
Gesetze und Normen formulieren Anforderungen, wie Medizinproduktehersteller den Entwicklungsprozess festlegen und dokumentieren müssen. Diese Anforderungen prüfen Benannte Stellen bei Audits.
Dieser Artikel zum Entwicklungsprozess gibt Ihnen Tipps zu dessen Gestaltung und zum Abgleich mit anderen Prozessen wie dem Risikomanagementprozess.
1. Entwicklungsprozess: Grundlagen
a) Definition
Mit dem Entwicklungsprozess wird festgelegt, auf welche Weise die an der Entwicklung beteiligten Rollen welche Tätigkeiten in welcher Reihenfolge ausführen, um die Inputs in die Outputs zu überführen.
| Elemente der Festlegung | Beispiele |
| Tätigkeiten | Systemanforderungen prüfen Architektur erstellen Testfälle ableiten Tests durchführen |
| Reihenfolge der Tätigkeiten | Sequenziell (Wasserfallmodell), parallel oder iterativ |
| Methoden und Verfahren (Auf welche Weise?) | Inspektion mit Checkliste zum Prüfen eines Dokuments Modellierung mit UML zum Erstellen einer Architektur Blackbox-Testverfahren zum systematischen Ableiten von Testfällen |
| Beteiligte Rollen | Entwicklungsleiterin Software-Architekt Programmiererin Tester |
| Inputs | Stakeholder-Anforderungen Organisatorische Vorgaben |
| Outputs | Baupläne (z. B. Architektur) Softwarecode Testergebnisse |
b) Ziele
Die Festlegung des Entwicklungsprozesses ist Teil der konstruktiven Qualitätssicherung, d. h. der Qualitätssicherung. Sie verfolgt das Ziel, Fehler zu vermeiden.
Insbesondere aus regulatorischer Sicht soll der Prozess bei Medizinprodukten sicherstellen, dass die Produkte die spezifizierte Sicherheit, Leistungsfähigkeit und Wirksamkeit haben.
Weiterhin soll der Prozess zur Effizienz beitragen:
- Beispielsweise werden die Ressourcen bestmöglich genutzt.
- Die am Entwicklungsprozess beteiligten Personen sind bestmöglich abgestimmt.
- Das Entwicklungsprojekt wird in der geplanten Zeit fertiggestellt.
- Nicht zuletzt werden Entwicklungsrisiken frühzeitig identifiziert.
2. Regulatorische Anforderungen an den Entwicklungsprozess
Die meisten Regularien und Normen folgen einem prozessorientierten Ansatz. Daher stellen sie Anforderungen an die Prozesse.
| Regulatorische Dokumente | Anforderungen an die Prozesse |
| Medizinprodukteverordnung (MDR) | Das QM-System beschreibt die Prozesse und Verfahren. Die Software-Lebenszyklusprozesse müssen festgelegt sein. |
| IEC 62304 | Die Norm verlangt die Einrichtung von fünf Prozessen, darunter einem Entwicklungsprozess. |
| ISO 13485 | Die Norm basiert auf prozessorientiertem Arbeiten. Sie fordert insgesamt 25 Verfahren, u. a. für die Entwicklung. |
| ISO 14971 | Die Norm verlangt einen Risikomanagementprozess. Diese sollte mit dem Entwicklungsprozess abgestimmt sein (s. Kapitel 4). |
| IEC 62366-1 | Die Norm fordert einen Usability-Engineering-Prozess. Dieser sollte mit dem Entwicklungsprozess abgestimmt oder in diesen integriert sein. |
| IEC 60601-1 | Zumindest für programmierbare elektrische medizinische Systeme besteht die Norm auf einem Entwicklungsprozess. Im Anhang zeigt sie ein Prozessmodell, das dem V-Modell gleicht (s. Abb. 1). |
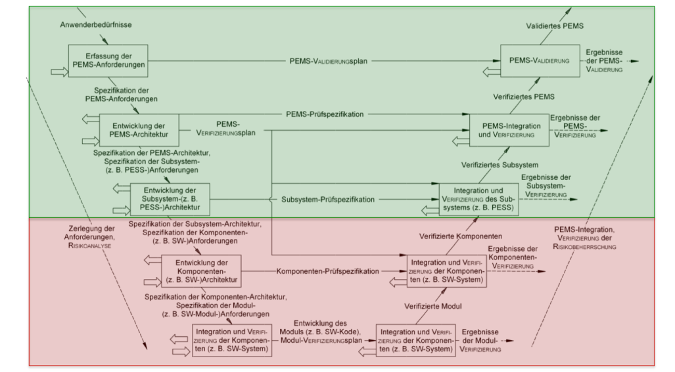
Keine(!) der Regularien fordert ein konkretes Prozessmodell.
