
Sie beschreibt darin, wann Sie eine erneute 510(k) Einreichung (Premarket Notification) benötigen und wann Sie die Änderungen „nur“ dokumentieren müssen.
1. Was ein Software Change ist
a) Software Changes im „Scope“ des Guidance Documents
Unter einem „Software Change“, der in den Anwendungsbereich des Guidance Documents fällt, versteht die FDA jede Änderung an einer Software, die als standalone Software oder als Teil eines Produkts (embedded Software) bereits legal nach einem 510(k) Verfahren zugelassen wurde.
D.h. für Änderungen der Software während der Entwicklung ist das Guidance Document ebenso wenig anwendbar wie für Software, die ein anderes Zulassungsverfahren (z.B. PMA) durchlaufen hat.
b) Beispiele für Software Changes
Beispiele für Software Changes sind:
- Geänderter Algorithmus, um Nutzen für Patienten weiter zu erhöhen
- Verbesserung der GUI (Benutzerschnittstelle) z.B. um Risiken zu minimieren
- Austausch einer Komponente eines Drittherstellers auf die neuste Version
- Implementierung einer neuen risikominimierenden Maßnahme
- Umsetzung eines neuen Kundenwunsches z.B. in Form zusätzlicher Funktionalität
- Fehlerbehebung (Bug Fix)
- Refactoring, um Wartbarkeit zu verbessern
- Umstieg auf nächste Version der Programmiersprache
c) IEC 62304 und Software Changes
Die IEC 62304 legt ebenfalls fest, was Hersteller bei Software Changes beachten sollten. Diese Vorgaben, zu denen Sie weiter unten mehr erfahren, betreffen aber nicht die Fragestellung, ob eine Neueinreichung (z.B. bei einer Benannten Stelle) notwendig ist.
2. Wann Sie bei der FDA neu einreichen müssen
a) Die Regeln
Die FDA verlangt bei Änderungen immer eine Neueinreichung, es sein denn eine der folgenden Bedingungen ist erfüllt (die Nummern beziehen sich auf die Grafik unten):
- (1) Die Änderung dient ausschließlich der Verbesserung der IT-Sicherheit
- (2) Die Änderung ist eine reines Bug-Fixing.
- Es spricht auch sonst nichts dagegen.
Den dritten Punkt können Sie dann als erfüllt sehen, wenn alle(!) der folgenden Bedingungen zutreffen
- (3) Es gibt keine neuen Fehlerursachen, d.h. Elemente in einer Ursachenkette, die zu einem nennenswerten Schaden führen können.
- (4) Es entstehen dadurch keine neuen Gefährdungen oder Gefährdungssituationen, die zu einem nennenswerten Schaden führen können.
- (5) Die Änderung betrifft keine bereits bestehende risikominimierende Maßnahme oder bedingt eine solche Maßnahme.
- (6) Die (klinische) Leistungsfähigkeit und die Erfüllung der Zweckbestimmung können durch die Änderung nicht beeinträchtigt werden.
- (7) Sonstige Änderungen sind beherrscht.
Erneut gibt es einen „Gummiparagraphen“ (Punkt 7.), auf den wir gleich zu sprechen kommen.
