
Die MDR und die IVDR verwenden die Begriffe Produktkategorie und generische Produktgruppe, ohne diese vollständig zu definieren. Die ISO 13485:2016 führt die Medizinproduktegruppe ein. Schließlich verwendet die MDCG den Begriff Device Range. Von den Definitionen dieser Begriffe hängt die Vergabe der UDIs und das Sampling der Produktakten bei der Zertifizierung ab.
Somit ist für Hersteller, Behörden und Benannte Stellen ein gemeinsames Verständnis dessen wichtig, was Produktkategorien, generische Produktgruppen, Medizinproduktegruppen und „Device Ranges“ sind. Das erspart unnötige Dispute bei Audits, bei Bewertungen der technischen Dokumentation und bei der Entscheidung, ob Hersteller für zwei Produkte die gleiche Basis-UDI-DI bzw. UDI-DI vergeben dürfen.
1. Definitionen: Was Produktgruppen und Produktkategorien sind
Die EU-Medizinprodukteverordnung MDR definiert zwar den Begriff generische Produktgruppe. Was eine Produktkategorie ist, legt sie nicht fest. Das Guidance Document MDCG 2019-13 schafft zum Glück mehr Klarheit.
a) Generische Produktgruppe – Generic device group
Definition MDR
Die MDR definiert eine generische Produktgruppe wie folgt:
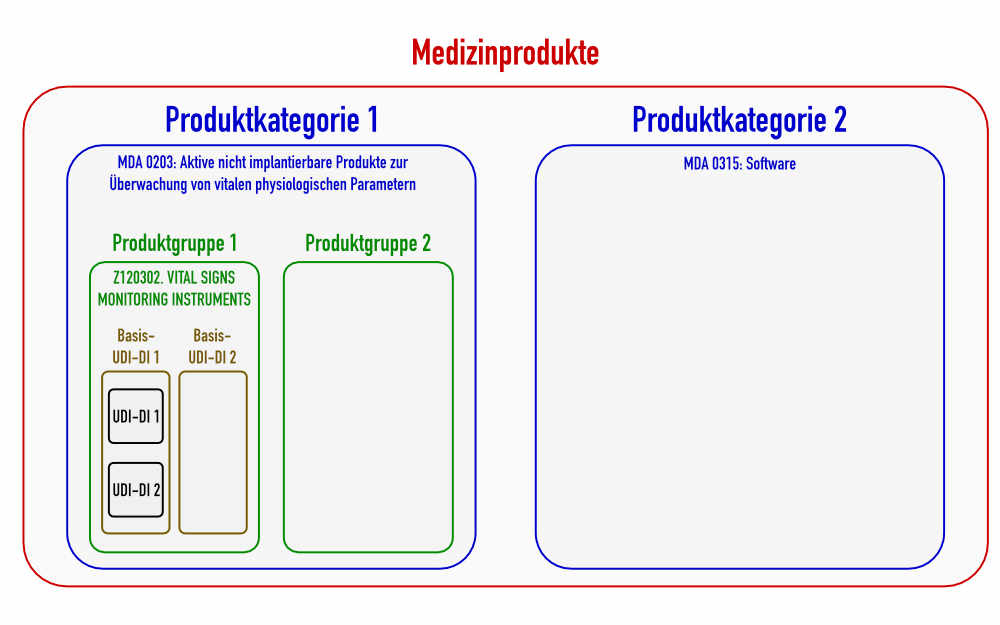
Festlegung der MDCG: Generische Produktgruppe entspricht der 4. Ebene der EMDN
Diese Definition würde sicher zu ständigen Diskussionen über die korrekte Klassifizierung führen. Daher ist es sehr begrüßenswert, dass die Medical Device Coordination Group MDCG die generische Produktgruppe verstanden haben will …
„as the 4th level of the European Nomenclature on Medical Devices (EMDN) (i.e. combination of one letter plus 6 digits) [in respect of the MDR], and in respect of the IVDR as the 3rd level of the EMDN (i.e. combination of one letter plus 4 digits respectively) in combination with the most appropriate IVP code.”