Lesen Sie weiter unten, wie Sie redundante Aufwände und Probleme im Audit vermeiden können, wenn Sie den Entwicklungsprozess und den Risikomanagementprozess aufeinander abstimmen.
3. Praxistipps für den Entwicklungsprozess
Tipp 1: V-Modell als Dokumentationsmodell nutzen
Die meisten Firmen arbeiten entweder nach einem Wasserfall- bzw. V-Modell oder nach einem agilen Prozessmodell.
Lesen Sie mehr zur agilen Software-Entwicklung und zum V-Modell.
Gleich, für welches Prozessmodell Sie sich entscheiden, Sie müssen
- sich auf ein Modell festlegen (zumindest pro Projekt) und Ihre Entscheidung dokumentieren,
- nach diesem Prozessmodell arbeiten und dabei
- die von den Regularien vorgeschriebenen Aktivitäten durchführen und dokumentieren.
Diese Aktivitäten müssen zu Dokumenten führen, wie sie beim V-Modell entstehen würden (s. Abb. 2). Das heißt aber nicht, dass Hersteller nach dem V-Modell entwickeln müssen.

Beachten Sie, dass Sie alle Aktivitäten auf der linken Seite in Abb. 2 (d. h. alle Dokumente) mit einer Verifizierung abschließen und dann freigeben.
Tipp 2: Agilen Entwicklungsprozess nutzen und Fallen vermeiden
In der Praxis bewähren sich iterativ-inkrementelle Ansätze. Denn es gelingt meist nicht, die Ergebnisse einer Phase vollständig und korrekt zu erzielen. Das zeigt sich beispielsweise beim Erheben der Stakeholder-Anforderungen und dem Erstellen der Architektur. So ist es meist nicht möglich, das Zeitverhalten und den Ressourcenverbrauch (z. B. CPU) ohne Experimente zu ermitteln.
Allerdings darf der Hersteller auch bei einem iterativen und inkrementellen Vorgehen nicht darauf verzichten, die Ergebnisse einer vorangegangenen Tätigkeit zu verifizieren, bevor er die nächste beginnt. Auch ist es nicht effizient, ein systematisches Requirements Engineering durch einen Try-and-Error-Ansatz zu ersetzen.
Lesen Sie mehr zu den Fallen in diesem Artikel zur agilen (Software-)Entwicklung.
Tipp 3: Den Entwicklungsprozess richtig „scopen“
Die Abbildung 2 suggeriert, dass der Entwicklungsprozess alle Aktivitäten umfasst. Zwar muss der Hersteller alle Aktivitäten durch einen Prozess abdecken; aber es ergibt durchaus Sinn, dass die Entwicklungsabteilung nur für die Tätigkeiten verantwortlich ist, die nach dem Erheben und Validieren der Stakeholder-Anforderungen folgen.
Zudem kann die Entwicklungsabteilung die Beschreibung des Entwicklungsprozesses auf ihren Verantwortungsbereich eingrenzen (s. Abb. 3). Dann würde das Erheben der Anforderungen durch einen anderen Prozess geregelt.
Tipp 4: Forschung und Entwicklung trennen
Es ist hilfreich, das Forschungsteam möglichst weitgehend von regulatorischen Einschränkungen zu befreien. Denn dessen Ziel sollte auch darin bestehen, die Entwicklungsrisiken zu minimieren, in dem es Lösungen und Technologien ausprobiert (erforscht).
4. Zusammenspiel mit dem Risikomanagementprozess
a) Problemstellung
Die meisten für die Entwicklung von Medizinprodukten relevanten Normen wie die ISO 13485, die ISO 14971, die IEC 62304 und die IEC 62366-1 stellen Anforderungen an Prozesse.
Um diese Anforderungen zu erfüllen, formulieren viele Medizinproduktehersteller Prozess- und Verfahrensbeschreibungen (SOPs) – und zwar für jeden dieser Prozesse getrennt. Dies führt dazu, dass die an der Entwicklung Beteiligten gleichzeitig mehrere dieser SOPs beachten müssen. Dann sind Fehler fast vorprogrammiert.
b) Lösung
Die folgende Abbildung zeigt zwar ein V-Modell, die Überlegungen sind davon aber unabhängig.
