
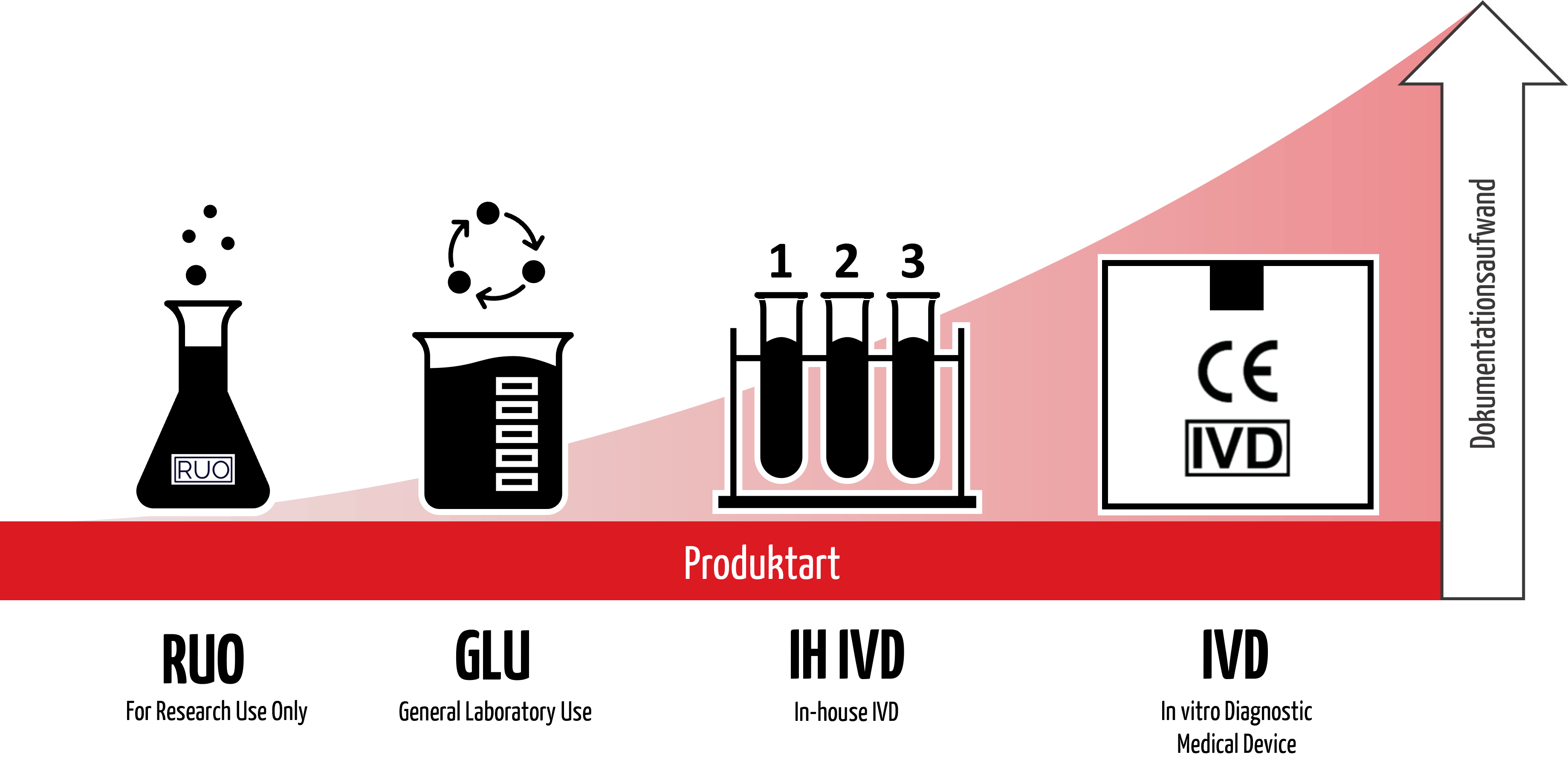
Mit dem Label „For Research Use Only“ (RUO) erklären Hersteller, dass ihre Produkte nicht in diagnostischen Verfahren eingesetzt werden sollen. Sie vermeiden damit die aufwendige Dokumentation für In-vitro-Diagnostika (CE-IVD-Produkte). Dennoch verwenden z. B. medizinische Labore RUO-Produkte in diagnostischen Verfahren, teilweise sogar mit Wissen der Hersteller. Das kann Konsequenzen haben – nicht nur für Hersteller und Betreiber, sondern auch für Patientinnen und Patienten.
Erfahren Sie in diesem Beitrag,
- was das Label „For Research Use Only“ (RUO) bedeutet,
- welche Anforderungen an RUO-Produkte bestehen,
- wie Sie rechtliche Probleme vermeiden und
- welche Alternativen es zu RUO-Produkten gibt.
1. „For Research Use Only” – Was bedeutet das?
Erhalten Produkte das Label „For Research Use Only“, bedeutet das, dass diese Produkte kaum regulatorischen Kontrollen unterliegen. Die Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR) möchte sich klar von RUO-Produkten distanzieren:
Der Geltungsbereich dieser Verordnung sollte klar vom Geltungsbereich anderer Rechtsvorschriften abgegrenzt werden, die Produkte wie Medizinprodukte, allgemeine Laborprodukte und allein für Forschungszwecke bestimmte Produkte betreffen.
IVDR Vorwort (7)
a) Betroffene Institutionen
Berührungspunkte mit RUO-Produkten haben vor allem:
- Medizinische Labore
- Medizinische Labore können zwar RUO-Produkte verwenden, werden dann aber zum Eigenhersteller, mit allen daraus folgenden Konsequenzen.
- Nutzen medizinische Labore RUO-Produkte zu medizinischen Zwecken, erfüllen sie aber nicht die Anforderungen an Inhouse-IVD, machen sie sich haft- und/oder strafbar.
Mehr Informationen zu „Lab Developed Tests“ finden Sie in unserem Beitrag: Die EU reguliert medizinische Labore. Sind Laboratory Developed Tests noch erlaubt?
- Hersteller
- Hersteller können RUO-Produkte als Komponenten für ihre IVD einsetzen, sind dann aber für die Konformität des Endprodukts mit der IVDR verantwortlich. Eine RUO-Kennzeichnung der Komponente entfällt.
- Kennzeichnen Hersteller ihre Produkte als „RUO“, ist der Zweck dieser Produkte entsprechend auszulegen und ggf. nachzuweisen. So sollten z. B. vorhersehbare Fehlanwendungen berücksichtigt werden. Das RUO-Label darf nicht bloß als „Schutzbehauptung“ auf dem Produkt angebracht werden; das kann rechtliche Konsequenzen haben.
b) Definition
Eine einheitliche Definition von Produkten „For Research Use Only“ existiert nicht. Wie der Name sagt, lassen sich darunter allgemein Produkte verstehen, die für wissenschaftliche Analysen im Rahmen von Forschungszwecken vorgesehen sind. Diese Produkte dürfen dabei keinem medizinischen Zweck dienen; dadurch grenzen sie sich von IVD und Inhouse-IVD ab.
Das Verständnis von „For Research Use Only“ ist allerdings in Europa und den USA unterschiedlich.
