
Seit 25. Mail 2017 ist die In-Vitro Diagnostic Medical Device Regulation (IVDR) in Kraft, welche die bisherige IVD-Richtlinie (98/79/EC, IVDD) inzwischen abgelöst hat. Die Unterschiede zwischen der IVDR und IVDD sind groß.
Dieser Artikel stellt diese Unterschiede vor. Damit verschafft er Herstellern, die ihre Produkte noch unter einer EU-Richtlinie (IVDD) in den Markt gebracht haben, einen Überblick über die zu schließenden „Gaps“. Er hilft auch Organisationen, die bisher noch nicht von der IVDR betroffen waren, beispielsweise Labore.
Beachten Sie auch den Hauptartikel zur IVDR sowie zu den Artikel zu den Übergangsfristen.
1. Unterschiede zwischen der IVDR und IVDD: Wie es dazu kam
a) Auslöser
Vielfach wird kolportiert, dass der Brustimplantate-Skandal Auslöser für die Überarbeitung des Medizinprodukterechts gewesen sei. Doch das bestreiten inzwischen die meisten Akteure. Damit ist weitgehend unklar, wer die Neuregulierung aus welchem Grund initiiert hat.
b) Unterschiede bezüglich der Verbindlichkeit
Die EU-Medizinprodukteverordnungen (MDR, IVDR) lösen die Medizinprodukte-Richtlinien (MDD, AIMD, IVDD) ab.
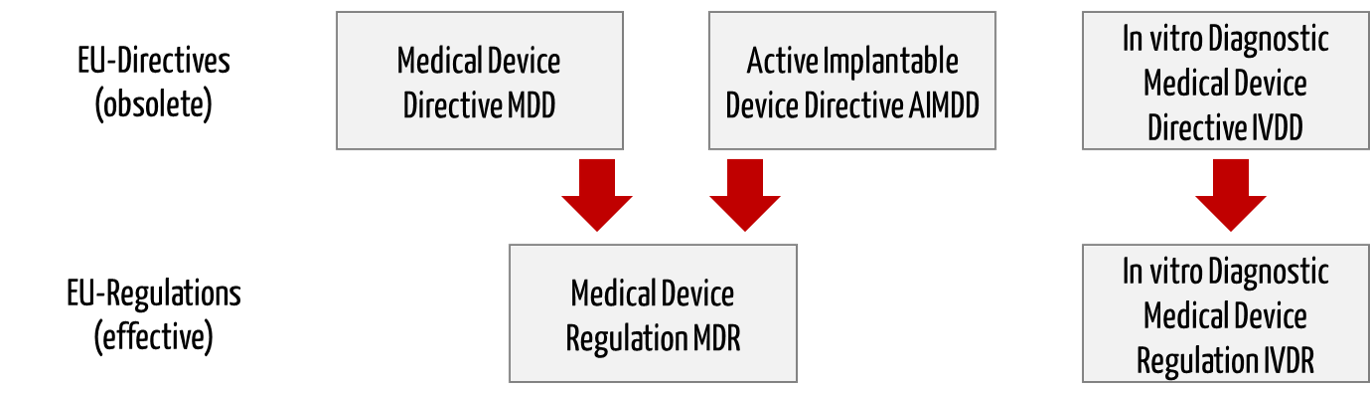
Die Richtlinie 98/79/EG über In-vitro-Diagnostika (IVD) wird durch die eine eigene Verordnung ersetzt (In vitro Diagnostic Medical Device Regulation (IVDR), Nummer 2017/746).
Lesen Sie hier mehr zum Thema Medical Devices Regulation, MDR.
c) Umfang und Aufbau der IVDR
Die EU-Verordnung ist in zehn Kapitel und 14 Anhänge aufgeteilt. Während die IVDD 24 Artikel enthielt, sind es bei der IVDR 113 Artikel. Das macht deutlich, dass die IVDR von Grund auf neu geschrieben wurde.
Die Kapitelstruktur und die einzelnen Kapitel stellt der Hauptartikel zur IVDR vor.