MDCG 2019-13
Beispielsweise würden Blutdruckmessgeräte (Blood Pressure Monitoring) keine generische Produktgruppe bilden (s. Abb. 2), da sie bereits auf der fünften Ebene anzusiedeln sind. Sie zählen zur generischen Produktgruppe „Vital Signs Monitoring Instruments“.
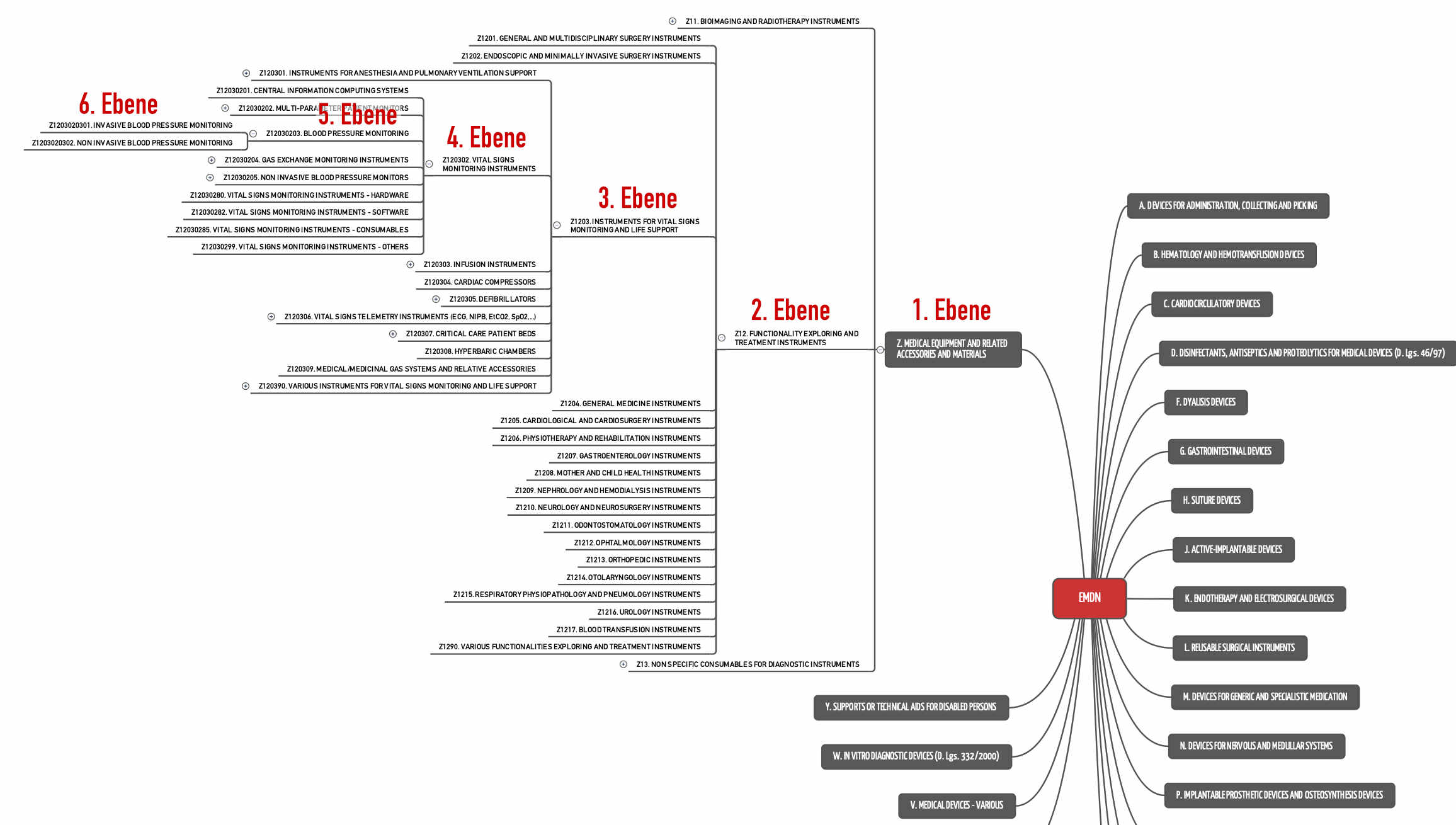
Das CND/EMDN-System umfasst 1681 Codes auf der vierten Ebene – ergo 1681 generische Produktgruppen.