Definition in Europa
In Europa liefert derzeit die Leitlinie MDCG 2024-11 einen Anhaltspunkt für die Definition von RUO-Produkten; sie bezieht sich auf die IVDR. Das Dokument dient der Klärung der Qualifizierung von Produkten als IVD bzw. als Zubehör zu einem IVD.
„Products intended for research use only cannot be intended by their manufacturers for a medical purpose (as outlined in Article 2(2) IVDR).”
Quelle: MDCG 2024-11
Demnach darf ein RUO-Produkt also nicht einmal ansatzweise einen medizinischen Zweck erkennen lassen.
Dies gilt auch für inhouse-entwickelte Tests (Home Brew Tests, Laboratory Developed Tests), die nur in einer Gesundheitseinrichtung für Forschungszwecke verwendet werden.
Die IVDR geht in Artikel 1 (3) a) auf RUO-Produkte ein. Diese werden aus dem Geltungsbereich ausgeschlossen:
Diese Verordnung gilt nicht für
a) Produkte für den allgemeinen Laborbedarf oder allein für Forschungszwecke bestimmte Produkte, es sei denn, sie sind aufgrund ihrer Merkmale vom Hersteller speziell für In-vitro-Untersuchungen bestimmt“
Quelle: IVDR, Artikel 1 (3) a)
Weiterhin wird in Artikel 2 (45) spezifiziert:
„Ein Produkt, das für eine Verwendung zu Forschungszwecken bestimmt ist und keine medizinische Zweckbestimmung hat, gilt nicht als Produkt für Leistungsstudien”
IVDR, Artikel 2 (45)
Produkte für Leistungsstudien sind hierbei:
„bezeichnet ein Produkt, das von einem Hersteller zur Verwendung in einer Leistungsstudie bestimmt ist.“
IVDR, Artikel 2 (45)
Die IVDR grenzt RUO-Produkte somit von IVD und Produkten für Leistungsstudien ab. Auch die EU-Verordnung macht hierbei das Fehlen einer medizinischen Zweckbestimmung deutlich.
Definition in den USA
Die FDA verfasste 2013 ein Guidance-Dokument zu RUO mit dem Titel „Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only“.
Diese Leitlinie definiert RUO folgendermaßen:
„An RUO product is an IVD product that is in the laboratory research phase of development and is being shipped or delivered for an investigation that is not subject to part 812 [Anm.: In part 812 geht es um Bereitstellung von Produkten für Leistungsbewertungszwecke als Vorstufe zum IVD.]“
Quelle: FDA Guide „Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only“, Kapitel III A
Einige Beispiele für Produkte, die nach Ansicht der FDA in diese Forschungsphase fallen, sind:
- Tests, die sich in der Entwicklung befinden, um die Methodik des Testkits, die erforderlichen Komponenten und die zu messenden Analyten zu ermitteln
- Instrumente, Software oder andere elektrische/mechanische Komponenten, die sich in der Entwicklung befinden, um die korrekten Einstellungen, Unterkomponenten, Untergruppen, grundlegenden Betriebseigenschaften und möglichen Verwendungsmethoden zu bestimmen
- In der Entwicklung befindliche Reagenzien, um Produktionsmethoden, Reinigungsgrade, Verpackungsanforderungen, Haltbarkeit, Lagerungsbedingungen usw. zu bestimmen
Die FDA spezifiziert allerdings im Weiteren:
„FDA also recognizes that there are certain products, such as instruments, systems, and reagents that are labeled for research use only and intended for use in the conduct of nonclinical laboratory research with goals other than the development of a commercial IVD product […].”
FDA Guide „Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only“, Kapitel III A
Nachfolgend werden Beispiele für derartige Forschungszwecke benannt, in denen das Produkt selbst nicht Forschungsgegenstand ist.
Die FDA sieht somit zwei „Typen“ von RUO-Produkten: Zum einen IVD-Produkte, deren Entwicklung noch nicht abgeschlossen ist und die selbst den Gegenstand der Forschung darstellen, und zum anderen Produkte für nichtklinische Forschung.
In beiden Fällen muss laut FDA eine deutlich sichtbare RUO-Kennzeichnung auf den Produkten angebracht werden. Die RUO-Kennzeichnung soll hierbei die Verwendung zur klinischen Diagnostik, zum Patientenmanagement und zu anderen Untersuchungen mit medizinischem Zweck verhindern.
c) Was das Label „For Research Use Only“ für Folgen hat
Normalerweise unterliegen IVD, abhängig von ihrer Risikoklasse, unterschiedlichen regulatorischen Anforderungen (etwa nach IVDR oder FDA).
RUO-Produkte fallen jedoch nicht unter die Definition von „In-vitro-Diagnostikum“ der IVDR oder der einschlägigen FDA-Vorschriften. Für RUO gelten die regulatorischen Anforderungen der IVDR daher nicht. In den USA sind sie von den cGMP und den FDA-Qualitätsvorschriften ausgenommen.
bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System – einzeln oder in Verbindung miteinander – vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
- a) über physiologische oder pathologische Prozesse oder Zustände,
- b) über kongenitale körperliche oder geistige Beeinträchtigungen,
- c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
- d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
- e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
- f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten auch als In-vitro-Diagnostika.
IVDR, Artikel 2 (2)
„In vitro diagnostic products are those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.”
21 CFR 809.3
RUO-Produkte fallen in der EU damit allerdings nicht automatisch völlig aus dem regulatorischen Geltungsbereich. Je nach Produkt müssen ggf. Anforderungen erfüllt werden, die nicht speziell für IVD vorgesehen sind (etwa die REACH-Verordnung oder die Maschinenrichtlinie).
Lesen Sie hier mehr zum Thema Maschinenrichtlinie: Was bei Medizinprodukten gilt.
Dennoch unterliegen RUO-Produkte demnach erheblich weniger Kontrollen als IVD. Daher ist es notwendig, ihren Einsatz entsprechend zu begrenzen. Betreiber müssen von deren Verwendung absehen, wenn sie
- Untersuchungen mit medizinischem Zweck durchzuführen und
- Leistungsstudien zu betreiben.
2. Gebrauch und Missbrauch des „Research-Use-Only“-Labels
a) Wofür RUO-Produkte eigentlich gedacht sind
Wie der Name „For Research Use Only“ besagt, sind diese Produkte ausschließlich für Forschungszwecke vorgesehen. Die vereinfachte Handhabung und die niedrigen Hürden beim Inverkehrbringen machen RUO-Produkte für diesen Bereich attraktiv.
Die Leitlinie MEDDEV. 2.14/2 rev.1 (IVD Guidance: Research Use Only products; A Guide For Manufacturers And Notified Bodies) listet mögliche Anwendungsbereiche von RUO-Produkten genau auf. Diese Leitlinie ist zwar im Rahmen der inzwischen veralteten Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD) entstanden, kann jedoch mangels eines aktuellen Ersatzes noch immer als Stand der Technik betrachtet werden (MDCG Guidance Dokument Status: Ongoing).
