess und die bestehende Risikomanagement-Akte zu Überarbeiten. Die Leistungsbewertung baut darauf auf, daher empfehle ich mit dem Risikomanagement zu beginnen.
Ich hoffe, das hilft Ihnen weiter.
Falls sie Anleitungen und Vorlagen benötigen, können Sie gerne eine kostenlose Demonstration unseres Auditgarants buchen: https://www.johner-institut.de/auditgarant/
Herzliche Grüße,
Sebastian Grömminger
Dear Dr. Grömminger,
What is the deadline for IVDR-manufacturers of legacy devices to register in Eudamed?
Thank you!
Best regards, Rony
Dear Rony, Article 113 states: Article 26(3) shall be applied 18 months after the date of application, provided EUDAMED is fully operational on May 26, 2022. Under these prerequisites, the deadline for registering the products and economic operators under IVDR is Nov. 26, 2023.
With best regards,
Sebastian Grömminger
Sehr geehrter Herr Dr. Grömminger,
vielen Dank für Ihre ausführliche Zusammenfassung. Eine kurze Frage habe ich jedoch: In welchem Artikel der IVDR sind die oben beschriebenen Abverkaufsregeln geregelt?
Über eine kurze Rückmeldung würde ich mich sehr freuen.
Vielen Dank
Mit freundlichen Grüßen
Leonie Ackermann
Liebe Frau Ackermann, vielen Dank für Ihr positives Feedback zum Artikel. Die betreffende Stelle in der IVDR ist Artikel 110 Absatz (4). die korrekte Begrifflichkeit für die umgangssprachliche „Abverkaufs-Regelung“ ist „Bereitstellung und Inbetriebnahme“.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Dr. Grömminger,
Vielen Dank für Ihre ausführlichen Erklärungen.
Wir sind Hersteller von Kontrollmaterialien im Bereich Toxikologie und Forensische Chemie (Drogenmissbrauch, Fahreignung, etc… unsere Produkte fallen per Definition unter der Klasse B. Wir können und werden die IVDr nicht umsetzen können. Wir werden die Übergangsfristen bis zum letzten Abverkauf unserer CE-Produkte nutzen und die Akkreditierung anstreben.
Meine Frage müssen wir uns (und die Produkte) trotzdem bei EUDAMED registrieren? Lieben Dank im Voraus für Ihre Rückmeldung. Mit freundlichen Grüßen, Natacha Valois
Liebe Frau Valois,
bitte bedenken Sie noch bei Ihren Überlegungen, dass die forensischen Produkte nicht zwingend einen medizinischen Zweck erfüllen und damit ggf. nicht unter die IVDR fallen. Für Ihre IVDs mit eindeutiger IVD-Zweckbestimmung ist die Antwort JA, auch wenn diese nach der Übergangsfrist nicht gemäß IVDR in Verkehr gebracht werden, müssen Sie die Registrierungspflichten in EUDAMED für die Firma und die „Legacy Devices“ erfüllen. Mehr dazu finden Sie hier: https://www.johner-institut.de/blog/johner-institut/legacy-devices/ und in der darin zitierten MDCG 2019-5, die zwar für die MDR geschrieben ist aber ausdrücklich auch für die IVDR gilt.
Ich hoffe, ich konnte Ihnen damit weiterhelfen,
wenn Sie mehr Unterstützung benötigen, melden Sie sich gerne bei uns.
Mit besten Grüßen,
Sebastian Grömminger
Sehr geehrter Dr. Grömminger,
seit einigen Monaten ändern viele Hersteller Name und/oder Adresse des legal Manufacturers auf deren Konformitätserklärungen.
Nun bekommen wir häufig Konformitätserklärungen zur IVDD, die nach dem 26.05.22 geändert und unterschrieben wurden.
Ist das rechtlich in Ordnung?
Darf ein Hersteller eine geringfügige Änderung der Konformitätserklärung (wie Addresse), die nichts mit dem Produkt an sich zu tun haben, auch nach 26.05.22 durchführen?
Oder müsste das in einem Sideletter zur Original-DoC passieren?