b) Medizinproduktegruppe gemäß ISO 13485: Granularer als die „generische Produktgruppe“
Nicht zu verwechseln mit der generischen Produktgruppe ist die Medizinproduktegruppe gemäß ISO 13485:2016:
Der Begriff Medizinproduktegruppe ist enger gefasst als die generischen Produktgruppen gemäß MDR, da er nur Produkte mit dem gleichem bestimmungsgemäßen Gebrauch und v.a. den gleichen Design- und Leistungseigenschaften zusammenfasst. Beispielsweise würde zwei Unterarm-Blutdruckmessgeräte für den Consumer-Markt, die sich in der Systemarchitektur unterscheiden, in die gleiche generische Produktgruppe fallen, nicht aber in die gleiche Medizinproduktegruppe.
Die ISO 13485 erlaubt Herstellern für Produkte einer Medizinproduktegruppe, eine gemeinsame technische Dokumentation zu erstellen. Die MDR bzw. MDCG erlauben dies für Produkte mit der gleichen Basis-UDI-DI, nicht notwendigerweise aber für Produkte aus der gleichen generischen Produktegruppe.
c) Produktkategorie – Category of device: Entsprechen den MDA/MDN-Codes
Die MDR lässt die Definition Produktkategorie vermissen. Laut MDCG sind damit die „Codes der ihnen entsprechenden Produktarten“ gemäß der EU-Durchführungsverordnung (EU) 2017/2185 gemeint.:
Diese Verordnung unterscheidet 71 Codes für die durch die MDR regulierten Produkte und 79 Codes für IVD-Produkte. Die Kategorien teilen die Medizinprodukte in größere Klassen ein als die Produktgruppen (siehe auch Abb. 1).
d) Device Range: Der Überbegriff
Die MDCG führt mit Device Range einen weiteren Begriff ein:
Device Range ist also der Überbegriff, der je nach Klasse der Produkte einmal eine Produktkategorie (Klasse IIa) und einmal die generische Produktgruppe (Klasse IIb) bezeichnet.
Lesen Sie hier mehr zum Thema Codes, insbesondere zu den UMDNS-, GMND-, CND/EMDN-Codes. Mit Download!
2. Relevanz: Weshalb Produktgruppen und Produktkategorien wichtig sind
a) Sampling von Medizinprodukten bei Audits und Bewertungen der technischen Dokumentation
Das Sampling hängt von der Klasse ab
Die Einteilung von Produkten in Produktgruppen und Produktkategorien ist v.a. für das Sampling, d.h. die Auswahl von Stichproben, bei QM-Audits und Bewertung der technischen Dokumentation relevant:
- Bei den weniger kritischeren Produkten der Klasse IIa genügt es, einen repräsentativen Vertreter pro Produktkategorie zu wählen (Artikel 52(6)).
- Bei den kritischeren Produkten der Klasse IIb und III muss sogar ein repräsentativer Vertreter pro generische Produktgruppe gewählt werden (Artikel 52(4)).
Ausnahmen und Einschränkungen von diesen Regeln
Allerdings gibt es laut MDCG 2019-13 Einschränkungen und Ergänzungen zu diesen Regeln:
- Wenn ein Hersteller sehr viele Geräte hat, müssen mindestens 15 % dieser Geräte für jede Produktgruppe bzw. Produktkategorie innerhalb der Gültigkeitsdauer des Zertifikats (typischerweise 5 Jahre) geprüft werden.
- Auch wenn ein Hersteller nur wenige Produkte hat, sollte die Benannte Stelle jedes Produkt nur einmal während der Gültigkeit des Zertifikats prüfen. Wenn es an Geräten mangelt, soll sie den Fokus z.B. auf die Post-Market Surveillance legen.
- Bei der Wahl der Stichprobe sollten die Benannten Stellen nicht (nur) das Zufallsprinzip walten lassen, sondern beispielsweise die Priorität auf Produkte mit neuen Technologien legen.
- Die Regel zum Sampling betrifft nichtimplantierbare Produkte der Klasse IIb und nicht die Produkte der Klasse III. Bei diesen müssen die Benannten Stellen die Prüfung der technischen Dokumentation für jedes Produkt durchführen.