Anwendungsbereiche:
- Grundlagenforschung
- Pharmazeutische Forschung
- Bessere Identifizierung und Quantifizierung einzelner chemischer Substanzen oder Liganden in biologischen Präparaten
- Inhouse-Herstellung von sogenannten „Homebrew-Kits“ zu Forschungszwecken
Explizit nicht als RUO sehen die Autoren:
- Die Verwendung von Rohstoffen, die als „For Research Use Only“ gekennzeichnet sind, die in einem IVD-Endprodukt verarbeitet werden
- Sogenannte „Forschungsprodukte“, die gegen ein IVD-Vergleichsprodukt getestet werden, das die CE-Kennzeichnung trägt
- Produkte für Marktstudien/Durchführbarkeitsstudien
Diesen Produkten kann ein medizinischer Zweck zugeordnet werden.
b) Wofür RUO-Produkte außerdem oft verwendet werden
Die geringen Hürden sind jedoch auch der Grund, weswegen RUO-Produkte für Zwecke verwendet werden, für die sie eigentlich nicht vorgesehen sind. Dies birgt sowohl für Hersteller als auch für Betreiber und Patient:innen Gefahren.
Verkauf von RUO-Produkten an medizinische Labore
RUO-Produkte werden von Herstellern teilweise auch an medizinische Labore vertrieben. Zwar forschen auch Mediziner:innen, doch dies ist kaum der Hauptzweck eines medizinischen Labors.
Daher muss man annehmen, dass im Vertriebsgespräch mit Ärztinnen und Ärzten stets ein medizinisches Interesse hinter der Anwendung des Produkts steht. Wer also wissentlich RUO-Produkte an medizinische Labore vertreibt, steht potenziell unter dem Verdacht, eine medizinische Zweckbestimmung hinter dem Vorwand „For Research Use Only“ zu verstecken, um sich der Verantwortung für ein Medizinprodukt zu entziehen.
Vermeiden Sie in den Werbematerialien für Produkte, die offensichtlich keinen medizinischen Zweck erfüllen, den Bezug zu konkreten diagnostischen Verfahren. Bleiben Sie immer auf der technischen bzw. analytischen Ebene.
Anwendung von RUO-Produkten in medizinischen Laboren
Nicht nur aus Herstellersicht ergibt sich ein Problem mit dem Vertrieb von RUO an medizinische Labore. Auch die Labore selbst können sich als Betreiber falsch verhalten und damit ggf. haftbar werden.
- Medizinische Labore haben die Freiheit, Inhouse-IVD selbst zu entwickeln. In diesen Fällen werden häufig RUO-Produkte in diagnostischen Verfahren eingesetzt. Das Labor trägt die volle Verantwortung für diese Tests.
Bereits im Rahmen der IVDD hat dies die MEDDEV. 2.14/2 kritisch gesehen. Mit der IVDR schränkt die EU den Routineeinsatz von solchen Inhouse-IVD explizit stärker ein.
Lesen Sie hier mehr zum Thema: Die EU reguliert medizinische Labore. Sind Laboratory Developed Tests noch erlaubt?
- Wegen der geringen regulatorischen Hürden sind RUO-Produkte im Einkauf sehr günstig. Medizinische Labore bevorzugen die günstigeren RUO-Produkte gegenüber teuren CE-IVD-Produkten, wenn sie damit dasselbe Leistungsniveau erreichen können. Der Einsatz von RUO-Produkten zu anderen Zwecken als Forschungszwecken ist, auch wenn sie ähnliche Ergebnisse liefern wie ein IVD, dennoch nicht gestattet.

Die Verwendung von RUO/General‑Laboratory‑Use‑Komponenten im diagnostischen Workflow kann dann vom vorgesehenen Gebrauch gedeckt sein, wenn ein IVD‑Hersteller in Zweckbestimmung/IFU die Kombination ausdrücklich vorsieht und Sicherheit, Leistung und Kompatibilität für diese Kombination belegt (z. B. Extraktion + PCR‑Cycler/Software + CE‑IVD‑PCR‑Kit) – entweder als definierte Kombination (Closed System) oder über kritische Spezifikationen für zulässige Kombinationen (Open System).
Team‑NB (Verband der Benannten Stellen für Medizinprodukte) betont: Bezieht ein Hersteller solche Produkte in Kombinationsangaben ein, trägt er die volle Nachweisverantwortung; zudem sollte die Leistung über die Überwachung nach dem Inverkehrbringen (PMS) kontinuierlich überwacht werden. Wo verfügbar, sind CE‑gekennzeichnete Geräte/Komponenten zu bevorzugen.
3. Konsequenzen einer falschen Einordnung
Fehlende Kontrollen können sich nachteilig auf die Qualität auswirken. Ob ein Produkt tatsächlich „For Research Use Only“ gedacht ist, sehen sich die einschlägigen Stellen (z. B. Behörden bei Inspektionen) daher genauer an.
Hersteller sollten außerdem beachten, dass es nicht ausreicht, eine RUO-Kennzeichnung an ein Produkt anzubringen, damit es nicht mehr die ansonsten gültigen Anforderungen an IVD erfüllen muss.
Einzig die tatsächlich vorgesehene Verwendung eines Produkts qualifiziert dieses als RUO, oder eben nicht. Dazu ziehen Behörden (sowohl europäische als auch die FDA) auch Marketingmaterial oder andere Informationen als Beweis heran.
Herstellern und Betreibern, die Produkte mit RUO-Label zweckentfremden, drohen empfindliche Strafen, da Patient:innen oder sogar der Allgemeinheit gravierende Schäden entstehen können.
a) Konsequenzen für Hersteller und Betreiber
IVD fälschlicherweise unter dem RUO-Label zu vertreiben oder zu anderen als Forschungszwecken einzusetzen, ist kein Kavaliersdelikt. Hersteller, die nachweislich einen medizinischen Zweck hinter der RUO-Kennzeichnung verstecken oder verstecken wollen, müssen in der EU mit rechtlichen Konsequenzen rechnen. Gleiches gilt für Betreiber, die RUO-Produkte zweckentfremden. Es drohen Geld- oder sogar Gefängnisstrafen. Hinzu kommt eine mögliche Haftung bei Schäden, die Patient:innen entstehen.
b) Konsequenzen in den USA
Auch in den USA drohen empfindliche Strafen. Wenn ein Produkt fälschlicherweise das RUO-Label trägt, ist das Produkt gemäß Abschnitt 502(a) und 502(o) des Gesetzes 21 U.S.C. 352(a), 352(o) falsch ausgewiesen und gilt gemäß Abschnitt 501(f) des Gesetzes 21 U.S.C. 351(f) als verunreinigt.
c) Konsequenzen für Patient:innen
Am härtesten kann es jedoch Patient:innen treffen. Die regulatorischen Anforderungen an IVD-Produkte sind schließlich keine aus der Luft gegriffenen Schikanen für Hersteller und Betreiber. Die Regelungen sollen die Patient:innen vor falschen Ergebnissen und daraus folgenden Fehlentscheidungen bewahren. Falsch-negative Ergebnisse können Patient:innen in falscher Sicherheit wiegen, und eine bestehende Erkrankung kann sich unbemerkt verschlimmern. Ein Beispiel ist die Metastasierung einer nicht festgestellten Krebserkrankung aufgrund mangelnder Leistungsfähigkeit des Tests.
Mängel können sogar so schwerwiegend sein, dass sie den Tod vieler Menschen verursachen: Eine nicht erkannte Virus-Infektion kann in der frühen Phase einer Pandemie viele Leben kosten, wie die Corona-Pandemie bedauerlicherweise gezeigt hat.
