
Eine Annahme hält sich hartnäckig: Allgemeiner Laborbedarf wie PCR-Cycler, NGS-Geräte, Fragment-Analyzer, Zentrifugen, Pipettier-Roboter und Extraktionskits müssen eine CE-IVD-Kennzeichnung tragen. Stimmt das?
Es fehlt oft Klarheit darüber,
- was als allgemeiner Laborbedarf zählt und
- ob dieser die Anforderungen der Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR) erfüllen muss.
Daher gibt es Unsicherheiten,
- was die Hersteller von IVD und die Hersteller von allgemeinem Laborbedarf bewerben dürfen,
- welche Anforderungen die jeweiligen Hersteller erfüllen müssen und
- für welche Zwecke die medizinischen Labore diese Produkte einsetzen dürfen,
um sich nicht strafbar zu machen.
Dieser Artikel verschafft Klarheit und gibt Herstellern und Betreibern (z. B. medizinischen Laboren) konkrete Tipps, um regulatorischen Ärger und unnötige Aufwände zu vermeiden.
1. Allgemeiner Laborbedarf: Eine regulatorische Einordnung
Zuerst gilt es zu klären, wie der allgemeine Laborbedarf zu qualifizieren ist. Fällt er in die Kategorie IVD, ist auch eine Klassifizierung notwendig.
a) Qualifizierung (Übersicht)
Die IVDR unterscheidet mehrere „Typen“ an Produkten:
- In-vitro-Diagnostika (IVD), die eine CE-Kennzeichnung tragen müssen (CE-IVD-Produkte)
- Produkte für Leistungsstudien
- Produkte, die ausschließlich innerhalb von in der Union ansässigen Gesundheitseinrichtungen hergestellt und verwendet werden (Inhouse-IVD)
- Produkte für den allgemeinen Laborbedarf (General Laboratory Use, GLU)
- Produkte ausschließlich für Forschungszwecke (Research Use Only, RUO)
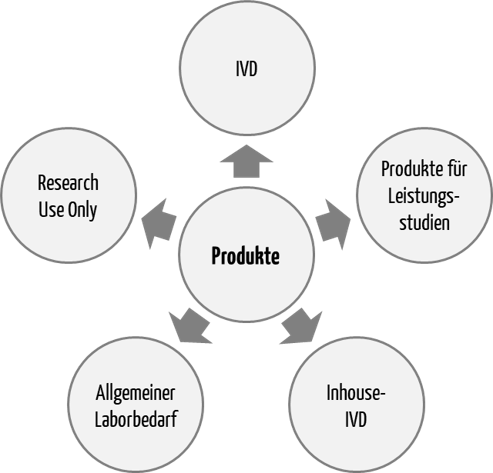
Die IVDR reguliert die ersten drei Typen. Die letzten beiden, also GLU und RUO, fallen gemäß Artikel 1 (3) a) IVDR nicht in den Geltungsbereich der Verordnung.
„Diese Verordnung gilt nicht für
Artikel 1 (3) a) IVDR
a) Produkte für den allgemeinen Laborbedarf oder allein für Forschungszwecke bestimmte Produkte, es sei denn, sie sind aufgrund ihrer Merkmale vom Hersteller speziell für In-vitro-Untersuchungen bestimmt;“
Eine Ausnahme liegt vor, wenn Produkte für den allgemeinen Laborbedarf aufgrund ihrer Merkmale vom Hersteller speziell für In-vitro-Untersuchungen bestimmt sind.
Produkte für den allgemeinen Laborbedarf fallen entweder nicht in den Geltungsbereich der IVDR (Regelfall) oder sie zählen als „CE-IVD“ und sind der Klasse A zuzuordnen (dazu später mehr).
Als RUO-Produkte gelten Produkte, die ausschließlich zur Analyse im Rahmen von wissenschaftlichen Forschungszwecken gedacht sind.
Lesen Sie hier mehr zum Thema: Laborprodukte „For Research Use Only (RUO)“ – oft eine gefährliche Behauptung. Sie finden hier weitere Tipps für Betreiber wie Labore.
b) Qualifizierung als „Allgemeiner Laborbedarf“
Wie bei RUO-Produkten gibt es auch bei Produkten für den allgemeinen Laborbedarf (General Laboratory Use, GLU) keine einheitliche Definition.
Allerdings liefert die IVDR mit dem oben genannten Artikel 1 (3) a) Anhaltspunkte: Besitzt das betreffende Produkt keine charakteristischen Eigenschaften, die es für eine spezifische IVD-Untersuchung anwendbar machen, ist es auch kein IVD. Solche Produkte sollten als allgemeine Laboranwendungen vorgesehen sein.