- Auch für die „aktiven Produkte der Klasse IIb, die dazu bestimmt sind, ein Arzneimittel […] an den Körper abzugeben und/oder aus dem Körper zu entfernen“ gibt es Einschränkungen (Artikel 52(2)).
Das betrifft auch unangekündigte Audits
Das Sampling bezieht sich auf die geplanten Audits ebenso wie auf die ungeplanten:
Im Rahmen solcher unangekündigten Vor-Ort-Audits prüft die Benannte Stelle eine angemessene Stichprobe der hergestellten Produkte oder eine angemessene Stichprobe aus dem Herstellungsprozess um festzustellen, ob das hergestellte Produkt mit der technischen Dokumentation übereinstimmt (mit Ausnahme der in Artikel 52 Absatz 8 Unterabsatz 2 genannten Produkte).
MDR Anhang IX 3.4
b) Vergabe der Basis-UDI-DI
Die Basis-UDI-DI gruppiert verschieden Produkte oder Modelle. Man könnte auch von den Produkten einer Produktfamilie sprechen. Diese Produkte dürfen aber nur dann eine gemeinsame Basis-UDI-DI haben, wenn
- sie über die gleiche Zweckbestimmung verfügen,
- deren Auslegung, d.h. das Design, im Wesentlichen gleich ist und
- sich auch die Herstellung, also die Produktion, im Wesentlichen nicht unterscheiden.
Nur wenn diese Bedingungen erfüllt sind, erlaubt die MDR bzw. die MDCG es, Produkte mit einer gemeinsamen Basis-UDI-DI zusammenzufassen.
Weil wie oben aufgeführt verschiedene Produkte einer generischen Produktgruppe sich in der Auslegung und Herstellung unterscheiden können, dürfte die Basis-UDI-DI am ehesten auf Ebene der Medizinproduktegruppen oder sogar auf Ebene von Produktvarianten vergeben werden.
Lesen Sie hier mehr zum Thema UDI.
Das folgende Video verschafft Ihnen das Verständnis über die Hierarchie aus Produktkategorien, generischer Produktgruppe und Medizinproduktegruppe, das Sie benötigen, um zu entscheiden, auf welcher Hierarchieebene Sie Ihre Basis-UDI-DI vergeben.
c) Weitere Anwendungsbereiche
Wie die Produkte in Gruppen und Kategorien einzuteilen sind, wirkt sich nicht nur bei den Audits und Prüfungen der technischen Dokumentation aus:
- Ein EU-Bevollmächtigter muss mindestens für alle Produkte einer generischen Produktgruppe benannt werden. Es ist also nicht möglich, für zwei Produkte einer Produktgruppe zwei EU-Bevollmächtigte zu benennen.
- Die EU-Kommission behält sich das Recht vor, weitere Klassifizierungsregeln für Produktgruppen und Produktkategorien festzulegen (Artikel 51(3)).
- Die Hersteller müssen je nach Klasse des Produkts den Periodic Safety Update Report (PSUR) „für jedes Produkt und gegebenenfalls für jede Produktkategorie oder Produktgruppe“ erstellen (Artikel 86(1)).
- Hersteller müssen der Benannten Stelle alle ihre Produkte oder Produktgruppen nennen, wenn sie dort einen Antrag zur Bewertung des QM-Systems stellen (Anhang IX 2.1).
- Die Bescheinigung der Benannten Stellen müssen die Angabe zum Produkt bzw. Produktgruppe enthalten.
3. Kritik und Fazit
Endlich ist Klarheit geschaffen, mag man denken. Endlich ist klar, wie Produkte ganz konkret in Produktkategorien und dann in generische Produktklassen zu unterteilen sind. Doch ganz so einfach ist es nicht:
Die Regeln zur Klassifizierung von Medizinprodukten in Kategorien und Gruppen haben anscheinend verschieden Arbeitsgruppen erstellt. Die Folge ist, dass keine konsistente Taxonomie entstanden ist: Die Codes der EU-Kommission für die Produktkategorien entsprechen nicht(!) stringent den höheren Ebenen der CND/EMDN-Klassifizierung.