4. Alternative zu „Research Use Only”
Um rechtliche Probleme und Gefahren für Dritte zu vermeiden, sollten Hersteller und Anwender auf allgemeinen Laborbedarf als Alternative zu RUO-Produkten zurückgreifen.
Es gibt Laborprodukte, die offensichtlich keinen konkreten medizinischen Zweck haben, z. B.:
- Reine Chemikalien
- Nährmedien
- Reaktionsgefäße
- Puffer
- Waschlösungen
- qPCR-Cycler
- Sequenziergeräte
- Zentrifugen
Lesen Sie hier mehr zum Thema: Allgemeiner Laborbedarf: Was Hersteller und Labore wissen müssen, um sich Ärger und unnötige Aufwände zu ersparen
5. Möglichkeiten, um sich abzusichern
Um rechtliche und andere negative Konsequenzen bei der Nutzung von RUO-Produkten zu vermeiden, können Hersteller, Betreiber und Patient:innen die folgenden Schritte unternehmen:
a) Hersteller
Für Hersteller ist es besonders wichtig, die Zweckbestimmung ihres Produkts entsprechend eng festzulegen.
Analyten-spezifische Reagenzien sollten nur für konkrete nichtmedizinische Zwecke als RUO-Produkt gekennzeichnet werden.
SARS-CoV-2 und seine Mutationen: Ein Testkit, das bei bereits existierendem positivem Ergebnis individueller Patienten mit spezifischen Primern und Sonden die Varianten B.1.1.7 (Alpha-Variante) und B.1.351 (Beta-Variante) von der Ausgangsvariante unterscheidet, kann ein RUO-Produkt sein, wenn damit lediglich die Verbreitung der Variante in der Bevölkerung ermittelt werden soll.
Eine konkrete Zweckbestimmung wäre in diesem Fall: „Ausschließlich für die epidemiologische Forschung zum Zwecke der Erhebung der Verbreitung von SARS-CoV-2-Varianten in der Bevölkerung bestimmt.“
Wenn nun ein medizinisches Labor aufgrund neuer Erkenntnisse zur spezifischen Behandlung einer Varianten-Infektion diesen Test nutzt, um die bestmögliche Behandlung zu gewährleisten, wäre dies eine Nutzung außerhalb der Zweckbestimmung (Off-Label-Use). Das betreibende Labor bietet ein Inhouse-IVD an und ist für die Konformität des Tests verantwortlich.
Hinweis: Der Hersteller hätte sich jedoch ausreichend abgesichert, solange er diesen klinischen Nutzen nicht mit dem Produkt bewirbt.
b) Betreiber
Betreiber sollten genau verzeichnen, wozu sie IVD und RUO-Produkte nutzen.
Medizinische Labore sind Betreiber von Medizinprodukten bzw. IVD und stehen daher in der Verantwortung, Medizinprodukte nur ihrer Zweckbestimmung entsprechend und gemäß den allgemein anerkannten Regeln der Technik anzuwenden. Das fordert § 4 der Medizinprodukte-Betreiberverordnung MPBetreibV.
Um sich abzusichern, sollten Labore ein Verzeichnis führen, das auflistet, welche Medizinprodukte und IVD sich im Betrieb und der Routineanwendung befinden. Dieses Verzeichnis sollte einen Verweis auf das zutreffende Untersuchungsverfahren und die Zweckbestimmung der IVD enthalten.
Außerdem kann dieses Verzeichnis auch dazu dienen, Untersuchungsverfahren zu nennen, bei denen kein adäquates CE-IVD-Produkt am Markt verfügbar ist. Der Mangel an Alternativen rechtfertigt den Einsatz von RUO-Produkten im Rahmen von eigenentwickelten und validierten Verfahren als Inhouse-IVD, sofern das Labor prüfen und belegen kann, dass die grundlegenden Sicherheits- und Leistungsanforderungen und die zusätzlichen Anforderungen von Artikel 5 (5) der IVDR erfüllt werden.
Lesen Sie hier mehr zum Thema: Die EU reguliert medizinische Labore. Sind Laboratory Developed Tests noch erlaubt?
c) Patient:innen
Patient:innen fehlt das erforderliche Fachwissen, um selbst eine Abgrenzung vorzunehmen. Oft erhalten sie wenig bis keine Information über den Test, der durchgeführt wird. Für Patient:innen gilt daher: Fragen Sie Ihre Ärztin, Ihren Arzt oder in Ihrer Apotheke!
- Patient:innen können sich den kompletten Testbericht vom Labor aushändigen lassen, damit sie sich im Zweifel eine Zweitmeinung einholen können. Darin sollte auch ausgewiesen sein, welcher konkrete Test durchgeführt wurde.
- Patient:innen sollten sich informieren, wie „gut“ oder „schlecht“ ein Test funktioniert, sowie das Verhältnis von Nutzen und Risiko klären.
- Künftig können sich Patient:innen und Ärzt:innen auch über die EUDAMED zu den Produkten informieren und rückschließen, ob der jeweilige Test mit zertifizierten und damit rechtskonformen IVD-Produkten durchgeführt wurde oder nicht.
6. Fazit
Produkte „For Research Use Only“ haben nach Meinung der EU-Kommission und der FDA in der Diagnostik nichts verloren. Um für diagnostische Zwecke eingesetzt zu werden, müssen Produkte die nötigen Kontrollmechanismen durchlaufen. Doch gerade diese gelten für RUO-Produkte nicht.
Wer RUO-Produkte dennoch für andere Zwecke als die reine Forschung nutzt oder vertreibt, spielt mit dem Feuer. Hersteller und Betreiber laufen Gefahr, in rechtliche Schwierigkeiten zu geraten, und gefährden möglicherweise sogar die Gesundheit von Patient:innen. RUO-Produkte sollten daher ausschließlich für die Forschung genutzt werden.
Sollten Sie als Hersteller oder medizinisches Labor feststellen, dass ein RUO-Produkt sich für die In-vitro-Diagnostik besonders gut eignet, überlegen Sie, ob sich die Weiterentwicklung und die Konformitätsbewertung zum IVD bzw. Inhouse-IVD lohnt.
Vielen Dank an Dr. Boris Handorn, Rechtsanwalt und Partner der PRODUKTKANZLEI Augsburg für seinen wertvollen Input zu diesem Beitrag.
Wenn Sie sich einen noch besseren Überblick über die Anforderungen der IVDR an die Verwendung von Inhouse-IVD verschaffen wollen, dann besuchen Sie unser Seminar IVDR für medizinische Labore.
Wenn Sie bereits wissen, dass Sie künftig zum CE-IVD-Hersteller und Inverkehrbringer werden wollen, empfehlen wir Ihnen das Seminar Technische Dokumentation nach IVDR.
Oder Sie holen sich die Unterstützung des IVD-Teams des Johner Instituts.
- Es hilft Ihnen bei der Qualifizierung Ihrer Produkte bzw. Untersuchungsverfahren, beispielsweise mit Inhouse-Workshops zur Zulassungsstrategie und zu Inhouse-IVDs.