Dennoch dürfen diese Produkte im Rahmen von in-vitro-diagnostischen Untersuchungsverfahren Anwendung finden, auch wenn sie selbst keine IVD sind. Sobald derartige Kombinationen beansprucht werden, erwarten Benannte Stellen einen Nachweis, dass die Kombination sicher ist und die vorgesehene Leistung nicht beeinträchtigt. Der Schwerpunkt liegt hierbei auf Risiko- und Leistungsbewertung (Team-NB Position Paper on Demonstration of Safety and Performance for Combinatorial Use of Reagent Devices with other devices or Equipment).
Leitlinien
Die MDCG 2024-11 dient der Klärung der Qualifizierung von Produkten als IVD bzw. als Zubehör zu einem IVD. Des Weiteren enthält die Leitlinie konkrete Beispiele für allgemeinen Laborbedarf.
Neben der Leitlinie MDCG 2024-11 sollte zukünftig das Manual on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices zur Klärung beitragen. Ein Kapitel zu „Borderline between IVDs and general laboratory equipment” ist bereits angelegt, jedoch leider noch ohne Inhalt.
Die MDCG 2024-11 liefert Beispiele
Die MCDG 2024-11 liefert eine Gegenüberstellung, wann bestimmte Produkttypen als GLU und wann als IVD qualifiziert werden:
| General laboratory use product | Covered by IVDR* | |
| Centrifuges | General centrifuges, cytospin | Hematocrite centrifuge |
| Pipettes | General purpose pipettes (e.g. single or multiple pipettes, plastic pipettes, Pasteur pipettes) | Blood coagulation pipettes with automatic timing (accessory for coagulometer) |
| Tubes and flasks | Erlenmeyer flasks, general plastic tubes | Blood collection tubes, urine specimen containers |
| Plates | General empty ELISA plates, general empty Petri dishes | Coated microtiter plates for the diagnosis of Lyme’s disease |
| Nucleic acid extraction products | DNA and RNA extraction/purification kits that only extract/purify nucleic acids | DNA and RNA extraction kits intended to prepare an extract to be used with a specific IVD device or of specifically intended to be used for in vitro diagnostic examination |
| General equipment | Scales, balances, microtomes, incubators, sterilizers for laboratory equipment, paraffin embedding machine, water baths, ELISA plate washers | |
| HPLC products | Size-exclusion HPLC columns | HPLC columns for IVD purposes: e.g. HbA1c |
| PCR machines | General PCR machine | PCR machine intended for IVD purposes: e.g. detection and differentiation of influenza/SARS-CoV-2 RNA |
| Detection equipment | Mass spectrometers, spectrophotometers, ELISA readers providing raw data which is not readily readable and understandable by the user (e.g. peaks, OD) | McFarland bacteria density reader specifically intended for IVD examination of human specimens |
| Others | Foetal calf serum, cell culture media, fixation solution, mounting media, buffers (e.g. PBS), chemicals (e.g. sulphuric acid, formol, water) | Wash solution intended to be used with a specific IVD analyser, culture media intended for amplification and detection of infectious agents in human specimens |
*Insofern die vom Hersteller angegebenen Zweckbestimmung die Definition eines IVD erfüllt.
Quelle: MDCG 2024-11
Diese Gegenüberstellung bestätigt, dass es von den spezifischen Eigenschaften der Produkte für den allgemeinen Laborbedarf abhängt, ob sie sich als IVD qualifizieren.
Weitere Beispiele
Eine unspezifische Zentrifuge fällt als allgemeiner Laborbedarf nicht unter die IVDR.
Hingegen ist eine Hämatokrit-Zentrifuge, deren Rotor speziell dafür ausgelegt wurde, dass er mit Hämatokrit-Kapillaren gemäß EN ISO 12772 beladen werden kann und die das Ablesen des Hämatokritwertes direkt am Gerät ermöglicht, ein IVD.
Produkte für den allgemeinen Laborbedarf werden allerdings zum IVD, wenn sie vom Hersteller für ein konkretes in-vitro-diagnostisches Verfahren vorgesehen sind. Dies ist insbesondere in Klassifizierungsregel 5 der IVDR (Anhang VIII) verankert.
Das Extraktionskit A zur Extraktion von RNA und DNA aus EDTA-Blut ist vom Hersteller X speziell für das qPCR-basierte In-vitro-Diagnoseverfahren in Kombination mit qPCR-Kit B auf dem C-System von Hersteller X vorgesehen. Dieses Extraktionskit ist ein IVD und trägt ein CE-Kennzeichen.
Ein qPCR-Cycler ohne spezifische Eigenschaften, der mit jedem beliebigen qPCR-Kit von jedem beliebigen Hersteller verwendet werden kann (insofern der Channel für die entsprechende Sondenmarkierung vorliegt) und als Output Rohdaten liefert (bspw. Schmelzkurvendiagramme), ist ein Produkt für den allgemeinen Laborbedarf. Eine CE-IVD-Markierung ist nicht gestattet (s. Kapitel 2.a).