2. Was die IVDR nicht geändert hat
Einige Konzepte hat die IVDR beibehalten. Beispielsweise kennt die Verordnung weiterhin „grundlegende Anforderungen“, die jetzt in der deutschen Version grundlegende Sicherheits- und Leistungsanforderungen heißen.
Auch die nachzuweisenden Leistungsparameter unterscheiden sich kaum zwischen der IVDD (Anhang I, Abschnitt 3) und der IVDR (Anhang I, 9.1.) kaum. Es wird lediglich besser unterschieden zwischen analytischen Leistungsmerkmalen (Anhang I, 9.1.a) und klinischen Leistungsmerkmalen (Anhang I, 9.1.b).
Hersteller müssen auch künftig mittels eines Konformitätsbewertungsverfahrens nachweisen, dass sie und ihre Produkte diese Anforderungen erfüllen. Die IVDR hat auch nichts daran geändert, dass die Konformität durch ein CE-Zeichen ausgedrückt werden muss.
3. Wichtige Änderungen durch die IVDR
a) Unterschiede beim Anwendungsbereich
Die IVDR reglementiert im Gegensatz zur IVDD auch die Eigenherstellungen und Verwendung von sogenannten In-house IVD (auch Lab-Developed Tests oder LDT genannt). Damit müssen medizinische Labore den Anforderungen der IVDR genügen.
b) Unterschiede bei der Klassifizierung und Zulassung
Klassifizierung
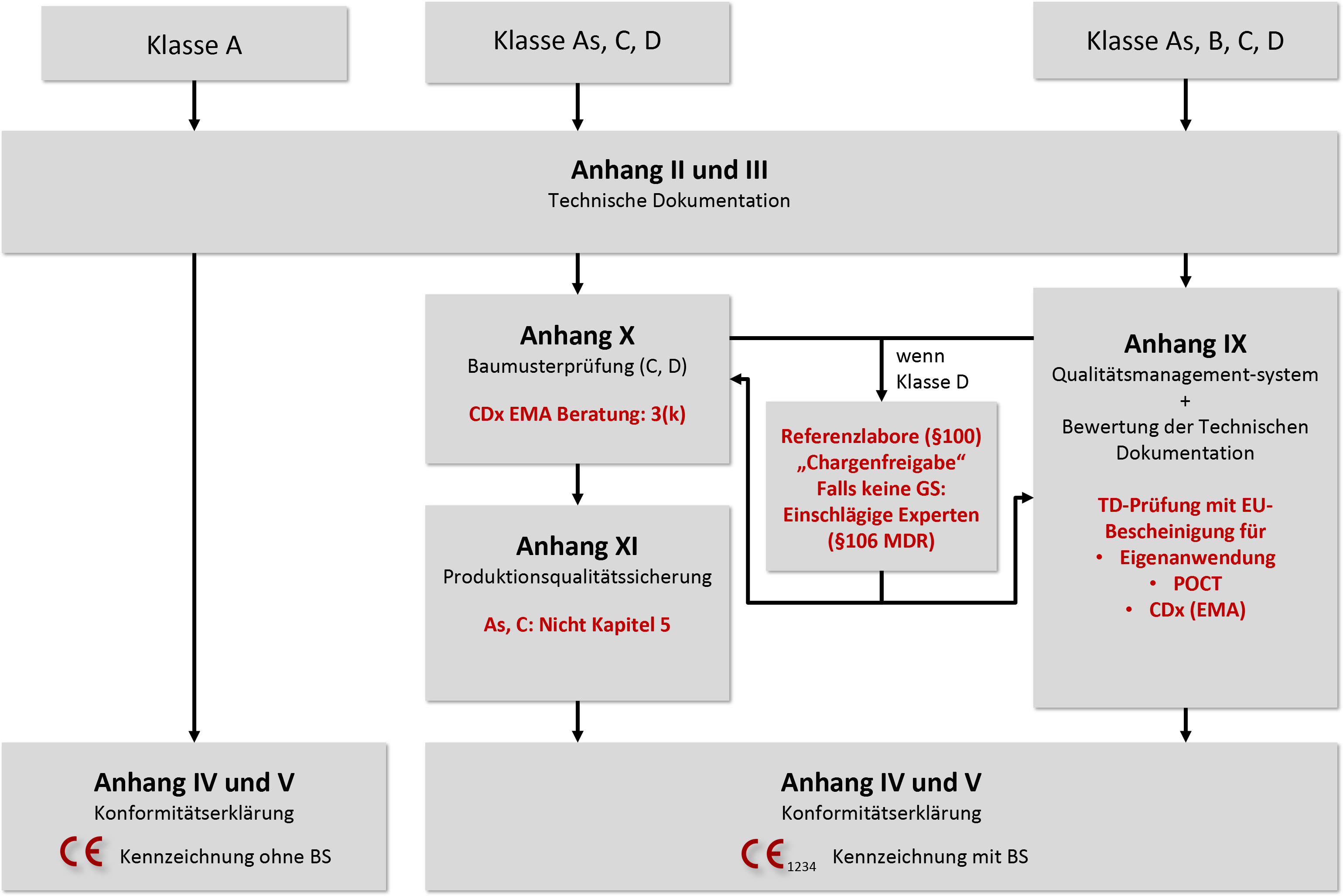
Für die Klassifizierung von IVD führt die IVDR risikobasierte Klassifizierungsregeln ein. (Bei der IVDD beruhte die Klassifizierung auf einem Listensystem.) Ein IVD-Hersteller muss für jedes Produkt alle Durchführungsvorschriften und Regeln gemäß Anhang VIII durchgehen, um ein Produkt einer der vier Klassen zuzuordnen:
- Klasse D: Hohes individuelles und hohes öffentliches Risiko, lebensbedrohliches Risiko bei falschem Ergebnis für mehrere Individuen; z. B. IVD für die Transfusionsmedizin oder zur Bestimmung lebenskritischer und gleichzeitig hoch ansteckender Krankheiten
