
Die Klassifizierung von In-vitro-Diagnostika (IVD) kann weitreichende Folgen haben, da diese Einfluss auf das Konformitätsbewertungsverfahren, auf Zertifizierungsaudits und somit auf die Markteinführung hat.
Damit Ihr IVD nicht unnötig einer hohen Risikoklasse zugeordnet wird, verschafft Ihnen dieser Artikel einen Überblick darüber,
- wie sich Ihr Produkt als IVD qualifiziert,
- wie IVD nach der IVDR klassifiziert werden,
- wie Sie eine unnötig hohe Klassifizierung vermeiden können und
- wie Ihnen der IVD-Klassifikator des Johner Instituts dabei hilft.
1. Was qualifiziert ein Produkt als ein IVD?
a) Medizinischer Zweck und Qualifizierung als Medizinprodukt
Wenn man die Definition eines IVDs gemäß Artikel 2 (2) der IVDR betrachtet, dann steht dort, dass jedes IVD auch ein Medizinprodukt ist: „‚In-vitro-Diagnostikum‘ bezeichnet ein Medizinprodukt, dass […]“. Daher muss man für die Überlegungen zur Qualifizierung eines Produkts als IVD zunächst die Definition eines Medizinprodukts berücksichtigen:
„Medizinprodukt“ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper – auch aus Organ-, Blut- und Gewebespenden – stammenden Proben,
und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
(MDR Art. 2 (1))
Der Definition eines Medizinprodukts folgend muss jedes Medizinprodukt einen „spezifischen medizinischen Zweck“ haben. Der 4. Spiegelstrich der Definition eines Medizinprodukts benennt die „Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper […] stammenden Proben“ als einen medizinischen Zweck und verweist damit auf die In-vitro-Diagnostika (IVD), die eine Untergruppe der Medizinprodukte darstellen.
Gemäß Art. 1 (6) der MDR gilt die MDR allerdings „nicht für a) In-vitro-Diagnostika im Sinne der Verordnung (EU) 2017/746“; für IVD ist die IVDR als spezielle Verordnung anzuwenden.
b) Qualifizierung als IVD
Die In-vitro-Diagnostika sind somit eine Untergruppe der Medizinprodukte, die medizinische bzw. diagnostische Informationen aus menschlichen Proben gewinnen. Die Art der durch ein IVD bereitgestellten Informationen wird in der Definition eines IVD weiter ausgeführt:
‚In-vitro-Diagnostikum‘ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern:
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten als auch In-vitro-Diagnostika;
(IVDR Art. 2 (2))
Demnach sind IVD Produkte, die vom Hersteller für die „in-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben“ vorgesehen sind „und ausschließlich oder hauptsächlich dazu [dienen], Informationen“ zu einem in-vitro-diagnostischen Zweck, wie in IVDR, Art. 2 (2) Unterpunkte a) bis f) aufgelistet, zu liefern.
c) Grenzfälle bei der Qualifizierung als IVD
In manchen Fällen ist die Entscheidung, ob ein Produkt ein IVD ist oder nicht, jedoch nicht eindeutig.
Bei Unsicherheiten soll das Manual on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices helfen. Allerdings enthält die Version von September 2023 noch zahlreiche leere Kapitel. Eine wertvolle Ergänzung sind das Manual on Borderline and Classification in the Community Regulatory Framework for Medical Devices in der Version 1.22 von 2019 und die MEDDEV 2.14/1 „IVD Medical Device Borderline and Classification issues“, die IVD-Herstellern Klarheit verschaffen kann. Das Manual und die Leitlinie beschreiben Grenzfälle der Qualifizierung von IVD. Die MEDDEV 2.14/1 betrachtet in Abschnitt 1.4 die Abgrenzung von IVD und „products for general laboratory use“.
Ebenso betrachtet das MDCG 2024-11 Guidance on qualification of in vitro diagnostic medical devices verschiedene Fragestellungen hinsichtlich der Qualifizierung von Produkten als IVD und nennt unterstützende Beispiele.