Übersicht
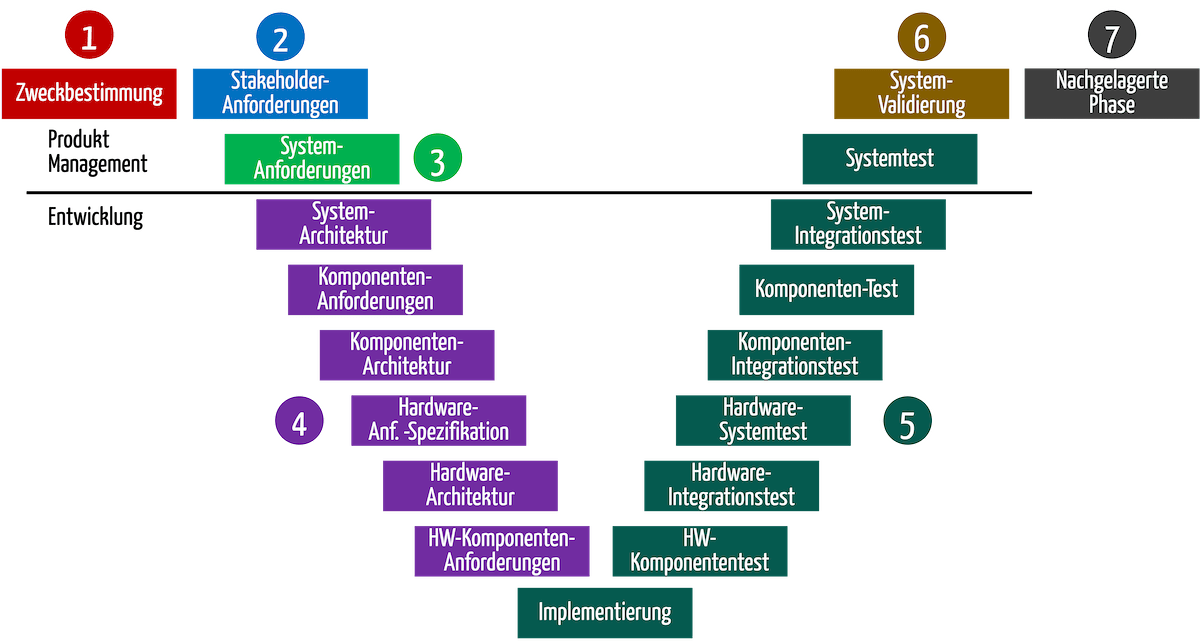
Die Beschreibung des Entwicklungsprozesses sollten in jeder Phase die Referenzen auf die jeweiligen Tätigkeiten im Risikomanagementprozess enthalten. So wird sichergestellt, dass das Risikomanagement nicht vergessen wird.
Einzelne Phasen
| # | Phase im Entwicklungsprozess | Tätigkeiten im Entwicklungsprozess | Tätigkeiten im Risikomanagementprozess |
| 1 | Kontext und Geschäftsmodell | Für das Produkt die Geschäftserwartungen und den Kontext definieren Zweckbestimmung formulieren | Risikoanalyse mit einer Preliminary Hazard Analysis starten. Bereits aus der Zweckbestimmung wird offensichtlich welche Gefährdungen und Risiken das Produkt verursacht. |
| 2 | Stakeholder-Anforderungen | Stakeholder-Anforderungen identifizieren Stand der Technik ermitteln | Risikopolitik und der Risikoakzeptanzkriterien |
| 3 | System-Spezifikation | Systemanforderungen und Systemspezifikation ableiten. Dabei Anforderungen an das Produkt aus einer Blackbox-Sicht und damit dessen Inputs und Outputs festlegen. | Die Input- und Output-Analyse hilft, weitere Gefährdungen und Risiken zu identifizieren. Mit diesem Teil der Risikoanalyse sollten die Hersteller die Preliminary Hazard Analysis abschließen können und damit die Identifikation und Bewertung der wichtigsten Gefährdungen bzw. Risiken. |
| 4 | System-Entwurf | Produkt- bzw. System entwerfen, z. B. Architektur erstellen, Software Design erstellen, Isolationsdiagramme entwerfen | Die Risikoanalyse kann abgeschlossen werden, wenn Anforderungen in eine Lösung überführt wurden. Die Art, wie das Produkt aufgebaut sein soll, führt zusätzliche Risiken ein oder hilft, diese zu minimieren. Risiken lassen sich mit einer eine FMEA identifizieren. Gleichzeitig beginnt spätestens in dieser Phase die Planung der Maßnahmen, die das Risiko minimieren, und mit der die funktionale Sicherheit des Produkts erreicht werden muss. |
| 5 | Umsetzung und Verifizierung | Software programmieren Erste Produkte (prototypisch) bauen Beides mit Tests verifizieren, z.B. Software-System-Test | Wirksamkeit der risikominimierenden Maßnahmen verifizieren. |
| 6 | Validierung | Produkt validieren, z. B. mit einer summativen Evaluation (Usability-Tests) und klinischer Bewertung | Die Validierung dient dazu, nicht nur die Sicherheit der Produkte, sondern auch ihren Nutzen nachzuweisen und damit, dass das Nutzen-Risikoverhältnis akzeptabel ist. |
| 7 | Nachgelagerte Phase | Nicht mehr Gegenstand der Entwicklung | Ermitteln der Risiken bei Produktion, Versand und Inbetriebnahme Im Rahmen der Post-Market Surveillance fortlaufende Prüfung, ob alle Risiken vollständig und richtig bewertet und im Vergleich zum Nutzen noch akzeptabel sind |
5. Zusammenspiel mit dem Usability-Engineering-Prozess
Das Qualitätsmanagement und das Usability Engineering spielen eng zusammen: Während die ISO 13485 allgemeine Konzepte und Anforderungen formuliert, stellt die IEC 62366-1 konkrete Anforderungen im Kontext Usability (Gebrauchstauglichkeit). Dieser Beitrag zeigt das Mapping beider Normen.