Ist der qPCR-Cycler nun vom Hersteller für die in-vitro-diagnostische Untersuchung zum Nachweis von C. difficile mit einem konkreten IVD-Kit und zugehöriger Auswertesoftware vorgesehen, wird der Cycler zum CE-IVD-Produkt.
Zu beachten ist: Wird auf diesem CE-IVD-Cycler ein anderes als das vom Hersteller deklarierte Kit verwendet, wenngleich dieses auch zum Nachweis von C. difficile dient, wird der Cycler off-label verwendet und die CE-IVD-Markierung bietet dem Betreiber keinen Vorteil mehr (s. Kapitel 2.b).
c) Klassifizierung
Qualifiziert sich ein Produkt als In-vitro-Diagnostikum, muss der Hersteller seinem Produkt eine Klasse zuweisen.
Klassifizierungsregel 5 der IVDR besagt:
„Die folgenden Produkte werden der Klasse A zugeordnet:
IVDR, Anhang VIII, Regel 5
- Erzeugnisse für den allgemeinen Laborbedarf, Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen sowie allgemeine Nährmedien und histologische Färbungen, die vom Hersteller dafür vorgesehen sind, die Produkte für In-vitro-Diagnoseverfahren im Zusammenhang mit einer spezifischen Untersuchung einsetzbar zu machen;
- Instrumente, die vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind; […]“
Der oben erwähnte qPCR-Cycler für die in-vitro-diagnostische Untersuchung zum Nachweis von C. difficile mit einem konkreten IVD-Kit und zugehöriger Auswertesoftware fällt in die Klasse A.
Weitere Beispiele zu Klasse A-Produkten liefert die MDCG 2020-16:
„Rule 5a applies to general laboratory products like pipettes, stain powders, glass microscope slides, centrifuges, pipette tips or instrument liquid collection containers, buffers which usually do not fall under the definition of an IVD medical device.”
MDCG 2020-16
Die Leitlinie nennt Beispiele für Produkte, nicht aber im Zusammenhang mit konkreten Anwendungen. Sobald diese Produkte für eine spezifische Anwendung genutzt werden, gelten sie nicht mehr als allgemeiner Laborbedarf.
- “Solutions like cleaners, buffer solutions, lysing solutions, diluents specified for use with an IVD.
- Pipette with a specific fixed one volume specifically intended for a particular IVD test with specified human sample […].
- Kits for Isolation and purification of nucleic acids from human specimens.
- Nucleic acid quantitation kits.“
Die letzten beiden Beispiele (Extraktionskit und Quantifizierungskit) führen weder spezifische Produkteigenschaften auf noch wird ein Bezug zu einer spezifischen in-vitro-diagnostischen Untersuchung hergestellt. Gleichwohl besagt Regel 5a des Anhangs VIII der IVDR:
“[…] die vom Hersteller dafür vorgesehen sind, die Produkte für In-vitro-Diagnoseverfahren im Zusammenhang mit einer spezifischen Untersuchung einsetzbar zu machen.”
IVDR, Anhang VIII, Regel 5a
Ohne diesen Zusammenhang handelt es sich um Produkte für den allgemeinen Laborbedarf.
Die MDCG 2024-11 ist in ihrer Gegenüberstellung (s. Tabelle 1) deutlich klarer als das Dokument MDCG 2020-16.
Zu beachten sind allerdings in diesem Zusammenhang auch gewisse Abhängigkeiten von Produkten. Sollte ein Gerät eine integrierte Auswertesoftware besitzen, kann eine Höherklassifizierung notwendig sein.
Ein Beispiel hierfür sind qPCR-basierte Point-of-Care-Geräte. Das Gerät ist nicht nur in der Lage, die qPCR durchzuführen und Schmelzkurvendiagramme zu erzeugen, sondern liefert über die integrierte Auswertesoftware direkt diagnostische Informationen, z. B. die Menge eines oder mehrerer spezifischer Pathogene in der untersuchten humanen Probe.
Lesen Sie hier mehr zum Thema: Klassifizierung von In-vitro-Diagnostika: Wie Sie eine zu hohe Einstufung vermeiden
2. Tipps für Hersteller
Tipp 1: Legen Sie die Zweckbestimmung präzise fest
Hersteller sollten zunächst ihre Produktbeschreibung inklusive Zweckbestimmung genau kennen bzw. festlegen. Denn davon hängt die Qualifizierung ab.
Tipp 2: Wägen Sie dabei ab, welchen Nutzen eine Zertifizierung hat
Bei Produkten im „Graubereich“ ist es denkbar, mit der gewünschten Qualifizierung zu starten und dann die Zweckbestimmung entsprechend zu formulieren. Sie können sich von der Frage leiten lassen, ob die CE-IVD-Markierung tatsächlich einen Mehrwert für den Kunden bietet oder ob sie höhere Aufwände und Kosten für die Betreiber mit sich bringt.