Weiterführende Informationen zum Thema Allgemeiner Laborbedarf finden Sie auch in unserem Blog-Beitrag Allgemeiner Laborbedarf: Was Hersteller und Labore wissen müssen, um sich Ärger und unnötige Aufwände zu ersparen.
Werden mit einem Produkt aus dem menschlichen Körper stammende Proben zu einem in-vitro-diagnostischen Zweck analysiert, ist dieses Produkt gemäß IVDR als IVD zu qualifizieren und nachfolgend in die Risikoklassen A, B, C oder D einzuordnen.
2. Warum die richtige IVD-Klassifizierung wichtig ist
Bei einer falschen Klassifizierung kann es zu Abweichungen im Audit oder bei der Produktprüfung durch die Benannte Stelle kommen. Bei Meinungsverschiedenheiten zwischen Hersteller und Benannter Stelle wird an die zuständige Behörde verwiesen. Im ungünstigsten Fall stuft diese das Produkt in eine unnötig hohe Klasse ein, was unweigerlich zu einer Verzögerung der Markteinführung führt. Zudem können Wettbewerber die Klassifizierung infrage stellen und mittels Unterlassungsklagen die Vermarktung der Produkte behindern.
Es ist daher entscheidend, dass IVD unter der IVDR richtig klassifiziert werden, um diese Probleme zu vermeiden.
Tipps dazu, wie Ihnen dies gelingen kann, finden Sie weiter unten in diesem Beitrag.
3. Klassifizierung nach IVDR
Die Klassifizierung von IVD richtet sich nach Anhang VIII der IVDR. Dieser unterteilt sich in zwei Abschnitte:
- Im ersten Abschnitt befinden sich allgemeine Regeln für die Durchführung der Klassifizierung, die sogenannten Durchführungsvorschriften. Diese betreffen etwa Abhängigkeiten von Produkten oder Regeln bei Konflikten.
- Der zweite Abschnitt umfasst sieben Klassifizierungsregeln für die Zuordnung von IVD zu den Risikoklassen A bis D.
3.1 Allgemeine Regeln für die Durchführung der Klassifizierung nach IVDR
Die IVDR gibt Regeln für die Durchführung der Klassifizierung vor, die sogenannten Durchführungsvorschriften (IVDR Anhang VIII 1. Abschnitt).
Zweckbestimmung
„Die Anwendung der Klassifizierungsregeln richtet sich nach der Zweckbestimmung der Produkte“ (IVDR Anhang VIII 1.1.). Mit dieser legt der Hersteller fest, zu welchem medizinischen Zweck sein Produkt genutzt werden soll. Die Zweckbestimmung entscheidet, je nach Risiko, letztlich über die Klassifizierung.
Die Zweckbestimmung findet sich in der Kennzeichnung, der Gebrauchsanweisung und den Werbematerialien des Produkts. Sie macht aus dem Produkt ein IVD.
Lesen Sie mehr zur Zweckbestimmung von Medizinprodukten und IVD in unserem Blog-Beitrag Zweckbestimmung und bestimmungsgemäßer Gebrauch: Folgenreicher als Sie denken!
Fällt Ihr Produkt in eine höhere Risikoklasse, als Sie erwartet haben, ist möglicherweise die Zweckbestimmung nicht korrekt festgelegt.
Abhängigkeiten von Produkten
Wenn ein IVD zusammen mit anderen IVD-Produkten verwendet wird, gibt es hinsichtlich der Klassifizierung zwei Möglichkeiten. Prinzipiell gilt, „wenn das betreffende Produkt dazu bestimmt ist, in Verbindung mit einem anderen Produkt angewandt zu werden, werden die Klassifizierungsregeln auf jedes Produkt gesondert angewendet“ (IVDR Anhang VIII 1.2.). Beispielsweise kann ein ELISA-Reader unabhängig von einem antikörperbasierten Reagenzien-Kit klassifiziert werden, wenn beide als separate Produkte in Verkehr gebracht werden.