Vielen Dank für Ihre Hilfe und mit freundlichen Grüßen,
Martina Mitterhuber
Liebe Frau Mitterhuber,
vielen Dank für Ihre interessante Frage. Zunächst bin ich sehr erfreut zu lesen, dass Sie Konformitätserklärungen mit einem Datum später als der 26. Mai 2022 sehr kritisch prüfen. Damit kommen Sie Ihrer gesetzlichen Verantwortung nach. Wenn bereits für das Produkt eine DoC mit Datum vom 26. Mai oder früher vorlag und die Änderung gemäß MDCG 2022-6 eine nicht-signifikante Änderung darstellt, ist das durchaus legitim. Es dürfen jedoch keine neuen Zertifikate einer Benannten Stelle ausgestellt worden sein (betrifft nur Liste A, Liste B und Selbst-Tests gemäß IVDD). Lassen Sie sich im Zweifel eine Begründung für die Einstufung als nicht-signifikante Änderung gemäß MDCG 2022-6 vom Hersteller geben, dann sind Sie Ihrer Sorgfaltspflicht nachgekommen. Wenn jedoch weitere Zweifel aufkommen sollten, müssten Sie ggf. auch Ihre zuständige Behörde informieren.
Ich hoffe, ich konnte Ihre Frage damit beantworten.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Herr Dr. Grömminger,
aus Sicht der Importeure und Distributeure von IVD ergeben sich mit dem neuen Rechtsrahmen zusätzliche Pflichten, u.a. die Kennzeichnungspflicht als Importeur und die diversen Prüfpflichten. Unter Artikel 110 para 4 IVDR ist auch der Abverkauf der „alten“ IVDD konformer Produkte geregelt. Dieser Zeitrahmen ändert sich nach meinem Verständis entsprechend der hier oben von Ihnen dargestellten Übergangsfristen je nach Produktklassiffizierung.
Infolge dieser sehr unterschiedlichen Übergangsfristen wird die Teilung der Warenlagerbestände bei Importeuren und Händlern in „alte“ (IVDD konforme) und „neue“ (IVDR konform) IVD noch länger anhalten.
Meine Frage hierzu: Welche IVD genau fallen unter den Geltungsbereich der IVDR, „alte“ und „neue“ oder nur die „neuen“? Oder anders gefragt: Treffen die Kennzeichnungs- und Prüfpflichten ausschließlich auf die nach IVDR konform erklärten Produkte oder auch auf die bereits nach IVDD zugelassenen Produkte zu, wenn ich sie auf dem Unionsmarkt in Verkehr bringe. Wie wird hier differenziert?
Ich danke Ihnen für eine kurze orientierende Antwort.
Mit freundlichen Grüßen,
Anne Schneider
Liebe Frau Schneider,
vielen Dank für Ihre spannende Frage. Die Pflichten für die Importeure und Distributoren gemäß der IVDR gelten grundsätzlich für Produkte, die auch eine IVDR-Konformitätserklärung besitzen. Allerdings müssen Sie sich bereits als Wirtschaftsakteur registrieren auch für Produkte in der Übergangsfrist, die noch eine IVDD-Konformitätserklärung besitzen. Die Anforderungen des Kapitel VII der IVDR, die Sie als Importeur oder Distributor betreffen, sind auch für IVDD-konforme Produkte in der Übergangsfrist für Sie bindend. Lesen Sie dazu mehr in unserem Artikel zu Legacy Devices oder direkt in der MDCG 2022-8.
Ich hoffe, ich konnte Ihnen damit weiterhelfen.
Mit herzlichen Grüßen,
Sebastian Grömminger
Sehr geehrter Herr Dr. Grömminger,
um meine Frage ein wenig einzugrenzen:
Sie schreiben unter 3. „Was sich nicht ändert!“
Die IVDR gilt ab 26. Mai 2022. Damit gelten auch die Überwachungspflichten der Produkte im Markt (Artikel 78 bis 81) sowie zur Vigilanz (Artikel 82 bis 87) und der Überwachung durch zuständige Behörden, ungeachtet der Übergangsfristen. Somit ist das gesamte Kapitel VII der IVDR anzuwenden, auch wenn die Produkte noch unter IVDD in Verkehr gebracht werden.
Hier kommen ja insbesondere die Herstellerpflichten zur Sprache.
Gilt dies aber ebenso für die unter Kapitel II genannten Allgemeinen Pflichten für Importeure (Artikel 13) und Händler (Artikel 14)? Müssen also auch die von der Übergangsfrist betroffenen IVDD konformen Produkte (z.B. IVD der Klasse B) nach GB gemäß IVDR Regeln gekennzeichnet und geprüft werden, soweit anwendbar?