| Qualitätsmanagement ISO 13485 | Usability IEC 62366-1 |
|---|---|
| Kapitel 6.1: Bereitstellung von Ressourcen | 4.3 Anpassen des Usability-Engineering-Aufwands |
| Kapitel 6.2: Personelle Ressourcen | 4.1.1 Gebrauchstauglichkeitsorientierter Entwicklungs-Prozess |
| Kapitel 7.1: Planung der Produktrealisierung | 4.3 Anpassen des Usability-Engineering-Aufwands |
| Kapitel 7.3.2 Entwicklungsplanung | 5.5 Auswählen der gefährdungsbezogenen Use Scenarios für die summative Evaluation 5.7 Erstellen eines Plans für die User Interface Evaluation |
| Kapitel 7.3.3 Entwicklungseingaben | 5.1 Erstellen der Use Specification 5.2 Ermitteln von Merkmalen des User Interface in Bezug auf Sicherheit und mögliche Use Errors 5.3 Ermitteln bekannter oder vorhersehbarer Gefährdungen und Gefährdungssituationen 5.4 Ermitteln und Beschreiben gefährdungsbezogener Use Scenarios 5.6 Erstellen der User Interface Specification |
| Kapitel 7.3.4 Entwicklungsergebnisse | 5.8 Durchführen des Designs des User Interface, Implementierung und formative Evaluation |
| Kapitel 7.3.5 Entwicklungsbewertung | 5.8 Durchführen des Designs des User Interface, Implementierung und formative Evaluation |
| Kapitel 7.3.6 Entwicklungsverifizierung | 5.8 Durchführen des Designs des User Interface, Implementierung und formative Evaluation 5.9 Durchführen der summativen Evaluation der Usability des User Interface |
| Kapitel 7.3.7 Entwicklungsvalidierung | 5.8 Durchführen des Designs des User Interface, Implementierung und formative Evaluation 5.9 Durchführen der summativen Evaluation der Usability des User Interface |
6. Fazit und Zusammenfassung
Hersteller sollten viel Energie aufuwenden, um ihren Entwicklungsprozess präzise und spezifisch zu beschreiben. Denn sonst drohen Probleme.
| Fehler | Mögliche Folgen |
| Im schlimmsten Fall wird der Prozess nicht oder nicht vollständig beschrieben. | Das führt zu einer Nicht-Konformität (bei nicht definierten und gelebten Prozessen reagieren Benannte Stellen sensibel). Es droht Chaos in der Entwicklung |
| Der Prozess wird ungenau beschrieben. | Regulatorische Anforderungen werden nicht erfüllt, und in Audits drohen Abweichungen. Das Team weiß nicht, wie es vorgehen soll, was zu unsicheren Produkten und verzögerten Projekten führt. |
| Der Prozess wird zwar präzise, aber unpassend beschrieben. | Das Team empfindet den Prozess (zu Recht) als bürokratisch und behindernd. Das Team weicht vom Prozess ab (Nicht-Konformität) oder es arbeitet ineffizient und ineffektiv. |
Der Aufwand rechnet sich: Mit einem guten Entwicklungsprozess führen die Entwicklungsprojekte schnell und planbar zu sicheren Produkten, zu zufriedenen Kunden und wirtschaftlichem Erfolg.
Das Johner Institut hilft!
Die Expertinnen und Experten des Johner Instituts helfen dabei, in kürzester Zeit, schlanke, maßgeschneiderte und gesetzeskonforme Entwicklungsprozesse zu beschreiben.
- Dadurch wird die Entwicklung schnell und planbar.
- Zudem liefert sie zuverlässig sichere und leistungsfähige Produkte.
- Schließlich wird Ärger bei Audits und Inspektionen vermieden.
Melden Sie sich, damit wir gemeinsam festlegen können, wie Sie diese Ziele am schnellsten erreichen.