Tipp 3: Verwenden Sie das CE-Zeichen nicht zu Marketingzwecken
Zusammenfassung
Hersteller sollten die CE-IVD-Kennzeichnung nicht zu Marketingzwecken verwenden. Die CE-IVD-Markierung hat weitreichende regulatorische und rechtliche Konsequenzen. Handelt es sich um ein IVD, muss der Hersteller auch den entsprechenden Nachweis erbringen. Kann er dies nicht, muss der Hersteller mit rechtlichen Konsequenzen rechnen.
Herleitung Teil 1: Spezifische Anwendungen
Handelt es sich um ein IVD, so muss dieses IVD spezifische Eigenschaften besitzen, die es für eine oder mehrere spezifische in-vitro-diagnostische Anwendungen nutzbar machen. Die MDCG 2024-11 schreibt hierzu:
„Where, on the basis of its characteristics, the manufacturer specifically intends its product to be used for in vitro diagnostic examination, the product becomes an IVD or an accessory of an IVD and must comply with the applicable requirements of the IVDR.”
MDCG 2024-11
Die Leitlinie konkretisiert im Weiteren:
„If, however, the product for general laboratory use does not possess specific characteristics that makes it suitable for one or more identified in vitro diagnostic examination procedures, then the manufacturer should not qualify the product as an IVD. Merely adding the statement “for in vitro diagnostic use” to a product is not sufficient to qualify a product as an IVD.”
Demnach ist es dem Hersteller sogar untersagt, ein Produkt CE-IVD zu markieren, wenn es keine Eigenschaften aufweist, die es für ein oder mehrere spezifische in-vitro-diagnostische Untersuchungen tauglich machen.
Herleitung Teil 2: Probenvorbereitung
Konkret geht die MDCG 2024-11 auf Produkte ein, die im Zusammenhang mit der Vorbereitung humaner Probenmaterialien verwendet werden. Auch diese gelten nicht pauschal als IVD:
„Products for general laboratory use utilised in vitro in the preparation of specimens that have been obtained for examination (e.g. paraffin, stains) are considered neither as IVD nor as accessories and fall outside the scope of the IVDR unless such products, in view of their characteristics, are specifically intended by their manufacturer to be used for in vitro diagnostic examination or fulfil the definition of an accessory.”
MDCG 2024-11
Herleitung Teil 3: Regulatorische Anforderungen
Unabhängig davon, ob es sich um Reagenzien, Geräte oder sonstige Instrumente oder Apparate für den allgemeinen Laborbedarf handelt, gilt:
Behauptet der Hersteller, dass das Produkt für die in-vitro-diagnostische Anwendung vorgesehen ist, so muss er den Nachweis für diese spezifischen Eigenschaften und die Verbindung zum jeweiligen in-vitro-diagnostischen Verfahren erbringen.
Die MDCG 2020-16 schreibt hierzu:
„As a consequence, if such products are specifically intended by the manufacturer to be used for in vitro diagnostic examinations, then they are considered as IVDs and are captured by rule 5.”
MDCG 2020-16
Das bedeutet: Wenn es sich bei dem Produkt um ein IVD handelt, so muss der Hersteller die damit verbundenen regulatorischen Anforderungen erfüllen: Er muss den Nachweis erbringen, dass das Produkt unter Berücksichtigung seiner Zweckbestimmung die grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I erfüllt und eine Technische Dokumentation gemäß Anhang II und III erstellen und pflegen.
Der Nachweis der Verbindung mit einem oder mehreren konkreten In-vitro-Untersuchungsverfahren kann im Rahmen der Leistungsbewertung erfolgen. Die MDCG 2020-16 ergänzt für IVD-Geräte der Klasse A:
„Due to their interdependence, the performance of the reagent on this instrument will be part of the conformity assessment of the reagent.”
MDCG 2020-16
Fällt das Produkt hingegen nicht in den Geltungsbereich der IVDR, erspart das dem Hersteller viel Aufwand.
Tipp 4: Bevorzugen Sie GLU gegenüber RUO
Schaffen Sie Klarheit und verwenden Sie den Begriff „General Laboratory Use“ (GLU) anstelle von „Research Use Only“ (ROU).
Hersteller kommen dann nicht in Erklärungsnot, weshalb sie RUO-Produkte an medizinische Labore verkaufen. Denn allgemeine Laborreagenzien bzw. Geräte für den allgemeinen Laborbedarf dürfen für in-vitro-diagnostische Verfahren eingesetzt werden.