Mit besten Grüßen,
Anne Schneider
Liebe Frau Schneider,
danke für die Ergänzung. Die MDCG schreibt dazu: „In addition to the requirements set out in Chapter VII IVDR, also other IVDR requirements should apply to ‘legacy devices’, provided that those requirements relate to post-market surveillance, market surveillance, vigilance or registration of economic operators and devices“ (MDCG 2022-8). Da die Anforderungen der Artikel 13 und 14 im Wesentlichen mit Vigilanz und der Post-Market-Surveillance zu tun haben, würde ich diese als anwendbar betrachten.
Beste Grüße,
Sebastian Grömminger
Sehr geehrter Herr Grömminger,
vielen herzlichen Dank für Ihre Antwort.
Mit freundlichen Grüßen,
Martina Mitterhuber
Sehr geehrter Prof. Johner,
Wir stellen uns gerade ein sehr spezielle Frage:
Wir gehen davon aus, dass die technische Dokumentationen von „legacy IVDs“ auf Grundlage der 98/79 EG erstellt wurde. Wie stellt es sich nun aber dar wenn Änderungen an dieser TD nach dem 26.Mai 2022 durchgeführt werden müssen?
Die ISO 13485 verlangt Lenkungsdokumente für solche Vorgänge, diese werden aber natürlich auf die Anforderungen der neuen IVDR angepasst.
Müssen dann auch die geänderten TDs schon IVDR gemäß erstellt werden oder wäre es evtl. zulässig zwei SOPs zu erstellen einmal für Altprodukte (98/79 EG) und einmal für neue Produkte nach IVDR.
Sehr geehrte(r) Dr. Breß,
ob ein Produkt nach einer Änderung weiter „nur“ der IVDD oder der IVDR entsprechen muss, hängt davon ab, ob es sich um eine signifikante Änderung handelt oder nicht.
Das regelt die MDCG 2022-6 „Guidance on significant changes regarding the transitional provision under Article 110(3) of the IVDR“. Wenn die Änderung nach durchlaufen der darin enthaltenen Kriterien als nicht-signifikant eingestuft wird, kann die Technische Dokumentation weiterhin gemäß IVDD geführt werden. Wenn die Änderung jedoch signifikant ist, führt an der IVDR und der Technischen Dokumentation gemäß Anhang II/III kein Weg mehr vorbei. Und Ja, entsprechend sollten Sie auch zwei SOPs für die Erstellung der Technischen Dokumentation vorhalten.
Ich hoffe, ich konnte Ihre Frage adäquat beantworten. Gerne unterstützen wir bei der Qualifizierung von Änderungen gemäß MDCG 2022-6, wenn Sie Unterstützung benötigen.
Herzliche Grüße,
Sebastian Grömminger
Sehr geehrter Herr Grömminger,
Ich habe eine kurze Frage bezüglich folgenden Abschnittes in der IVDR:
Artikel 110
(3)… Produkte, die ab dem 26. Mai 2022 gemäß Absatz 3 rechtmäßig in Verkehr gebracht werden, dürfen bis zu folgenden Zeitpunkten weiterhin auf dem Markt bereitgestellt oder in Betrieb genommen werden:
a) 26. Mai 2026 für Produkte gemäß Absatz 3 Unterabsatz 2 oder Absatz 3 Unterabsatz 3 Buchstabe a;
b) 26. Mai 2027 für Produkte gemäß Absatz 3 Unterabsatz 3 Buchstabe b;
c) 26. Mai 2028 für Produkte gemäß Absatz 3 Unterabsatz 3 Buchstaben c und d.
Bedeutet dies man kann auch weiterhin neue Produkte nach IVDD auf den Markt bringen? Wie ist das Ihrer Meinung nach zu verstehen?
Gruß,
Fabina Stritt
Lieber Fabian Stritt,
bitte seien Sie vorsichtig mit dem Begriff „neue Produkte“. Wenn für diese neuen Produkte bereits eine IVDD-Konformitätserklärung vor dem 26. Mai 2022 vorlag, dürfen Produkte, die unter diese Konformitätserklärung fallen noch hergestellt und in Verkehr gebracht werden. Neu entwickelte Produkte dürfen Sie allerdings nicht mehr unter der IVDD in Verkehr bringen, da Sie dafür berets die IVDR-Konformität benötigen. Es sind lediglich nicht-signifikante Änderungen an IVDD-Produkten möglich (siehe MDCG 2022-6).