- Es erstellt Expertenmeinungen zur Qualifizierung Ihres Produkts, die Sie Ihren Kunden und/oder Benannten Stellen vorlegen können.
- Es unterstützt Sie bei allen Tätigkeiten bis zur „Zertifizierung“ Ihres Produkts (z. B. bei der Leistungsbewertung) und darüber hinaus (z. B. bei der Post-Market Surveillance).
Oder nutzen Sie unsere E-Learning-Plattform Auditgarant: Erfahren Sie, wie Sie die regulatorischen Anforderungen erfüllen, erhalten Sie Zugang zu unseren IVD-spezifischen Vorlagen sowie Templates und lernen Sie in Tutorials, wie Sie Ihr Produkt zur Zulassung bringen.
Versionshistorie:
- 2026-02-18 Abschnitt 2 b) Verwendung von RUO im diagnostischen Workflow ergänzt, Verweis auf Postitionspapier des Team-NB
- 2024-10-11 1. b) Verweis auf MDCG 2024-11 ergänzt, MEDDEV 2.14/2 rev. 1 als Quelle entfernt; 2. a) 2. Abschnitt Erläuterung zu MEDDEV 2.14/2 rev. 1 ergänzt
- 2024-02-01 Vollständige Überarbeitung; Abschnitt „Die Sache mit den Analyten-spezifischen Reagenzien“ entfernt; Kürzung von Kapitel 4 (Löschen von Unterkapiteln a) bis c)); Verweis auf Artikel zum Allgemeinen Laborbedarf
- 2021-11-16 Erstveröffentlichung



Sehr Interessant!
Vielen Dank!
Vielen Dank für das positive Feedback! Das freut uns.
Beste Grüße, Sebastian Grömminger
Vielen Dank für Ihren interessanten Artikel zu RUO. Ich vermisse jedoch noch eine Konkretisierung zu folgendem Punkt: Man liesst, dass ein RUO-Produkt kein CE-Label tragen darf. Ein spezielles Messgerät kann aber ein RUO sein. Um es zu nutzen muss es aber ein CE-Zeichen tragen welches zeigt, dass das Gerät die Normen elektrische Sicherheit, EMV usw erfüllt.
CE? kein CE?
Lieber Alexander, vielen Dank für diese spannende Frage und den Hinweis, dass dieser Aspekt zu Mess-Geräten fehlt.
Ein Mess- oder Analysegerät hat per se keinen medizinischen Zweck und fällt daher nicht zwingend unter die Definition eines IVD. Sie haben allerdings recht: Als Laborgerät muss es dennoch die Anforderungen der Niederspannungsrichtline (2014/35/EU) und ggf. auch die Maschinienrichtlinie (2006/42/EG) einhalten und damit auch unter diesen Richtlinien eine CE-Kennzeichnung tragen.
Mehr zu diesen beiden Themen finden Sie hier:
https://www.johner-institut.de/blog/systems-engineering/iec-61010-ivd/
https://www.johner-institut.de/blog/systems-engineering/maschinenrichtlinie-medizinprodukte/
Sollte das Mess- oder Analysegerät aber (zudem) einen medizinischen, besser gesagt einen in-vitro-diagnostischen Zweck besitzen, dann sollte es zudem das IVD-Symbol tragen bzw. klar als in-vitro-Diagnostikum gekennzeichnet sein und muss dann gemäß IVDD (98/79/EG) bzw. bald unter IVDR (2017/746/EU) konformitätsbewertet werden. Damit muss es natürlich auch ein CE oder besser ein CE-IVD Zeichen tragen.
Herzliche Grüße,
Sebastian Grömminger
Vielen Dank für den interessanten Artikel zum Thema RUO. Wir sind Hersteller von MR-Spulen die keine Medizinprodukte sind und nur für Forschungszwecke verwendet werden dürfen. Wir stellen uns immer wieder die Frage, wie wir diese „Research only devices“ korrekt labeln. Denken Sie man könnte das RUO-Symbol auch für Nicht-Medizinprodukte verwenden oder ist es ausschließlich für IVDRs vorgesehen? Wenn ja, gibt es ein gleichwertiges Symbol für die Kennzeichnung von Nicht-Medizinprodukten die nur für Forschungszwecken verwendet werden dürfen?
Vielen Dank & viele Grüße,
Marius Berthel
Lieber Herr Berthel,
vielen Dank für Ihre Frage.
Wenn Ihre Produkte tatsächlich nur für Forschungszwecke eingesetzt werden, dann ist das Research Use Only Label genau das Richtige. Wenn Sie allerdings wissen sollten, dass das Produkt auch für medizinische Zwecke eingesetzt wird, müsste abgegrenzt sein, wer der legale Hersteller des (IVD-)Medizinproduktes ist. Eine MR-Spule hört sich jedoch vielmehr nach einem Bauteil an, dass in MRT-Gerät eingebaut wird und damit eine rein technische Komponente darstellt. Im letzteren Fall sind sie in der Rolle eines Zulieferers und nicht in der eines Herstellers.
Herzliche Grüße, Sebastian Grömminger
Vielen dank für diesen erhellenden Einblick.
Die rechtlichen Rahmenbedingungen von IVDs mit medizinischem „intended pupose“ glaube ich anhand dieses Artikels zumindest grob verstanden zu haben. Jedoch frage ich mich nach wie vor wie es eigentlich um die Regulierung von In-Vitro assays mit nicht direkt medizinischem intended purpose steht. Also beispielsweise zur Untersuchung von Wirkstoffen. Zum einen in der Grundlagenforschung aber noch viel wichtiger und interessanter in der präklinischen Bewertung von neuen Wirkstoffen. Also nicht für eine Performances study des IVDs sondern für die eines anderen Produktes. Sind diese frei verkäuflich, ist ein Qualitätsmanagement vorgeschrieben, müssen diese für den intended purpose validiert sein und welche Rolle spielen IVDR und GLP hier für den Hersteller der In-Vitro Produkte. So viele Fragen und so viele unterschiedliche Informationen. Wenn sie mir auf den richtigen Pfad helfen könnten wäre ich ihnen sehr dankbar.
Beste Grüße,
Jakob Wolf
Lieber Herr Wolf,
ja in der Tat, hier tun sich viele Fragen auf an der „Bordeline“ zum IVD. Zur Identifizierung von Biomarkern in der Grundlagenforschung braucht das Produkt keinen CE-IVD-Status, da dies keine medizinische Zweckbestimmung (ZB) ist. Bei präklinischen Tests kommt es auf die ZB an. Ein RUO Reagenzien-Kit kann in diesen Fällen in der Regel durchaus verwendet werden, allerdings dürfen keinerlei Ergebnisse an Patienten mitgeteilt werden und das Probenmaterial muss für Forschungszwecke freigegeben sein (z.B. Left-over Material von anderen Untersuchungen). Hier ist in der Regel ein Ethik-Votum erforderlich. Bei klinischen Studien, egal ob interventionell oder nicht-interventionell muss das Produkt, wenn es einen medizinischen Zweck erfüllt der IVDR entsprechen. Anstelle eines CE-IVD-Produktes kann aber auch ein In-house-Test verwendet werden.