Tipp 5: Beachten Sie weitere gesetzliche Anforderungen
Auch unabhängig von der Qualifizierung als IVD müssen die Hersteller gegebenenfalls andere EU-Anforderungen einhalten, die nicht speziell für IVD vorgesehen sind. Insbesondere GLU-Geräte unterliegen der Maschinenrichtlinie bzw. Maschinenverordnung und sind daher mit einem entsprechenden CE-Kennzeichen zum Nachweis der Konformität zu markieren.
Lesen Sie hier mehr zum Thema: Maschinenrichtlinie: Was bei Medizinprodukten gilt
3. Tipps für medizinische Labore
Tipp 1: Vermeiden Sie Off-label-use
Generell gilt: Verwenden medizinische Labore Produkte für den allgemeinen Laborbedarf in Inhouse-Verfahren, so tragen sie die Verantwortung für die Konformität der Untersuchung mit der IVDR.
Das gilt auch, wenn sie Geräte außerhalb deren Zweckbestimmung bzw. außerhalb deren bestimmungsgemäßen Gebrauchs einsetzen.
Verwendet ein Labor ein CE-IVD-markiertes NGS-Gerät, so ist, gemäß Zweckbestimmung des Herstellers, die in-vitro-diagnostische Anwendung auf ein bestimmtes Untersuchungsverfahren bezogen, etwa auf den Nachweis von CFTR-Mutationen mit einem entsprechenden Kit und der zugehörigen Auswertesoftware desselben Herstellers.
Falls das Labor exakt dieses NGS-Gerät nun zur Detektion von Mutationen in völlig anderen Genen nutzt, (beispielsweise ein Brustkrebspanel untersucht mit einem anderen NGS-Kit (vielleicht sogar von einem anderen Hersteller) und einer inhouse entwickelten NGS-Pipeline) dann ist die CE-IVD-Markierung für das Labor wertlos.
Unter diesen Umständen hätte das Labor auch ein günstigeres GLU-NGS-Gerät des Herstellers kaufen können, denn die Verantwortung für die Konformität des Verfahrens trägt es ohnehin.
Daher sollten die Labore bei CE-IVD-markierten Produkten stets die genaue Zweckbestimmung des Herstellers prüfen. Dazu gehört auch die Information, in welcher Kombination Produkte zu verwenden sind, d. h. Gerät A mit Kit B und Software C.
Die CE-IVD-Markierung bezieht sich ausschließlich auf die vom Hersteller angegebene Zweckbestimmung.
Wird ein Produkt in einem anderen Zusammenhang eingesetzt (d. h. andere Produktkombination, andere Indikation o. ä.), wird das Labor schnell zum Eigenhersteller eines Inhouse-IVDs mit allen daraus folgenden Konsequenzen.
Daher ist die pauschale Verwendung von CE-IVD-markierten Produkten auch nicht immer sinnvoll.
Tipp 2: Lassen Sie Vorsicht bei Eigenherstellungen walten
Falls ein medizinisches Labor eine „Eigenherstellung“ eines IVD vornimmt, so muss es für dieses Inhouse-IVD die Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der IVDR nachweisen.
Das gilt unabhängig davon, ob das Labor ein „zweckentfremdetes“ CE-IVD-Produkt oder ein GLU-Produkt verwendet.
Hierbei sei insbesondere auf Anhang I der IVDR verwiesen:
„Wenn ein Produkt zur Verwendung in Kombination mit anderen Produkten oder Ausrüstungen bestimmt ist, muss die Kombination einschließlich der Verbindungen sicher sein und darf die vorgesehene Leistung der Produkte nicht beeinträchtigen. Jede Einschränkung der Anwendung im Zusammenhang mit solchen Kombinationen wird auf der Kennzeichnung und/oder in der Gebrauchsanweisung angegeben.“
Anhang I, Kapitel II, Abschnitt 13.1 der IVDR
Was aber, wenn die Kombination mit einem GLU oder sogar RUO-Produkt vom IVD-Hersteller vorgegeben wird?
Dann ist entscheidend, dass der Hersteller diese Kombination in Zweckbestimmung und IFU abgebildet und die Sicherheit, Leistung und Kompatibilität des Gesamtsystems belegt hat. Team-NB (der Verband der Benannten Stellen für Medizinprodukte) konkretisiert hierzu: Bei „Closed Systems“ sind vom Hersteller definierte Kombinationen zu validieren; bei „Open Systems“ müssen Hersteller die zulässigen Geräte/Komponenten über kritische Spezifikationen beschreiben und die Übertragbarkeit der Leistungsdaten begründen. Werden hierbei GLU/RUO-Produkte in Kombinationsangaben einbezogen, trägt der Hersteller nach Team-NB die volle Nachweisverantwortung.
Ist die Kombination im Workflow des Herstellers (Zweckbestimmung/IFU) spezifiziert, kann das dort genannte GLU/RUO‑Produkt im Rahmen dieser Vorgaben verwendet werden; außerhalb davon trägt das Labor typischerweise die Verantwortung im Sinne eines Inhouse‑Verfahrens.