Ich hoffe, ich konnte Ihnen damit Ihre Frage beantworten.
Beste Grüße,
Sebastian Grömminger
Sehr geehrter Herr Grömminger,
könnten Sie bitte mehr licht auf das sogennant sell-off IVDR werfen? Was genau bedeutet Abverkaufsfrist wegfallen?
Vielen Dank im Voraus!
MfG
Rony
Lieber Rony,
der Wegfall der Abverkaufs-Frist bedeutet, dass IVD-Produkte, die bereits unter der IVDD vor dem 26. Mai 2022 in Verkehr gebracht wurden (im Falle von sonstigen IVDs nach IVDD die in Klasse A fallen) bzw. noch innerhalb der geltenden Übergangsfrist in Verkehr gebracht wurden oder noch werden (im Falle von Klasse B, C D und A-steril), zeitlich uneingeschränkt an die Endverbraucher verkauft werden dürfen. Das setzt lediglich voraus, dass die Produkte noch konform sind, also die Haltbarkeit noch nicht überschritten ist, die Verpackung unversehrt, kein Rückruf vorliegt und die Konformitätserklärung noch gültig ist.
Die Produkte müssen also rechtsgültig nach IVDD in Verkehr gebracht worden sein. D.h. in Form von einem Besitzerwechsel, Nutzungsvertrag etc. an eine natürliche oder juristische Person, die in der EU ansässig ist. Dann kann das Produkt zeitlich unbegrenzt abverkauft werden, solange es die Produkthaltbarkeit erlaubt.
Ich hoffe, damit konnte ich ihnen die aktuell geplante Anpassung der IVDR hinsichtlich Abverkaufs-Frist bzw. sell-off ausreichend erklären.
haken Sie gerne spezifisch nach, wenn noch etwas unklar sein sollte.
Mit besten Grüßen,
Sebastian Grömminger
Liebes Johner-Team,
erstmal großen Dank für die wirklich informativen Artikel zur IVDR und UDI, das hilft sehr!
Ich habe eine Frage zu den Fristen zur Basis-UDI eines Klasse A non-sterile IVD:
1) Ab wann muss der Hersteller eine Basis-UDI zuweisen und in den entsprechenden Dokumenten (DoC, TD etc) ausweisen?
2) Ab wann muss der Hersteller diese Basis-UDI in der EUDAMED registrieren? Uns ist nicht klar, ob die IVDR einen Unterschied zwischen der Zuweisung der Basis-UDI und der Registrierung in der EUDAMED macht.
3) MDCG 2022-12 spricht in Bezug auf Artikel 1-3 davon, dass einerseits das Eintragen in die Datenbank freiwillig sei (Absatz 1), andrerseits „that obligation of UDI assignment … applies from 26. May 2022“ (Absatz 3); was etwas widersprüchlich klingt.
4) Auf der EU-Seite findet sich eine Frist ab „Funktionalität“ von 24 Monaten, hier im Artikel unter 2e) von 18 Monaten?
5) Bezieht sich die Funktionalität („fully functional“) auf die gesamte EUDAMED oder nur das entsprechende Modul?
Ich hoffe, Sie können etwas Licht ins Dunkel bringen ;-). Vielen Dank schonmal und herzliche Grüße
Liebe Frau Roeschard,
vielen herzlichen Dank für Ihre spannenden Fragen.
Zu welchem Zeitpunkt Sie die Basis-UDI in Ihren Dokumenten ausweisen, bleibt Ihnen überlassen. Jedoch muss gesagt, werden, dass der späteste Zeitpunkt bei einem Klasse A-Produkt der ist, an dem Sie die Konformitätserklärung gemäß IVDR ausstellen würden. Zu diesem Zeitpunkt muss die Basis-UDI in allen relevanten Dokumenten genannt sein.