Sobald Hersteller oder Benannte Stelle entscheiden müssen, zu welcher Produktkategorie eine generische Produktgruppe zählt, sind Diskussionen vorprogrammiert. Es wird langsam Zeit für unsere Behörden, eine Standardisierung der Standardisierung zu erwägen.
Doch konzentrieren wir uns auf das Gute: Wir verfügen nun für das Sampling von Produkten über zwei Klassifikationssysteme, die das Defizit fehlender bzw. ungenauer Definitionen der Begriffe Produktkategorie und generische Produktgruppe kompensieren.



Sehr geehrter Herr Prof. Johner,
danke für diese hilfreiche Zusammenfassung.
Allerdings kann ich Ihre Aussage, dass Hersteller „je nach Klasse des Produkts den „Periodic Safety Update Report (PSUR) „nur“ pro Produktgruppe bzw. Produktkategorie erstellen“ müssen, nicht ganz nachvollziehen. Im MDR Artikel 86(1) steht, dass für Produkte der Klassen IIa, IIb und III pro Produkt UND ggf. für jede Produktkategorie oder Produktgruppe ein PSUR zu erstellen ist. Was „gegebenenfalls“ hier meint, ist unklar. Ansonsten klingt das eher nach mehreren Berichten.
Wenn eine Zusammenfassung mehrerer Produkte nach Kategorie oder Klasse in einem PSUR möglich ist, was sind dann dafür die Voraussetzungen? Und wo ist dies definiert?
Danke und mit freundlichen Grüßen
Sehr geehrte Frau Stridde,
Sie haben Recht. Die Aussage ist missverständlich. Ich hatte die Produktvarianten mit gemeinsamer Basis-UDI-DI im Kopf. Das „gegebenenfalls“ ändere ich sofort.
Danke für den wertvollen Hinweis!
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
wir stecken gerade bei unserer Umstellung der TD gemäß der MDR fest und wären sehr dankbar, wenn Sie uns diesbezüglich weiterhelfen könnten!
Wir würden gerne nur eine einzige TD führen. Diese soll Portsysteme inklusive Einführbestecken umfassen und da Portsysteme implantierbare Medizinprodukte der Klasse III sind, wäre die Basis-UDI-DI entsprechend zu vergeben.
Unsere Kunden kaufen aber auch z. T. Ports und Einführbestecke separat, um – je nach Operation und Operationsverlauf – ein Einführbesteck optional nutzen zu können. Wir würden unsere Einführbestecke für den Verkauf auch mit dem Zusatz kennzeichnen „Nur in Kombination mit den Ports der Firma PHS-Medical zu verwenden“.
Da die UDI wohl auch z. B. auf Lieferscheinen und der Rechnung vorhanden sein muss, müssten auch die separat verkauften Einführbestecke dann zwangsläufig unter der Basis-UDI der implantierbaren Portsysteme verkauft werden, auch wenn die Einführbestecke selber ja keine implantierbaren Medizinprodukte sind, da laut MDR nur Produkte mit der gleichen Basis-UDI-DI eine gemeinsame TD besitzen dürfen. Haben wir das so richtig verstanden?
Als Alternative stellte sich uns die Frage, ob wir die Portsysteme mit Einführhilfen als ein „Konfigurierbares Produkt“ (MDR Anhang VI Teil C) betrachten könnten? Da würde wohl die UDI-DI einer Konfigurationsgruppe und nicht einzelnen Konfigurationen innerhalb einer Gruppe zugeteilt. Eine Konfigurationsgruppe ist scheinbar die Zusammenstellung der in der TD beschriebenen möglichen Konfigurationen für ein bestimmtes Produkt, so dass das Einführbesteck eine Konfiguration der verbauten Komponenten ist und der Port mit Einführbesteck eine andere Konfiguration der verbauten Komponenten – wenn wir das denn so richtig verstanden haben?
Wir wären für eine Beantwortung dieser Fragen sehr dankbar!!!
Mit freundlichen Grüßen
Katrin Münster
Qualitätsmanagement
Sehr geehrte Frau Münster,
danke für Ihre spannende Fragen!