Lesen Sie hier mehr zum Thema: Die EU reguliert medizinische Labore. Sind Inhouse-IVD (LDT) noch erlaubt?
Tipp 3: Prüfen Sie die Marketing-Claims der Hersteller genau
Medizinische Labore sollten die Angaben von Herstellern CE-IVD-markierter Produkte genau prüfen:
Ist die CE-IVD-Markierung unspezifisch?
Liegen weder spezifische Produkteigenschaften vor noch wird auf ein konkretes Untersuchungsverfahren vom Hersteller verwiesen? Das Produkt ist widerrechtlich als CE-IVD markiert.
Was sagen die Gebrauchsanweisung bzw. Produktinformation aus?
Bezieht sich die in-vitro-diagnostische Anwendung auf ein konkretes Untersuchungsverfahren mit spezifischen Produktkombinationen? Entsprechen diese nicht der Laborroutine, ist die CE-IVD-Markierung für das Labor wertlos.
Tipp 4: Führen Sie ein Verzeichnis, wozu Produkte verwendet werden
Führen Sie ein Verzeichnis über alle Produkte (Medizinprodukte, IVD, GLU, RUO) und verweisen Sie auf die zugehörigen Zweckbestimmungen und Untersuchungsverfahren, die im Zusammenhang mit dem jeweiligen Produkt in Ihrem Labor zur Anwendung kommen.
Damit verschaffen Sie sich nicht nur einen Überblick über alle angewandten Verfahren, sondern können auch prüfen, ob die vom Hersteller angegebenen Zweckbestimmungen mit Ihrem Verwendungszweck übereinstimmen. Falls nicht, handelt es sich um eine Eigenentwicklung.
Für das Verfahren – als Inhouse-IVD – müssen Sie prüfen und belegen, dass das Verfahren die grundlegenden Sicherheits- und Leistungsanforderungen und die zusätzlichen Anforderungen des Artikels 5 (5) der IVDR erfüllt.
Weiterhin kann dieses Verzeichnis dazu dienen, Untersuchungsverfahren zu identifizieren, für die kein adäquates CE-IVD-Produkt am Markt verfügbar ist. Es liefert Ihnen somit eine Grundlage, um Begründungen aufzubauen hinsichtlich der Anforderung, die sich aus Art. 5 (5) d) der IVDR ergibt.
Lesen Sie hier mehr zum Thema: Die EU reguliert medizinische Labore. Sind Inhouse-IVD (LDT) noch erlaubt?
4. Zusammenfassung und Fazit
a) Produkte ohne spezifischen Eigenschaften zählen oft als GLU
Viele Produkte, die im Rahmen in-vitro-diagnostischer Anwendungen eingesetzt werden, fallen in den Bereich der Produkte für den allgemeinen Laborbedarf (GLU).
Besitzen diese Produkte jedoch spezifische Eigenschaften, die sie nachweislich für ein bestimmtes in-vitro-diagnostisches Untersuchungsverfahren anwendbar machen, müssen die Hersteller diese mit einer CE-IVD-Markierung versehen, mit den daraus folgenden regulatorischen Konsequenzen.
Produkte für den allgemeinen Laborbedarf ohne diese spezifischen Eigenschaften fallen nicht in den Geltungsbereich der IVDR, dürfen aber dennoch bei in-vitro-diagnostischen Verfahren angewendet werden.
b) Die Zweckbestimmung hat Konsequenzen für Hersteller und Labore
Bei der Kombination von Produkten für den allgemeinen Laborbedarf sowie IVD im Rahmen eines laborspezifischen Untersuchungsverfahrens ist stets auf die Zweckbestimmung der einzelnen Produkte zu achten: Möglicherweise hat das Labor (unwissentlich) ein Inhouse-IVD entwickelt und trägt die volle Verantwortung für das Verfahren.
Medizinische Labore sollten die Zweckbestimmung der Produkte, die sie verwenden, kennen und diese mit der Zweckbestimmung ihrer eigenen Untersuchungsverfahren abgleichen. So können sie eruieren, ob die Verwendung eines CE-IVD-markierten Produkts überhaupt sinnvoll bzw. nützlich ist.
c) Ein zertifiziertes Produkt ist nicht per se besser
Hersteller sollten unter Einbeziehung der Kundenanforderungen überlegen, ob eine Qualifizierung als IVD-Produkt tatsächlich einen Mehrwert liefert.
Handelt es sich nicht um ein IVD, trägt es zur Klarheit und Rechtssicherheit bei, wenn die Hersteller anstelle der RUO-Markierung von einem „General Laboratory Use“-Produkt sprechen.
Wenn Sie sich einen besseren Überblick über die Anforderungen der IVDR an die Verwendung von Inhouse-IVD verschaffen wollen, dann besuchen Sie unser Seminar IVDR für medizinische Labore.