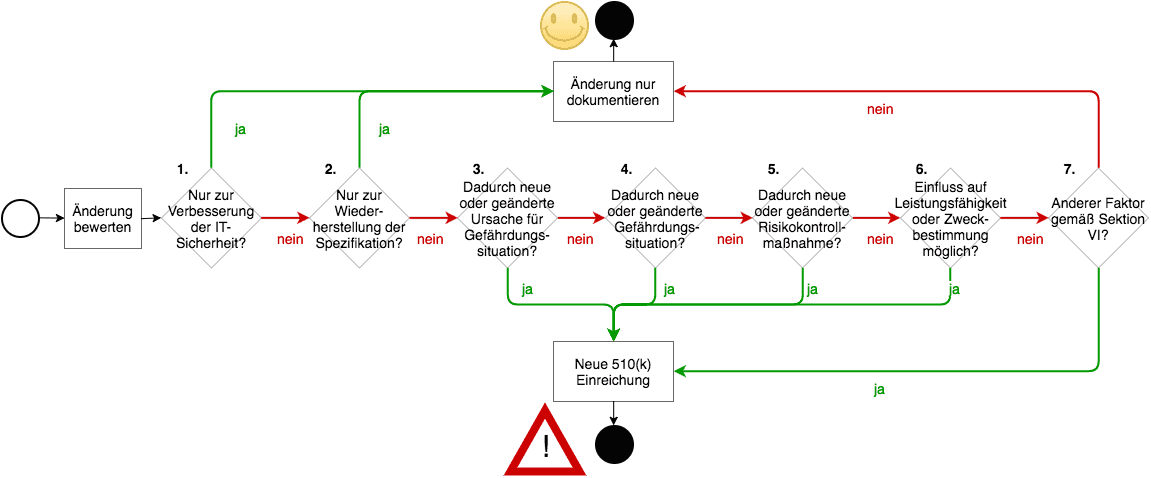
Die Darstellung macht klar, dass die FDA die Notwendigkeit eines erneuten Einreichens v.a. davom abhängig macht, ob zusätzliche oder höhere Risiken durch den Software Change entstehen können. Gleichzeitig möchte die FDA Verbesserungen der IT-Sicherheit nicht durch bürokratische Hürden erschweren.
b) Beispiele
Die Nummerierung in der Liste oben entspricht der der FDA. Zu jedem dieser Punkte nennt die FDA Beispiele, die wir hier aufgreifen, ergänzen oder in verständlicherer Sprache wiedergeben.
- Verbesserung der IT-Sicherheit
- Prüfen von Benutzereingaben einer Anwendung auf Cross-side Scripting
- Austausch des Verschlüsselungsalgorithmus durch eine sicherere Version
- Bug-Fixing (Herstellen der spezifizierten Funktionalität)
- Austausch des fehlerhaften Kleiner-Zeichens (<) durch das korrekte Kleiner-Gleich-Zeichen (≤).
- Ergänzung der vergessenen Maskierung von Sonderzeichen beim Einlesen von HL7 Nachrichten
- Neue Fehlerquellen
- Hinzufügen eines neuen Behandlungsparameters bei einem EEG-Gerät
- Software für ein zusätzliches Betriebssystem anbieten
- Neue Gefährdungen oder Gefährdungssituationen
- Ein implantiertes aktives Medizinprodukt, das Software enthält, soll über kabellose Verbindung „von außen“ konfiguriert werden können. Diese neue Übertragung kann eine Energiezufuhr und damit eine neue bzw. geänderte Gefährdung(ssituation) bedeuten.
- Neue oder geänderte Risikokontrollmaßnahmen
- Die Alarmgrenzen eines Chirurgie-Roboters werden aufgeweitet, um in der Praxis auftretende Fehlalarme zu minimieren.
- Bei der Überprüfung einer Probennummer verlangt die Software vom Bediener nicht mehr eine explizite Freigabe der Messung, sondern gibt nur eine Warnung aus und protokolliert das Problem.
- Möglicher Einfluss auf Leistungsfähigkeit oder Erreichen der Zweckbestimmung
- Um den Durchsatz eines IVDs zu erhöhen, ändert der Hersteller die Software dahingehen, dass sie die Messung verkürzt.
- Der Hersteller möchte durch einen neuen Software-Algorithmus die Detektion von Arhythmien aus EKG-Daten verbessern.
- Weitere Faktoren: Es gibt Faktoren, bei denen keines der o.g. Kriterien greift, bei denen aber dennoch eine Neueinreichung abgewogen werden muss. Die FDA ermutigt die Hersteller in diesem Graubereich (wie sie es selbst nennt), Kontakt mit ihr aufzunehmen. Als Beispiele nennt sie:
- Änderungen an Programmiersprache
- Portierung auf neue Hardware, Betriebssysteme, Middleware usw.