- Klasse C: Hohes individuelles bzw. moderates öffentliches Risiko, lebensbedrohliches Risiko bei falschem Ergebnis für ein Individuum; z. B. Gentests, Test zum Bestimmen von Medikamentenspiegeln, ansteckenden Krankheiten oder angeborenen Krankheiten bei Föten oder Embryonen. Auch die meisten Selbsttests bzw. Produkte zur Eigenanwendung (durch Patienten) fallen in diese Klasse.
- Klasse B: Moderates individuelles Risiko bzw. geringes öffentliches Risiko, kein lebensbedrohliches Risiko bei falschem Ergebnis. Die Klasse B ist auch die „Default-Klasse“ für IVD, die unter keine der genannten Klassifizierungsregeln fallen.
- Klasse A: Geringes individuelles und geringes öffentliches Risiko, Produkte ohne patientenspezifische Informationen. In diese Klasse fallen unkritische Produkte wie Waschlösungen oder Nährböden, aber auch Probenbehältnisse.
Die IVDR unterscheidet noch weitere Typen von In-vitro-Diagnostika:
- Produkte für patientennahe Tests. Diese werden für sich allein unter Berücksichtigung aller Klassifizierungsregeln klassifiziert.
- Produkte zur Eigenanwendung (durch Patienten/Laien) fallen mit einigen explizit aufgeführten Ausnahmen immer in Klasse C.
- Therapiebegleitende Diagnostika, die für die sichere und wirksame Verwendung eines dazugehörigen Arzneimittels wesentlich sind, werden der Klasse C zugeordnet.
Konformitätsbewertungsverfahren
Die Konformitätsbewertung erfolgt unter der IVDR nicht mehr selbst durch den Hersteller, sondern durch eine Benannte Stelle. Nur Hersteller von unsterilen Klasse-A-Produkten dürfen die Konformität noch selbst erklären. Eine zentrale Zulassungsbehörde (wie die EMA bei Arzneimitteln) gibt es nach wie vor nicht.