Wenn Sie bereits wissen, dass Sie künftig zum CE-IVD-Hersteller und Inverkehrbringer werden wollen, empfehlen wir Ihnen das Seminar Technische Dokumentation nach IVDR.
Das IVD-Team des Johner Instituts unterstützt Sie:
- Es hilft Ihnen bei der Qualifizierung Ihrer Produkte bzw. Untersuchungsverfahren, beispielsweise mit Inhouse-Workshops zur Zulassungsstrategie und zu Inhouse-IVD.
- Es erstellt Ihnen Expertenmeinungen zur Qualifizierung Ihres Produkts, die Sie Ihren Kunden und/oder Benannten Stellen vorlegen können.
- Es unterstützt Sie bei allen Tätigkeiten bis zur „Zertifizierung“ Ihres Produkts (z. B. bei der Leistungsbewertung) und darüber hinaus (z. B. bei der Post-Market Surveillance).
Versionshistorie:
- 2026-02-18 Ergänzung in Abschnitt 1 b) und 3, Tipp 2 hinsichtlich Kombination von Produkten und Verweis auf Team-NB Positionspapier
- 2024-10-10 Überarbeitung hinsichtlich MDCG 2024-11: Im gesamten Fachartikel MEDDEV 2.14/rev. 2 ersetzt durch MDCG 2024-11; Abschnitt 1.b) und Abschnitt 2, Tipp: 3 Zitate entsprechend angepasst; Abschnitt 2, Tipp 3: Herleitung Teil 3 Manual on borderline and classification in the community regulatory framework for medical devices als Quelle entfernt und Zitate aus MDCG 2020-16 hinzugefügt
- 2024-02-06 Erstveröffentlichung



Sehr geehrte Frau Bartsch,
Sie geben das Beispiel, dass für den qPCR-Cycler keine CE-Kennzeichnung notwendig ist, da er nicht speziell für Durchführung ein oder mehrere bestimmter Produkte konzipiert ist.
Ist es nicht aber so, dass es sich hierbei um eine Maschine handelt, die unter die Maschinenrichtlinie fällt und damit ebenfalls ein CE-Kennzeichen tragen muss?
In der Maschinenrichtlinie gibt es zwar die Ausnahme, dass sie nicht gilt für „Maschinen, die speziell für Forschungszwecke konstruiert und gebaut wurden und zur vorübergehenden Verwendung in Laboratorien bestimmt sind“. Wenn aber IVDs mit der Maschine durchgeführt werden, ist es kein Gerät mehr für Forschungszwecke. Mal ganz davon abgesehen, dass die „vorübergehende Verwendung“ zeitlich definiert sein müsste.
Für alle Produkte, die in der EU vertrieben werden, muss eine CE-Kennzeichnung angebracht werden, außer Lebensmittel, Arzneimittel, Chemikalien und Kosmetika.
Nur weil für den allgemeinen Laborbedarf nicht zwangsweise die IVDR gilt, müsste mindestens das Produktsicherheitsgesetz gelten. Das sieht ebenfalls eine CE-Kennzeichnung vor.
Jetzt steht in vielen Produkten u.a. PCR-Cycler oder Extraktionskit für „Research use only“ drin. Diese Produkte tragen daraufhin kein CE-Kennzeichen. Sie werden aber für die In-Vitro-Diagnostik genutzt. Wie kann das ohne CE-Kennzeichnung und ohne, dass diese Produkte als Zubehör gelten, funktionieren?
Lieber Herr Handt,
da Frau Bartsch noch im Urlaub ist, darf ich Ihnen die Antwort ihrer Kollegin weiterleiten:
Lieber Herr Handt,
vielen Dank für Ihre Nachfrage.
Grundsätzlich beschreibt die EU auf ihrer Webseite: „Viele Produkte benötigen eine CE-Kennzeichnung, bevor sie in der EU verkauft werden dürfen. Das CE-Zeichen ist ein Hinweis darauf, dass ein Produkt vom Hersteller geprüft wurde und dass es alle EU-weiten Anforderungen an Sicherheit, Gesundheitsschutz und Umweltschutz erfüllt. Es ist Pflicht für alle weltweit hergestellten Produkte, die in der EU vermarktet werden […]„. Dies bestätigen Hersteller in der zugrundeliegenden EU-Konformitätserklärung.
Wenn wir in diesem Fachartikel von einem CE-Kennzeichen sprechen, dann beziehen wir uns ganz spezifisch auf die CE-Konformitätskennzeichnung gemäß IVDR, Artikel 18. Dieses CE-Kennzeichen zeigt an, dass das Produkt als den Anforderungen der IVDR entsprechend betrachtet werden kann. Je nach Klassifizierung eines IVDs wird der CE-Kennzeichnung die Kennnummer der für das Konformitätsbewertungsverfahren gemäß Artikel 48 zuständigen Benannten Stelle hinzugefügt.