Auf diese Flut an Fragen hat die MDCG sehr aktuell (vor einem Monat) reagiert mit einem Q&A-Dokument: Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
zu Clinical Trials hilft Ihnen auch diese Seite weiter: https://ec.europa.eu/health/medicinal-products/eudralex/eudralex-volume-10_en
Ich hoffe, ich konnte Ihnen damit den Pfad aufzeigen an der Schwelle zum Arzneimittel.
Beachten Sie auch unseren Artikel zu CDx, falls das für Sie relevant ist: https://www.johner-institut.de/blog/regulatory-affairs/companion-diagnostics-cdx/
Mit besten Grüßen,
Sebastian Grömminger
Guten Tag,
eine kurze Frage ! Sind RUO-Produkte legacy Products?
Danke im Voraus!
Rony
Lieber Rony,
das ist eine rechtliche Grauzone. Es wäre aber kein „Legacy Device“ sondern eher ein „old device“. Die MDCG 2022-6 sagt: ‘Old’ devices are those devices that were placed on the market or put into service before 26 May 2022 in accordance with the IVDD or the applicable national rules before the IVDD had become applicable and which are still on the market or in use after 26 May 2022. Es gibt auch Produkte, die außerhalb des Anwendungsbereiches der IVDD lagen aber nun unter die IVDR fallen. Diesen speziellen Fall betrachtet die Leitlinie leider nicht. Andererseits muss man auch anerkennen, dass ein RUO-Produkt ja auch nie für diagnostische Zwecke gedacht war, außer es war eine Schutzbehauptung des Herstellers. Daher ist ein RUO-Produkt meiner Ansicht nach weder ein Legacy Device noch ein Old Device sondern ein „new Device“ das die Anforderungen an die IVDR erfüllen muss oder ein RUO das im tatsächlichen Sinne rein für wissenschaftliche Zwecke vorbehalten sein sollte.
Mit besten Grüßen,
Sebastian Grömminger
Hallo Herr Dr. Grömminger,
vielen Dank für den spannenden Beitrag.
Können Sie mir sagen, welche Auswirkungen es auf IVD-Hersteller hat, wenn sie bei der Herstellung von IVD-Reagenzien RUO- bzw. R&D-gekennzeichnete Inhaltsstoffe mit verwenden? Gibt es hier Konsequenzen für den Hersteller der IVD-Reagenzien oder ist es eher ein Problem des Herstellers des RUO-Produkts?
Mit freundlichen Grüßen
Maria
Liebe Maria,
vielen Dank für die spannende Frage. Wenn RUO-Materialien für die Herstellung eines IVD eingesetzt werden, liegt die Verantwortung beim IVD-Hersteller. Der RUO-Hersteller kann ja nicht für etwas belangt werden, was er nicht für das Produkt vorgesehen hat, solange er die o.g. Spielregeln einhält. Der IVD-Hersteller muss den RUO-Hersteller, von dem er die Eingangsware bezieht, im Rahmen der Lieferantenqualifizierung gemäß ISO 13485 bewerten und die Produktqualität sicherstellen. Das kann der Hersteller über verschiedene Arten vornehmen. Entweder mit Fokus auf die Produktprüfung (z.B. funktionale Wareneingangs QC jeder Charge) oder Prozesssicherheit durch Qualitäts-Sicherungs-Vereinbarungen mit dem Lieferanten (ggf. mit Audit). Bei kritischen Komponenten kann sogar beides sinnvoll sein.
Ich hoffe das beantwortet Ihre Frage.
Meine besten Grüße,
Sebastian Grömminger
Hallo Herr Dr. Grömminger,
Vielen Dank für ihr Beitrag.
I send my question in English because my German is a little poor.
What is the responsibility of a manufacturer if he sells a RUO component of a RUO kit (for example a box of calibrators) to a customer who incorporates this box into his own kit which he will use for clinical use? (in this example the calibrators are under a finished form, i.e. several vials of lyophilised product with various concentrations of reference standards)
Thanks for your advice
Best regards
Dear Francis,
thank you for your important question.
The responsibility of a RUO manufacturer is to keep the commercial claims for those products within the research scope only. In addition, if he learns that the customers buying those RUO products are misusing them for clinical purposes (i.e. incorporating them into an IVD reagent kit) I recommend to setup agreements with the customers, clarifying that the product shall not be used for medical purposes and any other use then for research purposes is unter the customer’s legal responsibility.
I hope this answers your question. Please let us know if you need further assistance.
With best regards,
Sebastian Grömminger
Dear Sebastian,
Many thanks, this confirms my thoughts.
Best regards
Hallo Herr Dr. Grömminger,
ich hätte eine Frage bezüglich ASRs und LDTs. Und zwar im Detail, wie es mit den regulatorischen Anforderungen aussieht wenn man ein ASR aus den USA bezieht und es in Europa in einen LDT einbaut. Darf das ASR hierbei bereits einen medizinischen Zweck haben oder muss es als RUO gekennzeichnet sein?
Vielen Dank für Ihre Hilfe. Mit freundlichen Grüßen,
Gabriel Wagner
Lieber Herr Wagner,
vielen Dank für ihre Frage.
Weist der Produzent dem ASR einen medizinischen Zweck, gemäß seiner Zweckbestimmung, zu, so fällt er unter die Anforderungen der IVDR und muss die grundlegenden Sicherheits- und Leistungsanforderungen erfüllen, bevor er sein Reagenz in der EU in Verkehr bringen darf. Verwendet das Labor dieses CE-IVD gemäß den Hersteller-Angaben in der Gebrauchsanweisung, so wird das Labor nicht zum Eigenhersteller (siehe Beitrag Die EU reguliert medizinische Labore. Sind In-house IVD (LDT) noch erlaubt?). Kennzeichnet der ASR-Produzent dagegen das Produkt als RUO, darf das Produkt nur für Forschungszwecke eingesetzt werden. Die Anforderungen der IVDR sind für den ASR-Produzenten in diesem Fall nicht anwendbar, wohl aber für das Labor. Bei der Verwendung innerhalb eines In-house IVD (LDT) ist dem RUO-Produkt bzw. ASR ein medizinischer Zweck zugewiesen, so dass die Bezeichnung RUO (die Bezeichnung ASR gibt es in Europa nicht) dann nicht mehr haltbar wäre. Das Labor muss folglich die Anforderungen der IVDR (Artikel 5 (5)) erfüllen.
Ich hoffe das beantwortet Ihre Frage.
Herzliche Grüße
Diana Gabriel
Sehr geehrte Frau Gabriel,
vielen Dank für Ihre Antwort. Mir ist noch nicht ganz klar wie das genaue Vorgehen ist wenn dem ASR bereits ein medizinischer Zweck zugeordnet wurde. Braucht die Firma aus den USA dann einen Bevollmächtigten in der EU welcher das ASR als IVD zertifizieren muss? Oder genügt es, wie von Ihnen angesprochen, dass der Hersteller die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt? Wäre die Klassifizierung als Risikoklasse A (abhängig des medizinschen Zwecks), da ein ASR nach Regel 5 kein Ergebnis liefert, hier eine Möglichkeit?
Vielen herzlichen Dank für Ihre Hilfe!