Diese Anforderung geht aus Art 24(3) der IVDR hervor und darauf bezieht sich auch der von Ihnen genannte Abschnitt in dem MDCG 2022-12 Guidance-Dokument. „Manufacturers should note that the obligation of UDI assignment (Basic UDI and UDI-DI) to a device applies from 26 May 2022 (Art. 24(3) IVDR).” Bitte beachten Sie, hier geht es um die Zuweisung der UDI, nicht die Produktregistrierung.
Im Gegensatz zum vorausgehenden Absatz: „The system may be used (on voluntary basis) for registration of devices even before the notice of full functionality of Eudamed has been published. Nevertheless, manufacturers should refer to the national provisions in Member States establishing product registration schemes.”
Da das Modul der Produktregistrierung in EUDAMED bereits funktionsfähig ist, verweist die MDCG 2022-12 darauf, dass Hersteller diese Funktion schon jetzt freiwillig nutzen dürfen, neben der Verpflichtung der UDI-Vergabe gemäß nationalen Anforderungen (BAnz AT 27.05.2022 B4).
Die nationale Vorschrift basiert auf dem MPDG – laut der Veröffentlichung des Bundesanzeiger für Hersteller mit Sitz in Deutschland (BAnz AT 27.05.2022 B4) gilt die Anforderung der Produktregistrierung (inkl. Basis-UDI des IVDR-konformen Produktes) seit dem 26. Mai 2022 über das DMIDS und hat für „legacy devices“ bis spätestens 26. November 2023 (18 Monate) zu erfolgen.
Wird im Amtsblatt der Europäischen Union die volle Funktionsfähigkeit von EUDAMED bekanntgegeben (alle Module funktionieren), so sind die Produkte bis mindestens zu dem späteren in Artikel 113 Absatz 3 Buchstabe f der IVDR genannten Datum wahrzunehmen. Das bedeutet, dass Sie Ihre Wirtschaftsakteure und Produkte erst frühestens 6 Monate nach Bekanntmachung der Funktionsfähigkeit von EUDAMED registrieren müssen, spätestens jedoch 24 Monate nach dieser Bekanntmachung (s. Art. 113 (3) a)).
Ich hoffe, ich konnte Ihnen damit Ihre Frage beantworten.
Beste Grüße,
Diana Gabriel
Sehr geehrter Frau Gabriel,
vielen Dank für den interessanten Fachartikel (Übergangsfristen der IVDR). Ich habe eine kurze Frage bezüglich was in dem Artikel geschrieben (Die verpflichtende Nutzung der ersten Module bezüglich der Wirtschaftsakteure, der UDI, der Benannten Stellen und Zertifikate könnte somit im vierten Quartal 2024 starten). Ich dachte gemäß dem Q&A-Dokument, Ende 2025 und nicht 2024.
Vielen Dank im Voaus!
MfG
Younis
Lieber Younis,
Sie haben völlig recht. Es ist Ende 2025. Vielen herzlichen Dank für Ihren Hinweis.
Ich habe es soeben im Artikel entsprechend angepasst.
Mit freundlichen Grüße
Diana Gabriel
Sehr geehrte Frau Gabriel,
ich hake bei der 2. Zeile ihrer wie immer sehr schönen Übersicht über die neuen Übergangsfristen. Es ist nachvollziehbar, dass der Gesetzgeber zwischen Produkten unterscheidet, die ein Zertifikat (insb. Liste A) haben, das zum Inkrafttreten der neuen Übergangsfristen noch gültig war, versus solchen, deren Zertifikat vorher (aber nach dem 26.5.2022) abgelaufen ist und in letzterem Fall die genannte Nebenbedingung fordert. Allerdings lautet die genaue Formulierung in dem Proposal, dass der Hersteller bereits VOR dem Ablauf des fraglichen Zertifikates einen entsprechenden Antrag zur IVDR Zertifizierung für dieses Produkt bei einer benannten Stelle gestellt hat. Das engt die Zahl vor allem der Liste A Produkte, die von der Regelung profitieren, doch erheblich ein, wenn ich das richtig verstehe. Die Wichtigkeit einer solchen Antragsstellung bei einer Benannten Stelle für eine Übergangsregelung konnte doch dem Hersteller zu dem Zeitpunkt (des Ablaufs des Zertifikats) doch gar nicht bewusst sein. Oder das Zertifikat war vielleicht sogar vor der offiziellen Benennung der Benannten Stelle , mit der ein Hersteller schon immer zusammengearbeitet hat, abgelaufen.