Es ist in der Tat so, dass nur Produkte eine gemeinsame TD haben können, die eine gleiche Basis-UDI-DI haben und das setzt u.a. gleiche Zweckbestimmungen voraus. Sie können allerdings Dokumente für mehrere Produkte gemeinsam erstellen (soweit das sinnvoll ist) und dann aus den verschiedenen TDs darauf verweise. Die TD wäre dann letztlich eine etwas ausführlichere „Link-Sammlung“.
Die Möglichkeit konfigurierbare Produkte aus den „Komponenten“ zu erstellen, wobei die Komponenten wieder Produkte sein dürfen. Allerdings ist das Konzept der konfigurierbaren Produkte eines mit Hinblick auf die UDI. Beachten Sie aber:
Es spricht aber nichts dagegen, eine TD zu führen, die alle Dokumente umfasst, die für mehr als ein „Produkt“ als Teil eines konfigurierbaren Produkts notwendig sind. Der Nachteil besteht aber darin, dass Sie damit die Lebenszyklen mehrerer Produkt aneinander „ketten“.
Beste Grüße, Christian Johner
Guten Tag, sehr geehrte Damen und Herren,
ich bin seit 01.09.2020 MDR Verantwortliche im Unternehmen TZMO Deutschland GmbH (Vertriebsgesellschaft für Inkontinenzprodukte PG 15).
Von Seiten unseres Herstellers wurde die Auflage erteilt, dass nicht nur die VE der Produkte mit allen notwendigen Daten „versehen“ werden, sondern auch die Umverpackungen (Kartons). Auf welchen § bzw. Artikel aus MDR bezieht sich hier der Hersteller bzw. ist das wirklich vom Gesetzgeber so vorgeschrieben. Wir haben viele Versicherte in der Häuslichkeit, die darauf bestehen und auch das Recht, ihre Inkontinenzartikel in neutralen Kartons zu erhalten.
Für eine kurze Rückantwort wäre ich Ihnen sehr dankbar.
Für Ihre Bemühungen vorab vielen Dank.
Freundliche Grüße aus Biesenthal sendet Ihnen
Kathrin Heimlich
Sehr geehrte Frau Heimlich,
möglicherweise liegt ein Missverständnis vor. Sie müssen auf den Umverkpackungen die UDI aufbringen, aber keine Patientendaten. Das wäre ein Verstoß gegen Datenschutzbestimmungen.
Sie sind zudem nicht verpflichtet, auf der Transportverpackung die UDI aufzubringen. D.h. sie können ganz neutrale Post verschicken. Nur sobald man die Transportverpackung öffnet, muss das Produkt und dessen UDI erkennbar sein. Patientendaten gehören aber nie drauf.
Viele Grüße, Christian Johner
CND Codes 3. Ebene:
Kann eine benannte Stelle darauf verweisen Codes auf der 3. Ebene zu wählen, wenn der Hersteller auf 4. Ebene sein Produkt nicht einkategorisieren kann oder muss dieser bei der Gesundheitsbehörde einen neuen Code anfordern?
Der Hersteller muss das Produkt einer Produktgruppe zuordnen. Andernfalls kann die Benannte Stelle nicht über das Sampling entscheiden. Laut MDCG 2019-13 entspricht die Produktgruppe bei Medizinprodukten der 4. Ebene, bei IVD der 3. Ebene der Codes. Das wäre auch die Ebene, die in der EUDAMED hinterlegt wird.
Sehr geehrter Hr. Johner!
Eine Frage zur Vergabe von Basic UDI-DIs. Wenn ich Produkte habe, die dieselbe Generische Produktgruppe (EMDN) teilen, aber aus unterschiedlichem Material hergestellt werden (z.B. ein Produkt aus Plastik, ein Produkt aus Metall…) – müssen hierfür dann unterschiedliche Basic UDI-DIs vergeben werden? Der Intended Use und auch das Design unterscheiden sich nicht.
Mit freundlichen Grüßen,
Michael
Guten Tag Michael,
für mich sprechen unterschiedliche Materialien für eine unterschiedliche Auslegung bzw. Design. Denn zum Design gehören auch Materialspezifikationen. Die Frage ist, ob dieser Unterschied in Ihrem Fall „essential“ ist. Ich würde das von den Risiken abhängig machen (gibt es z.B. unterschiedliche Risiken durch die unterschiedlichen Materialien).
Freundliche Grüße
Luca Salvatore