- Reengineering und Refactoring
Change Requests sind meist formalisierte und dokumentierte Anträge oder Wünsche an die Änderung eines Produkts bzw. einer Software. Bei Medizinprodukten gilt es, bei Change Requests die regulatorischen Anforderungen zu erfüllen.
3. Change Requests
a) Was sind Change Requests?
Wie der Name vermuten lässt, sind Change Requests, Bitten von Kunden, Anwendern oder eigenen Mitarbeitern, etwas zu ändern
- am Produkt bzw. am Code,
- an der Konfiguration / Parametrierung des Produkts,
- an der Umgebung, in der das Produkt läuft.
Die meisten Medizinproduktehersteller unterscheiden bei Change Requests, die von Anwendern kommen, nicht zwischen User Request und User Requirement. Meist entsprechen die User Requests konkreten (neuen bzw. geänderten) System-/Software-Anforderungen bzw. System-/Software-Spezifikationen und eben nicht User Requirements.
Lesen Sie hier mehr zum Unterschied zwischen User/Customer Request und User/Customer Requirement.
Hersteller sollten in ihren Ticket-Systemen beides unterscheiden, so wie auch die FDA und die ISO 13485 Stakeholder-Anforderungen (hierzu zählen User Requirements) und System-Anforderungen unterscheiden.
Ebenso sind Hersteller verpflichtet zu unterscheiden:
- Korrekturen
- Korrekturmaßnahmen
- Vorbeugemaßnahmen
- Sonstige Produktverbesserungen
Lesen Sie hier mehr zum Thema Korrekturen, Korrekturmaßnahmen und Vorbeugemaßnahmen.
b) Change Requests und Risikomanagement
Viele Firmen arbeiten mit Ticket- bzw. ALM-Systemen, mit denen sie den kompletten Workflow abbilden, vom ersten „Customer Request“ über das Release des Produkts bis zum späteren Change Request. Dabei fällt gelegentlich auf, dass es in den Tickets ein „Risikoklassifizierung“ in Form einer Dropdown-Liste gibt, mit Hilfe derer man abschätzen soll, ob ein „Request“ kritisch sei oder nicht. Der weitere Workflow hängt teilweise von dieser Einschätzung ab.

So gut es ist, dass man sich sehr früh über Auswirkungen von Änderungen am Produkt Gedanken macht, so sehr sei davor gewarnt, diese Klassifizierung nur am Anfang und nur auf Basis des Change Requests vorzunehmen.
Ohne Kenntnis des Nutzungskontexts, ohne Kenntnis der inneren Struktur (der System- oder Softwarearchitektur) ist so eine Abschätzung – letztlich bedürfte es einer Gefährdungs- und Risikoanalyse – nicht möglich. Daher darf es nicht einzig dem Produktmanager oder Support-Mitarbeiter überlassen sein, Risiken einzuschätzen. Das ist die Aufgabe eines Teams aus Risikomanager, Produktmanager und System- oder Softwarearchitekt(en). Wenn die Schnittstellen z.B. die GUI geändert wird, müssen Sie ggf. sogar die Kontextexperten und Ärzte involvieren.
Dieser Change Request findet aus Sicht Risikomanagements in der sog. nachgelagerten Phase statt und muss damit die Forderungen der ISO 14971 (Kapitel 9) erfüllen.
c) Change Requests und IEC 62304
Auch die IEC 62304 hat genaue Vorstellungen, mit Bezug zu diesen Change Requests. Zum einen erwartet Sie das Erfassen von Information,
- als Änderungsanforderungen für die Entwickler und
- für die Trendanalyse z.B. mit Hinblick auf den Problemlösungsprozess der IEC 62304
Dann geht es bei den Change Requests um die Freigabe, dass überhaupt etwas geändert werden darf. Die IEC 62304 spricht hier nicht von Change Requests sondern vom Konfigurationsmanagementprozess.