Nach den allgemeinen Pflichten für Hersteller gemäß Artikel 10 der IVDR müssen IVD-Hersteller ein Qualitätsmanagementsystem implementieren und umsetzen, welches die Anforderungen der IVDR, Artikel 10 sowie der ISO 13485 als harmonisierte Norm erfüllt. Für alle Produkte, unabhängig von ihrer Klasse, müssen Hersteller eine produktspezifische Technische Dokumentation gemäß Anhang II sowie die Technische Dokumentation über die Überwachung nach dem Inverkehrbringen (Post-Market Surveillance) gemäß Anhang III erstellen.
Die IVDR unterscheidet im Wesentlichen drei Konformitätsbewertungsverfahren:
- Die Selbsterklärung der Konformität durch den Hersteller für Klasse-A-Produkte ohne Anforderungen an die Sterilität
- Die Konformitätsbewertung auf Basis des QM-Systems und der Bewertung der Technischen Dokumentation (Anhang IX). Die IVDR differenziert beim Review der Technischen Dokumentation durch eine Benannte Stelle, ob diese pro Produktkategorie (Klasse B), pro Produktgruppe (Klasse C) oder pro Produkt (Klasse D) erfolgen muss.
- Alternativ dürfen die Hersteller ein Konformitätsbewertungsverfahren auf Basis einer Baumusterprüfung (Anhang X) oder einer Qualitätssicherung für die Produktion (Anhang XI) wählen.
Für Produkte, die als patientennahe Tests (Point-of-Care Tests, POCT) oder zur Eigenanwendung durch Laien gedacht sind, stellt die IVDR ebenso spezielle Anforderungen wie für die therapiebegleitenden Diagnostika (Companion Diagnostics oder CDx). Klasse-D-Produkte müssen Referenzlaboratorien und, sollten keine Gemeinsamen Spezifikationen (GS) verfügbar sein, Expert-Panels mit einbeziehen.
Gemeinsame Spezifikationen
Hersteller sollten auch weiterhin harmonisierte Normen nutzen. Die EU-Kommission behält sich zudem vor, sogenannte „Gemeinsame Spezifikationen“ (Common Specifications) festzulegen, die die Hersteller einhalten müssen. Sie dienen insbesondere bei Hochrisikoprodukten einer einheitlichen Spezifikation von Sicherheit- und Leistungsanforderungen.
c) Technische Dokumentation
Die Hersteller müssen sehr genau beschreiben,
- was das Produkt leisten soll,
- wie sie sicherstellen, dass die grundlegenden Sicherheits- und Leistungsanforderungen tatsächlich erbracht werden, und
- dass keine inakzeptablen Risiken bestehen.
Zweckbestimmung
Dafür ist zunächst eine präzise Zweckbestimmung erforderlich. Welche Informationen in einer Zweckbestimmung spezifiziert werden müssen, finden IVD-Hersteller wie Anwender in Anhang II, Abschnitt 1.1 sowie Anhang I, Abschnitt 20.4.1.c) beschrieben.
Hersteller und auch medizinische Labore, die In-house IVD entwickeln und verwenden, stellen schnell fest, dass ohne eine genaue Formulierung der Zweckbestimmung weder eine zielgerichtete regulatorische Strategie möglich ist noch der Umfang für die Leistungsbewertung bestimmt werden kann.
Leistungsbewertung
Doch gerade auf die Leistungsbewertung legt die IVDR ihren Fokus. Deutlich detaillierter als die IVDD beschreibt sie die Anforderungen an die Dokumentation der Leistungsbewertung. Sie widmet den drei Elementen der Leistungsbewertung, nämlich wissenschaftlicher Validität, analytischer und klinischer Leistung (Anhang XIII, Teil A).
Post-Market Follow-up
Die IVDR weist darauf hin, dass die Leistung – wie auch die Risiken – während des gesamten Lebenszyklus eines Produkts überwacht werden muss und dass die Leistungsbewertung keine einmalige Sache ist. Daher fordert die Verordnung eine Nachbeobachtung der Leistung nach dem Inverkehrbringen (Post-Market Follow-up, PMPF) gemäß Anhang XIII, Teil B.