Mit freundlichen Grüßen,
Gabriel Wagner
Lieber Herr Wagner,
Der US-Hersteller ist für die Zertifizierung seiner Produkte (Artikel 10, IVDR) verantwortlich. Das bedeutet, dass er in der Regel ein Qualitätsmanagementsystem etabliert und eine Technische Dokumentation erstellt. Je nach Klassifizierung ist in der Konformitätsbewertung eine Benannte Stelle involviert, die das Qualitätsmanagementsystem und die Technische Dokumentation des Herstellers prüft. Da sich der Hersteller außerhalb der Union befindet, muss er einen EU-Bevollmächtigten ernennen und sicherstellen, dass dem Bevollmächtigten die erforderliche Dokumentation durchgängig zugänglich ist, damit dieser die in Artikel 11 Absatz 3 genannten Aufgaben wahrnehmen kann. Unserer Erfahrung nach ist es sehr unwahrscheinlich, dass ein ASR gemäß Regel 5 klassifiziert werden kann. Um allerdings eine verlässliche Aussage bezüglich Klassifizierung und Konformitätsbewertungsverfahren treffen zu können, sind produktspezifische Angaben notwendig. Daher würde ich Sie gerne auf unser Kontaktformular verweisen.
Mit freundlichen Grüßen
Diana Gabriel
Hallo Frau Diana Gabriel,
vielen Dank für den sehr spannenden Beitrag.
Ich hätte noch eine weiterführende Frage dazu.
Was ist der Unterschied bei der Kennzeichnung meines Produktes zwischen RuO und „nichts“.
In welchen Fällen sollte ich das RuO Kennzeichen auf mein Produkt integrieren? Und in welchen praktischen Fällen vermarkte/etikettiere ich mein Produkt vollständig ohne eine Klassifizierung?!
Beste Grüße und Danke
Andreas Schmidt
Lieber Herr Schmidt,
vielen herzlichen Dank für Ihre Fragen.
Für RUO-Produkte gibt es in der EU keine regulatorischen Vorgaben an die Kennzeichnung. Generell kann man sagen, sobald die Gefahr besteht, dass das Produkt für diagnostische Zwecke verwendet wird, dies aber ausgeschlossen werden soll, die Kennzeichnung mit „Research use only“ erfolgen sollte. Achten Sie hier bitte auf ihre Zweckbestimmung, welche Art von Angaben Sie zum Beispiel auf ihrer Webseite tätigen und an wen Sie das Produkt vertreiben, so dass Ihr Produkt als RUO in Verkehr gebracht werden kann. Ohne jedoch Ihre Produkte genau zu kennen, ist eine Einschätzung, wann sie praktischerweise auf die Kennzeichnung verzichten können schwierig. Sollten Sie daher weitere produktspezifische Fragen bezüglich der RUO-Kennzeichnung haben, können Sie auch gerne unsere Micro-Consulting Anfrage nutzen.
Ich hoffe, ich konnte Ihnen damit schon etwas weiterhelfen.
Herzliche Grüße
Diana Gabriel
Hallo Frau Gabriel,
wie immer auf Ihrer Seite: Toller Beitrag.
Eine Health Institution nach IVDR (29) möchte unter Verwendung eines (vom Hersteller) mit RUO gekennzeichneten Antikörpers einen prognostischen LDT entwickeln. Ist dies möglich oder ist dies grundsätzlich ausgeschlossen? Wäre ein Quality Agreement mit- und/oder ein Audit von dem Hersteller von Nutzen. Ich frage dies, da mir aus meiner Arbeit viele pathologischen Labore bekannt sind, dei diesenn Weg gehen?!
Lieber Herr Oed,
vielen herzlichen Dank für ihr positives Feedback und ihre spannenden Fragen.
Gesundheitseinrichtungen können weiterhin RUO-Produkte erwerben. Verwenden medizinische Labore nun diese RUO-Produkte im Sinne der IVD-Definition (IVDR, Artikel 2 (2)), werden Sie zum Hersteller eines In-house IVD (auch LDT genannt). Als Hersteller eines In-house IVD müssen die medizinischen Labore den Artikel 5 (5) der IVDR erfüllen. Zu diesen Anforderungen zählt ebenso die Etablierung eines QM-Systems nach EN ISO 15189 oder nationalen Vorschriften und anwendbaren Teilen der ISO 13485 (Artikel 5 (5) b), c) und e). Gemäß (DIN) EN ISO 15189 müssen medizinische Labore sicherstellen, dass extern bereitgestellte Produkte und Dienstleistungen, die sich auf Labortätigkeiten auswirken, geeignet sind (Kapitel 6.8, Ähnliches im Kapitel 7.4 der ISO 13485). Hierzu gehören neben den Kriterien für die Qualifizierung der externen Anbieter auch die Bewertung der Leistung und Wiederbewertung dieser. Wie die medizinischen Labore im Speziellen nun diese Anforderungen an externe Anbieter umsetzen und belegen, legen Sie selbst in ihrem QM-System fest. Das können Qualitätssicherungsvereinbarungen (QSV) sein, aber auch Lieferantenaudits. Zum Beispiel können QSV von Nutzen sein, wenn ich als medizinisches Labor sicherstellen möchte, vor jeder Spezifikationsänderung der Rohware informiert zu werden. Ein Lieferantenaudit kann zum Beispiel von Nutzen sein, um zu überprüfen, ob der Lieferant tatsächlich die Anforderungen des medizinischen Labors initial und im weiteren Verlauf erfüllt. Ich empfehle Ihnen in diesem Zusammenhang auch unseren Artikel zu Inhouse IVD.
Herzliche Grüße
Diana Gabriel
Sehr geehrte Frau Daniel,
ich habe zwei Fragen zur Abgrenzung von RUO und allgemeinem Laborbedarf:
Weder RUO noch der allgemeine Laborbedarf besitzen eine medizinische Zweckbestimmung.
RUO-Produkte schließen mit der Bezeichnung RUO eine Verwendung in der Diagnostik explizit aus.
Der allgemeine Laborbedarf ist nicht für ein festgelegtes in-vitro-diagnostisches Untersuchungsverfahren geeignet (daher kein IVD, vgl. Meddev 2.14.1), schließen eine Anwendungen in der Diagnostik jedoch auch nicht aus.
Ist das die richtige Abgrenzung? Gibt es einen „Zwang“ für Hersteller, ein Produkt als RUO zu labeln?
Gemäß MEDDEV 2.14/2 fallen analyten-spezifische Reagenzien (ASR) ohne medizinischen Zweck unter die Kategorie allgemeiner Laborbedarf. Bei einer Verwendung innerhalb eines diagnostischen Tests in einer Gesundheitseinrichtung, wird dem ASR aber ein medizinischer Zweck zugewiesen, daher handelt es sich um einen LDT.
Wie ist das mit anderem allgemeinem Laborbedarf (z.B. eine Zentrifuge) – hier findet ja keine Umwidmung statt und keine Herstellung eines LDT. Wie lautet hier die Abgrenzung?
Vielen Dank für Ihre Expertise und mit freundlichen Grüßen,
Martina Mitterhuber
Liebe Frau Mitterhuber,
bitte entschuldigen Sie zunächst die verspätete Antwort aufgrund der Feiertage und Urlaubszeit und vielen Dank für Ihre Fragen.