d) Change Requests und ISO 13485 bzw. FDA
Sowohl die FDA als auch die ISO 13485 stellen Anforderungen an die Corrective and Preventive Actions. Den Fehler nur zu beheben, genügt nicht. Es geht auch um Vorbeugemaßnahmen.
e) Werkzeuge für das Verwalten von Change Requests
Zu den Vertretern von Werkzeugen für das Verwalten des Application Lifecycles zählen
- JIRA
- TeamFoundationServer
- Polarion mit dem speziellen Add-ons MedPack und RiskPack für die IEC 62304 und ISO 14971 konforme Dokumentation.
Mit einigen Ticketsysteme wie Bugzilla oder Mantis lässt sich der Software-Lifecycle nicht einfach IEC 62304-konform abbilden.
4. Software Change: Was Sie noch beachten sollten
Unabhängig von der Frage, ob ein Software Change eine Neueinreichung erforderlich macht, sollten Sie folgendes bedenken:
- Änderungen an der Software – auch während der Entwicklung – müssen einem Prozess folgen.
- Dazu zählt, dass Änderungen freigegeben sein müssen,
- dass die Software nach den Änderungen ganz oder teilweise neu geprüft (Regressionstests) wird und
- dass Sie Änderungsanfragen daraufhin untersuchen, ob weitergehende Maßnahmen wie eine Behördenmeldung oder ein Rückruf notwendig sind.
- Ihre Benannte Stelle kann unabhängig von der Vorstellung der FDA von Ihnen verlangen, Softwareänderungen (Software Changes) zu kommunizieren.



sehr geehrter Herr Johner
danke für diesen informativen Artikel.
Ich denke, dass SW Changes nicht nur für FDA, sondern auch unter der bestehenden Gesetzgebung MDD / IVDD und ganz besonders für die immer näher rückenden neuen Regularien MDR / IVDR notwendig sind? In Europa müssen die SW Changes an die Notizierten Stellen (NB) gemeldet oder zu mindest dokumentiert und bereitgehalten werden.
Herzliche Grüsse
Peter Pianegonda
Ich stimme Ihnen absolut zu, lieber Herr Pianegonda!
Die FDA hat nur genauere Vorgaben spezifiziert, weshalb ich diese im Artikel in den Fokus gestellt habe.
Durch die UDI-DI und UDI-PI-Abgrenzung bei Software wird aber implizit klar, welche „Größe der Änderung“ der Gesetzgeber als relevant sieht.
Danke für Ihren wichtigen Hinweis!
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner
Besten Dank für die wertvolle Zusammenstellung. Das bedeutet für Softwarehersteller eigentlich, dass sie nur sehr langsam und aufwändig neue Releases machen können, da wohl die wenigstens „nur“ zur Verbesserung der IT-Sicherheit oder reine Bugfix-Updates sind, sondern oft auch Verbesserungen z.B. von AI/ML-Modellen oder kleinen Funktionserweiterungen beinhalten. Im Endeffekt erlaubt dies im Idealfall somit maximal 2-3 Releases jährlich.
Ich habe gehört, dass die FDA sich dessen auch bewusst ist und deshalb eine verschnellerte Variante von 510(k) Einreichungen anbietet, die speziell für Software-Updates gedacht sind und innerhalb von 30 Tagen gereviewed sein sollen. Leider finde ich dazu keine Informationen. Können Sie mir bitte weiterhelfen?
Besten Dank und mfG,
Andre M
Sehr geehrter Herr M,
es ist genau, wie Sie sagen: Die FDA hat mit ihrem „Proposed Framework“ ihre Gedanken beschrieben.
Wir haben dazu im Artikel zur künstlichen Intelligenz in der Medizin im Kapitel 3.b) die relevanten Links.
Mit bestem Dank für Ihr Interesse und mit vielen Grüßen, Christian Johner