Dass die Leistungsbewertung und PMPF einen besonderen Stellenwert bei der Konformitätsbewertung eines IVDs einnehmen, zeigt auch der Fokus der Benannten Stellen während der Prüfung der Technischen Dokumentation.
Dies steht wiederum im Einklang mit der Anforderung, dass nur Produkte auf dem Markt sind, die auch ihren klinischen Nutzen nachweisen können. Und der liegt bei IVD insbesondere in der „Bereitstellung angemessener medizinischer Informationen über Patienten“ (IVDR, Vorwort (64)).
Der Begriff „angemessen“ wird in der englischen Version noch prägnanter benannt. Dort wird das Adjektiv „accurate“ verwendet. So schließt sich der Kreis, denn genau diese Anforderung finden wir in den Leistungsmerkmalen als Accuracy, also als analytische und klinische Genauigkeit, wieder.
d) Unterschiede bei den Produktanforderungen
Risikomanagement
Stammten bisher die Anforderungen an das Risikomanagement eher aus der ISO 14971, definiert die IVDR nun selbst detaillierter im Anhang I.
Leistungsstudien
Vergleichbar mit der MDR haben sich die Anforderungen an die Dokumentation und Durchführung von (klinischen) Leistungsstudien vervielfacht. Davon zeugen die Artikel 57 und der Anhang XIII, Abschnitt 2 bzw. Artikel 58 ff. und Anhang XIV. Letztere stellen weitere Anforderungen an bestimmte Leistungsstudien, die mit einem Risiko für den Patienten verbunden sind.
Für Leistungsstudien mit IVD gibt es seit 2019 auch eine spezielle Norm, die ISO 20916. Sie wird im Vorwort der IVDR referenziert.
Lesen Sie diesen Artikel zur Fallzahlplanung für Leistungsstudien mit IVD.
Labeling
Seitenweise beschreibt die IVDR in Absatz 20 des ersten Anhangs, was die Hersteller bezüglich des Labelings (Kennzeichnungen auf dem Etikett, Gebrauchsanweisungen, Warnungen und sonstige Hinweise) beachten müssen.
Software
Die IVDD nahm sich des Themas Software noch sehr stiefmütterlich an. Sie formulierte beispielsweise keine konkreten Anforderungen an den Software-Lebenszyklus. Das ändert die IVDR:
- Die Software muss gemäß den Software-Lebenszyklusprozessen einschließlich Verifizierung und Validierung entwickelt werden.
- Die IT-Sicherheit ist jetzt eine explizite Anforderung.
- Anforderungen an die Interoperabilität müssen Hersteller genau spezifizieren und deren Erfüllung prüfen. Apropos: Die neue ISO 13485:2016 fordert dies in der aktuellen Version ebenfalls.
- Das Risikomanagement muss Risiken auch aufgrund von Problemen mit der Interoperabilität betrachten.
- Die Laufzeitumgebung wie das Betriebssystem und die Hardware müssen Hersteller ebenso festlegen.
- Selbst auf mobile Plattformen, bei denen auch Bildschirmgrößen und die Benutzungsumgebung festzulegen sind, geht die Verordnung ein.
- Die Hersteller müssen eine Dokumentation bereitstellen, die Komponenten, Algorithmen und Technologien erkennen lässt.
- Die Software selbst unterliegt der Unique Device Identification (UDI).
- Das Personal muss auch in Hinblick auf Software kompetent sein.
Insgesamt sind das keine überraschenden Forderungen. Vielmehr reflektieren sie das, was die IEC 62304 und teilweise die ISO 13485 konkreter vorgeben.
UDI-System
Wie auch bei den anderen Medizinprodukten sind die Hersteller dazu verpflichtet, die Produkte eindeutig über eine UDI zu kennzeichnen und Informationen in der EUDAMED zu hinterlegen.
e) Unterschiede, die die Hersteller betreffen
Qualitätsmanagementsystem
Jeder(!) Hersteller eines IVD muss über ein Qualitätsmanagementsystem (QM-System) verfügen. Das gilt unabhängig vom Konformitätsbewertungsverfahren. Allerdings verlangt die IVDR nur bei den Konformitätsbewertungsverfahren gemäß Anhang IX und XI eine Bewertung des QM-Systems durch eine Benannte Stelle.