Sie haben es richtig zusammengefasst, während RUO-Produkte die Verwendung in der Diagnostik ausschließen (Produkte rein für Forschungszwecke), können Produkte für den allgemeinen Laborbedarf in einem medizinischen Labor und damit in der Diagnostik verwendet werden. Einen Zwang ein Produkt als RUO zu kennzeichnen gibt es nicht, jedoch dient dieses Label der Absicherung des Herstellers und deutet darauf hin, dass eine Anwendung in der Diagnostik nicht vorgesehen ist, da diese weder verifiziert noch validiert wurden ist. Jedoch ist für den Hersteller zu beachten, dass zum Beispiel Werbematerialien keinen Rückschluss auf einen medizinischen Zweck zulassen, denn dann wäre das Label „RUO“ nicht korrekt.
Ob es sich um allgemeinen Laborbedarf handelt oder um ein IVD-Produkt gemäß IVDR, hängt von der nachfolgenden in-vitro Untersuchung ab und ob spezifische Charakteristiken hierfür erforderlich sind. Es kommt also auf die Zweckbestimmung an, für die sie die Zentrifuge einsetzen. Sind zum Beispiel während des Untersuchungsverfahrens bestimmte Charakteristiken oder besondere Einstellungen der Zentrifuge notwendig, um ein diagnostisches Ergebnis zu erhalten, so ist davon auszugehen, dass die Anforderungen der IVDR erfüllt werden müssen (siehe Zweckbestimmung einer Hämatokrit-Zentrifuge). Bitte beachten Sie, dass für medizinische Labore die Anforderungen an das Qualitätsmanagement durch die ISO 15189 bzw. andere nationale Vorgaben unabhängig von der IVDR erfüllt werden müssen (Anforderung an die Ausrüstung, Verifizierung und Validierung von Untersuchungsverfahren, etc.). Dies bedeutet, dass auch der allgemeine Laborbedarf im Mindesten diese Anforderungen erfüllen muss.
Ich hoffe, ich konnte Ihnen mit der Antwort weiterhelfen.
Mit freundlichen Grüßen
Diana Gabriel
Sehr geehrte Frau Gabriel,
ich danke Ihnen sehr für Ihre Antwort und die logischen Erklärungen.
Mit freundlichen Grüßen,
Martina Mitterhuber
Guten Tag,
heute bin ich auf ihre interessante Seite zum Thema RUO gestoßen.
Ich würde mich sehr freuen, wenn sie mir bei nachfolgender Schilderung ihren Rat geben könnten.
Ich bin MTA , Bereich Immunhistochemie. Wir streben die Zertifizierung an, bzw. stecken mittendrin.
Folgender Fall : ich bestellte wie gewöhnlich ein Antikörperkonzentrat bei einer hier bekannten Firma.
Ich bekam die Antwort, dass der bisher verwendete Clon bzw. nicht mehr verfügbar wäre. Anbei wurde mir eine Alternative geschickt. Das an sich ist ja nichts Ungewöhnliches. Zum Glück habe ich genau geschaut – es handelte sich um ein RUO Antikörperkonzentrat. Ich war froh, dass gesehen zu haben, da im Arbeitsstress so etwas schon mal untergehen kann und man sich ja auch als langjähriger Kunde auf die Aplikationsspezialisten verlässt.
Wenn ich ihren Artikel richtig verstanden habe, sollte ich die Finger von RUO Antikörperkonzentraten lassen,oder ?
Liebe Frau Holl,
vielen Dank für Ihren Kommentar und Ihr Kompliment zu unserem Artikel.
Gerne versuche ich Ihnen zu antworten. Ich gehe davon aus, dass Sie normalerweise ein CE-IVD Antikörper beziehen und diesen in einem diagnostischen Verfahren einsetzen. Wenn Sie nun diesen CE-markierten IVD-Antikörper durch einen mit RUO gelabelten Antikörper ersetzen, führt dies zur Herstellung eines Inhouse-IVD. Daraus folgt, dass Sie vor Anwendung in der Routine die Anforderungen der IVDR, Artikel 5(5) erfüllen müssen. Dazu gehört, neben vielen weiteren Anforderungen, ausführliches Risikomanagement und eine Leistungsbewertung nach Stand der Technik. Sie dürften den RUO-Antikörper erst einsetzen, sobald Sie alle anwendbaren Anforderungen der IVDR erfüllt haben.
Ich hoffe, Ihre Frage zu Ihrer Zufriedenheit beantwortet zu haben. Sollte dies nicht der Fall sein, melden Sie sich gerne über unsere Kontaktseite.
Herzliche Grüße,
Ulrich Hafen
PS: Mehr zu den Anforderungen an Inhouse-IVD erfahren Sie in meinem Seminar „IVDR für medizinische Labore„.
Ich bin über die Maßen glücklich mit ihrer ausführlichen Antwort. Da ich bislang in der Tat nur CE- IVD’s benutzt habe, fehlte mir hier Fachwissen. Ich habe die zuständige Firma hierzu kontaktiert – bisher hielt man es nicht für nötig mir zu antworten.
Nochmals vielen herzlichen Dank,
I. Holl
Benötigen RUO-Produkte eine Konformitätserklärung? Ich bin der Meinung nein, da es keine Rechtsvorschriften gibt, mit denen sie konform sein können.
Die CE-Kennzeichnung erhalten sie nach IVDR nicht.
Gäbe es die Möglichkeit, dass sie trotzdem eine CE-Kennzeichnung nach einer anderen Rechtsvorschrift erhalten z.B. Maschinenrichtlinie bei Geräte?
Wie steht es hierum bei Allgemeinem Laborbedarf?
Lieber Herr Handt,
RUO-Produkte wie auch Produkte für den allgemeinen Laborbedarf können u. U. ein CE-Kennzeichen tragen, auch wenn sie nicht unter die IVDR fallen.
Dies gilt in der Regel insbesondere für Geräte (weniger für Reagenzien und reine Softwareprodukte).
Das CE-Kennzeichen ist eine verbindliche Konformitätskennzeichnung, die anzeigt, dass ein Produkt mit den Harmonisierungsvorschriften der Europäischen Union übereinstimmt. Mit der CE-Kennzeichnung erklärt ein Hersteller, dass sein Produkt mit allen anzuwendenden EU-Rechtsvorschriften konform ist. Je nach Produkt gelten neben IVDR (bzw. MDR) auch andere EU-Rechtsvorschriften, so zum Beispiel, wie Sie selbst schreiben, die Maschinenrichtlinie 2006/42/EG bzw. die Maschinenverordnung (EU) 2023/1230 oder die EMV-Richtlinie 2014/30/EU, die Niederspannungsrichtlinie 2014/35/EU u. a. Der Hersteller ist selbst dafür verantwortlich zu prüfen, welche EU-Rechtsvorschriften für sein Produkt anwendbar sind. Der Hersteller muss dann ggf. eine entsprechende Konformitätsbewertung seines Produktes durchführen, eine EU-Konformitätserklärung ausstellen, in der er erklärt, dass sein Produkt allen anwendbaren EU-Rechtsvorschriften entspricht und kann dann das CE-Label aufbringen.
Ich hoffe, Ihre Frage damit beantwortet zu haben. Sollte dies nicht der Fall sein, melden Sie sich gerne über unsere Kontaktseite.
Herzliche Grüße,
Sophie Bartsch