Bin ich hier auf der falschen Spur oder ist das wirklch so intendiert?
Lieber Herr Börchers,
vielen herzlichen Dank für Ihr positives Feedback.
Zu Ihrer Frage: Die EU scheint genau das beabsichtigt zu haben, denn sie schreibt im aktuellen Vorschlag: „These conditions will ensure that only manufacturers that are actively taking the necessary steps to transition to the new rules and continue to place on the market devices meeting high safety standards will benefit from the additional time.„. Sollte das Zertifikat bereits abgelaufen sein, so hatte der Hersteller unter Umständen nicht vor, das Produkt weiterhin in Verkehr zu bringen, denn gemäß Verordnung (EU) 2022/112 vom Januar 2022 endet die Übergangsfrist für diese Produkte im Mai 2025. Endeten IVDD-Zertifikate vor diesem Datum, so konnten Schritte zur Verlängerung des IVDD-Zertifikates mit der ausstellenden Benannten Stelle vereinbart werden. Produkte dürfen dementsprechend nicht signifikant geändert werden (Artikel 110, IVDR), da sonst das IVDD-Zertifikat seine Gültigkeit verliert. Sollte die Benannte Stelle, die das IVDD-Zertifikat ausgestellt hat, nicht gemäß IVDR ernannt sein, so sollte des Weiteren bereits ein Vertrag mit einer, unter IVDR ernannten, Benannten Stelle abgeschlossen sein (gemäß Anhang VII, IVDR) oder in naher Zukunft abgeschlossen werden, um die aktuelle Deadline vom Mai 2025 einhalten zu können. So ist aus Sicht der EU-Kommission erkennbar, dass der Hersteller aktiv Schritte einleitet, damit seine Produkte weiterhin in Verkehr gebracht werden können (siehe obiges Zitat und die resultierende Anforderung eines Vertrages). Erst dann profitiert der Hersteller von den neuen vorgeschlagenen Übergangsbestimmungen.
Ich hoffe, ich konnte Ihnen weiterhelfen.
Mit freundlichen Grüßen
Diana Gabriel
Sehr geehrtes Johner-Team,
Eine Frage zu den Übergangsvorschriften IVDR.
In VO (EU) 2024/1860 heisst es in Artikel 2(3): „Artikel 110 wird wie folgt geändert“
…3(c)d: „Der Hersteller hat spätestens bis 26. Mai 2025 ein QM-System gemäß Artikel 10 Absatz 8 eingerichtet;“
In VO (EU) 2023/603 Artikel 1(b) steht: „Absatz 3 erhält folgende Fassung“:
…3(c)d „Der Hersteller hat spätestens bis 26. Mai 2025 ein QM-System gemäß Artikel 10 Absatz 9 eingerichtet;“
Wir stellen Medizinprodukte und IVD her, unser ISO 13485 konformes QM-System besteht schon sehr lange und wurde 2024 rezertifiziert.
IVD-Produkte wurden bisher nicht durch eine Benannte Stelle konformitätsbewertet. Es fallen aber nunmehr einige unserer Produkte in die IVD-Risikoklasse A, die steril in den Verkehr gebracht werden und zukünftig zumindest hinsichtlich der Sterilisation und Aufrechterhaltung der Sterilität unter Hinzuziehung einer Benannten Stelle konformitätsbewertet werden müssen.
Ist davon auszugehen, dass eine positive Zertifizierung des QM-Systems nach ISO 13485 gemäß MDR (EU) 2017/745 Artikel 10 Absatz 9 durch eine für MDR zugelassene Benannte Stelle ohne weitere Konformitätsbewertung durch eine für IVDR zugelassene Benannte Stelle, die bis 26.05.2025 abgeschlossen sein müsste, akzeptiert wird -also einfach übertragen wird? Oder kann es sein, dass doppelt auditiert wird?
Es handelt sich um eine Benannte Stelle, die für die Zertifizierung beider Verordnungen zugelassen ist, die Konformitätsbewertung gemäß MDR bzw. IVDR aber in unterschiedliche Arbeitsgruppen aufgeteilt hat .
Lieber Herr Lichtenthal,
vielen Dank für Ihre detaillierte Anfrage bezüglich der Übergangsvorschriften der IVDR und der Bewertung Ihres Qualitätsmanagementsystems (QMS) unter der IVDR und MDR. Da Sie bereits ein nach ISO 13485 zertifiziertes System haben, sind Sie in einer guten Ausgangsposition, dennoch gibt es einige spezifische Anforderungen und Unterschiede zwischen MDR und IVDR zu beachten.