Ein qPCR-Cycler kann die CE-Kennzeichnung gemäß IVDR nur tragen, wenn es sich um ein IVD handelt. Dafür muss das Gerät gemäß IVDR, Artikel 2 als IVD qualifizieren und – im Fall von Klasse A – gemäß IVDR, Anhang VIII, Regel 5b) „vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen“ sein. Gemäß Anhang I, 13.1 folgt die grundlegende Sicherheits- und Leistungsanforderung: „Wenn ein Produkt zur Verwendung in Kombination mit anderen Produkten oder Ausrüstungen bestimmt ist, muss die Kombination einschließlich der Verbindungen sicher sein und darf die vorgesehene Leistung der Produkte nicht beeinträchtigen. Jede Einschränkung der Anwendung im Zusammenhang mit solchen Kombinationen wird auf der Kennzeichnung und/oder in der Gebrauchsanweisung angegeben.“ Die Kombination des qPCR-Cycler mit z.B. einem Assay für eine spezielle in-vitro diagnostische Untersuchung muss somit benannt und bzgl. Sicherheit und Leistung bewertet werden.
Für die CE-Kennzeichnung von Medizinprodukten gemäß Maschinenrichtlinie finden Sie weitere Informationen in unserem Fachartikel „Maschinenrichtlinie: Was bei Medizinprodukten gilt“
Herzliche Grüße,
Catharina Bertram
Ich hoffe, diese Antwort hilft Ihnen weiter.
Viele Grüße
Tea Bodrusic
Danke für Ihre Antwort.
Dennoch wird mir nicht ganz klar, ob Anhang I, 13.1 den qPCR-Cycler dann auch zum IVD-Zubehör macht.
Ich vermute nein.
Es muss geprüft werden, ob Produkte (von anderen Herstellern), die mit dem eigentlichen IVD in Kombination verwendet werden sollen, Einfluss auf Sicherheit und Leistung haben.
Nur wenn diese Produkte vom Herstellern speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind, werden sie zu IVD-Zubehör und müssen entsprechend gekennzeichnet werden (CE nach IVDR)?
Wenn geprüft wird, ob der qPCR-Cycler (kein IVD-Zubehör) Einfluss auf Sicherheit und Leistung hat, ist dies ausreichend. Der Hinweis auf diese Prüfung muss dann in die Gebrauchsanweisung aufgenommen werden? Es handelt sich damit um eine Einschränkung, dass nur der geprüfte qPCR-Cycler verwendet werden darf.
Wenn ich diese Einschränkung nicht angebe, dürfte theoretisch jeder qPCR-Cycler verwendet werden, korrekt?
Lieber Herr Handt,
vielen Dank, dass Sie nochmal nachhaken.
Ich möchte zunächst anmerken, dass sich ein qPCR-Cyclers vermutlich i. d. R. nicht als „Zubehör“ qualifiziert sondern eher selbst zum IVD-Gerät wird. Gerne können wir Ihren konkreten Fall auch im direkten Gespräch diskutieren und gemeinsam bewerten.
Es ist richtig, dass der Hersteller des Cyclers letztlich für dessen Zweckbestimmung verantwortlich ist. Der Hersteller des Cyclers legt fest, ob es sich beispielsweise um ein IVD handelt, dann sollte das Produkt auch die entsprechenden Eigenschaften aufweisen. Ist der Cycler ein IVD, so muss er die Anforderungen der IVDR erfüllen.
Wird für die Verwendung eines Assays von Hersteller X wiederum ein Cycler benötigt, so muss der Hersteller X gewährleisten, dass diese Kombination sicher ist und die vorgesehene Leistung des Assays nicht beeinträchtigt wird (IVDR, Anhang I, 13.1). Demnach ist Hersteller X dafür verantwortlich festzulegen, welcher konkrete Cycler für die Abarbeitung des Assays eingesetzt werden soll oder Hersteller X gibt an, welche (sicherheits- und leistungskritischen) Spezifikationen ein solcher Cycler erfüllen muss (z. B. Ramp Rate, Farbkanäle, Plattenformate etc.). Die Leistungsbewertung des Assays hat dann auf einem solchen Cycler zu erfolgen und ist damit Bestandteil der Koformitätsbewertung des Assays. So sieht es auch die MCGG: MDCG 2020-16 rev.3, Rule 5 (b) „Due to their interdependence, the performance of the reagent on this instrument will be part of the conformity assessment of the reagent.“
Wie oben erwähnt, gibt Hersteller X in der Gebrauchsanweisung des Assays an, welcher konkrete Cycler zu verwenden ist oder aber welche Spezifikationen dieser erfüllen muss.
Ich hoffe, dies beantwortet Ihre Frage.
Mit besten Grüßen
Sophie Bartsch