Die Anforderungen an das Qualitätsmanagementsystem sind umfassend und sowohl über die Pflichten als Hersteller in Artikel 10 der IVDR sowie durch die ISO 13485 als harmonisierte Norm vorgegeben. Das QM-System muss u. a. behandeln:
- Organisation, Prozesse, Verantwortlichkeiten (auch des Managements)
- Strategie, um die Konformität zu gewährleisten
- Erheben von Anforderungen
- Ressourcen-Management
- Umgang mit Zulieferern
- Risikomanagement
- Bewertung der Leistungsfähigkeit der Produkte
- Entwicklung
- Produktion
- Wartung
- Überwachung nach dem Inverkehrbringen
- UDI
- CAPA
Wenn Sie alle diese Verfahren beschrieben und implementiert haben, ist es kein großer Schritt mehr bis zur Konformität mit der ISO 13485:2016.
Die IVDR fordert bei einem gewählten Konformitätsbewertungsverfahren gemäß Anhang IX und Anhang XI, dass die Benannten Stellen mindestens alle fünf Jahre ein unangekündigtes Audit durchführen.
Post-Market Surveillance
Die Hersteller müssen die Post-Market Surveillance (als Teil des QM-Systems) präzise planen und durchführen. Die IVDR beschreibt die Anforderungen detailliert, u. a. in Artikel 78 ff. sowie im Anhang III.
Personal
Ebenso wie in der ISO 13485:2016 legt auch bei der IVDR verstärkt Gewicht auf eine ausreichende Qualifizierung von Personal. Die Verantwortliche Person ist eine neue, explizit geforderte Rolle.
4. Unterschiede zwischen der IVDR und IVDD überbrücken
Um die Unterschiede bei den Anforderungen von IVDR und IVDD zu überbrücken, sollten Sie als Hersteller folgende Schritte gehen:
- Stellen Sie sicher, dass das QM-System den Anforderungen der IVDR und der ISO 13485:2016 genügt.
- Formulieren Sie eine präzise Zweckbestimmung gemäß Anhang II, Abschnitt 1.1. bzw. Anhang I, Abschnitt 20.4.1.c) und dokumentieren Sie diese. Sie muss durch entsprechende Leistungsdaten belegt sein/werden. Auch für das Marketing dürfen keine abweichenden Formulierungen genutzt werden.
- Bestimmen Sie die Klassifizierung Ihrer Produkte.
- Erstellen Sie eine Gap-Analyse für die Technische Dokumentation und das Post-Market-System.
- Schließen Sie die dabei gefundenen Gaps.
- Legen Sie gemeinsam mit Entwicklung, Logistik und Produktion Ihre UDI-Strategie fest.
- Stimmen Sie mit Ihrer Beannnten Stelle den Zeit- und Umsetzungsplan ab.
5. Fazit
Die IVDR ist deutlich umfangreicher als die IVDD. Dies begründet sich vor allem durch die konkreteren Vorgaben zur Dokumentation in der IVDR.
Ein wesentlicher Unterschied sind die risikobasierten Klassifizierungsregeln und die Konformitätsbewertung durch Benannte Stellen für alle Produkte der Klassen B, C und D.
Dass die IVDR durch den nötigen klinischen Nachweis und die Leistungsbewertung der Produkte zu mehr Sicherheit für Patienten, Anwender und Dritte führt, lässt sich nicht bezweifeln. Es ist aber fraglich, ob die IVDR genau wie die MDR nicht mit weniger Bürokratie ausgekommen wäre, denn die hat eine Hemmung der Innovation zur Folge.
Unterstützung
Benötigen Sie Unterstützung bei der
- präzisen Formulierung Ihrer Zweckbestimmung,
- Zusammenstellung einer schlanken Technischen Dokumentation oder
- Herleitung einer produktspezifischen Leistungsbewertungsstrategie?
Dann nehmen Sie gleich Kontakt auf. Hier erhalten Sie schnell und unkompliziert Hilfe.



{kind=link}