Anhand der von Ihnen angeführten Verordnungen EU 2024/1860 und EU 2023/603 und Ihrem spezifischen Fall betreffend IVD-Produkte der Klasse A, die steril in den Verkehr gebracht werden sollen, ist eine Überprüfung durch eine für die IVDR benannte Stelle notwendig. Das QMS, obwohl es bereits nach ISO 13485 zertifiziert ist, muss speziell für die Aspekte der Sterilisation und Aufrechterhaltung der Sterilität gemäß den Anforderungen der IVDR konformitätsbewertet werden.
Die Tatsache, dass Ihre benannte Stelle für beide Verordnungen (MDR und IVDR) zugelassen ist, bedeutet nicht automatisch, dass eine Zertifizierung unter der einen Regulation direkt für die andere gültig ist. Die Unterschiede in der Spezifikation beider Verordnungen und die Aufteilung in verschiedene Arbeitsgruppen innerhalb der benannten Stelle weisen darauf hin, dass eine separate Überprüfung und möglicherweise ein spezifisches Audit für die IVDR-konforme Bescheinigung Ihres QMS notwendig sind. Die genauen Details dieser Bewertung sollten direkt mit Ihrer benannten Stelle besprochen werden.
Zusammenfassend lässt sich sagen, dass Sie mit einer zusätzlichen Auditierung durch Ihre benannte Stelle rechnen sollten, speziell ausgerichtet auf die Anforderungen der IVDR bezüglich sterilen IVDs der Klasse A. Eine direkte Übertragung der Zertifizierung ohne weiteres Audit erscheint unter den gegebenen Regularien und Ihrer spezifischen Produktkonstellation unwahrscheinlich.
Für eine genauere Abstimmung und zur Sicherstellung, dass alle Rechtsvorschriften und Spezifikationen korrekt eingehalten werden, empfehle ich, frühzeitig und regelmäßig das Gespräch mit Ihrer Benannten Stelle zu suchen. Nur so können Sie effizient auf die Fristen reagieren und eine rechtzeitige Konformitätsbewertung sicherstellen.
Sollten Sie weitere Unterstützung benötigen, stehen wir Ihnen gerne zur Verfügung. Ich freue mich von Ihnen zu hören.
Herzliche Grüße,
Kai Eder
Liebes Johner-Team,
unsere nationale Behörde fordert für die Ausstellung von Freiverkaufszertifikaten von uns als Hersteller von IVDs (sonstige IVDs gem. IVDD und unter der IVDR Klasse A steril), dass wir einen Nachweis erbringen, dass unser QM-System die Anforderungen von Artikel 10 der IVDR erfüllen. Derzeit ist es nur erforderlich, dass wir als Hersteller sicherstellen, dass das QM-System diese Anforderungen erfüllt – was es auch tut. Es muss aber noch nicht zertifiziert sein. Wie kann man daher jetzt einen Nachweis erbringen, dass die Anforderungen erfüllt sind, ohne dass wir die entsprechenden SOPs, etc. von unserem QM-System an die Behörde übermitteln? Wir als Hersteller können nur eine Bestätigung ausstellen, dass wir diese Anforderungen erfüllen. Das ist in meinen Augen aber kein Nachweis.
Vielen Dank im Voraus für Ihre Einschätzung.
Schöne Grüße,
Maximilian
Lieber Herr Fürhauser,
Vielen Dank für die interessante Frage und die Schilderung Ihrer Situation. Sie haben die aktuellen Gegebenheiten der IVDR Übergangsfristen für sterile Klasse A Prodkte und Anforderungen an die QM-Systeme der Hersteller richtig erfasst.
Leider kann ich Ihnen auf diese unternehmens- und produktspezifische Frage hier in unserem Fachartikelblog keine passgenaue Antworten geben. Sie können sich gerne über unser Kontaktformular bei uns melden und wir erarbeiten zusammen im Rahmen der Beratung eine Lösung, wie Sie an ihr Freiverkaufszertifikat kommen.
Ich freue mich von Ihnen zu hören und danke für das Verständnis,
Kai Eder