Ähnlich verhält es sich mit Zubehör. „Zubehör für ein In-vitro-Diagnostikum wird unabhängig von dem Produkt, mit dem es verwendet wird, gesondert klassifiziert“ (IVDR Anhang VIII 1.3.). An einem Mikroskop, das speziell für die Pathologie vorgesehen ist, kann als Zubehör eine Digitalkamera angeschlossen werden, damit gleichzeitig ein zweiter Pathologe auf das histologische Schnittbild sehen kann. Ist die Kamera als Zubehör definiert, wird sie getrennt vom Mikroskop klassifiziert.
„Zubehör eines In-vitro-Diagnostikums“ bezeichnet einen Gegenstand, der zwar an sich kein In-vitro-Diagnostikum ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten In-vitro-Diagnostika verwendet zu werden, und der speziell dessen/deren Verwendung gemäß seiner/ihrer Zweckbestimmung(en) ermöglicht oder mit dem die medizinische Funktion des In-vitro-Diagnostikums/der In-vitro-Diagnostika im Hinblick auf dessen/deren Zweckbestimmung(en) gezielt und unmittelbar unterstützt werden soll;“
(IVDR Art. 2 (4))
Werden Komponenten hingegen zu einem IVD-Produkt zusammengefasst, werden sie gemeinsam klassifiziert (gleiche Risikoklasse). Das trifft zum Beispiel bei einem PCR-Kit zu, das aus Puffer, Enzym, Primer und Sonden besteht. Gleiches gilt für Kalibratoren (IVDR Anhang VIII 1.5.) und Kontrollmaterialien mit quantitativen oder qualitativen zugeordneten Werten (IVDR Anhang VIII 1.6.), welche zur Verwendung mit einem IVD-Produkt vorgesehen sind; sie werden derselben Klasse zugerechnet wie das Produkt.
Software
Bei Software gilt grundsätzlich: Erfüllt eine Software einen eigenen medizinischen Zweck, wird sie separat klassifiziert. Steuert die Software hingegen ein Produkt oder beeinflusst dessen Anwendung, so wird die Software derselben Klasse zugerechnet wie das Produkt (IVDR Anhang VIII 1.4.). Eine Software, die ein Laborgerät ansteuert (Start, Stopp), überwacht, von remote die Plattenbelegung festlegt oder die Qualität von Messergebissen beurteilt, wird gemeinsam mit dem Gerät klassifiziert.
Beispiele und genaue Erläuterungen zur Klassifizierung von IVD-Software finden Sie in unserem Beitrag IVD-Software richtig klassifizieren.
Bitte beachten Sie weiterhin:
- Bei mehreren Zweckbestimmungen eines Produkts, die mit unterschiedlichen Risikoklassen assoziiert sind, wird das gesamte Produkt in die höchste Klasse eingestuft.
- Wenn mehrere Klassifizierungsregeln gelten, wird die Regel der höchsten Klasse angewendet.
- Jede Klassifizierungsregel gilt nicht nur für erstmalige, sondern auch für Bestätigungs- und Ergänzungstests.
- Es müssen immer alle Klassifizierungs- und Durchführungsregeln berücksichtigt werden, um herauszufinden, in welche Klasse das Produkt fällt.
3.2 Die sieben Klassifizierungsregeln zur Einordnung in die Risikoklassen A–D
Im zweiten Teil des Anhangs VIII finden sich die entscheidenden sieben Regeln für die Zuordnung der passenden Risikoklasse. Die IVDR unterteilt IVD in die vier Risikoklassen A, B, C und D. Die Einordnung ist abhängig davon, welche Gefahren von dem IVD ausgehen können.
Es müssen stets alle sieben Regeln berücksichtigt werden. Für eine erste Einschätzung kann man sich bei der Zuordnung nach der Faustregel des International Medical Device Regulators Forum (IMDRF) richten. Diese lautet:
Je höher die Gefahr für eine lebensbedrohliche Erkrankung und je größer der betroffene Personenkreis ist, desto höher ist die Risikoklasse.
Dies bedeutet für die einzelnen Risikoklassen:
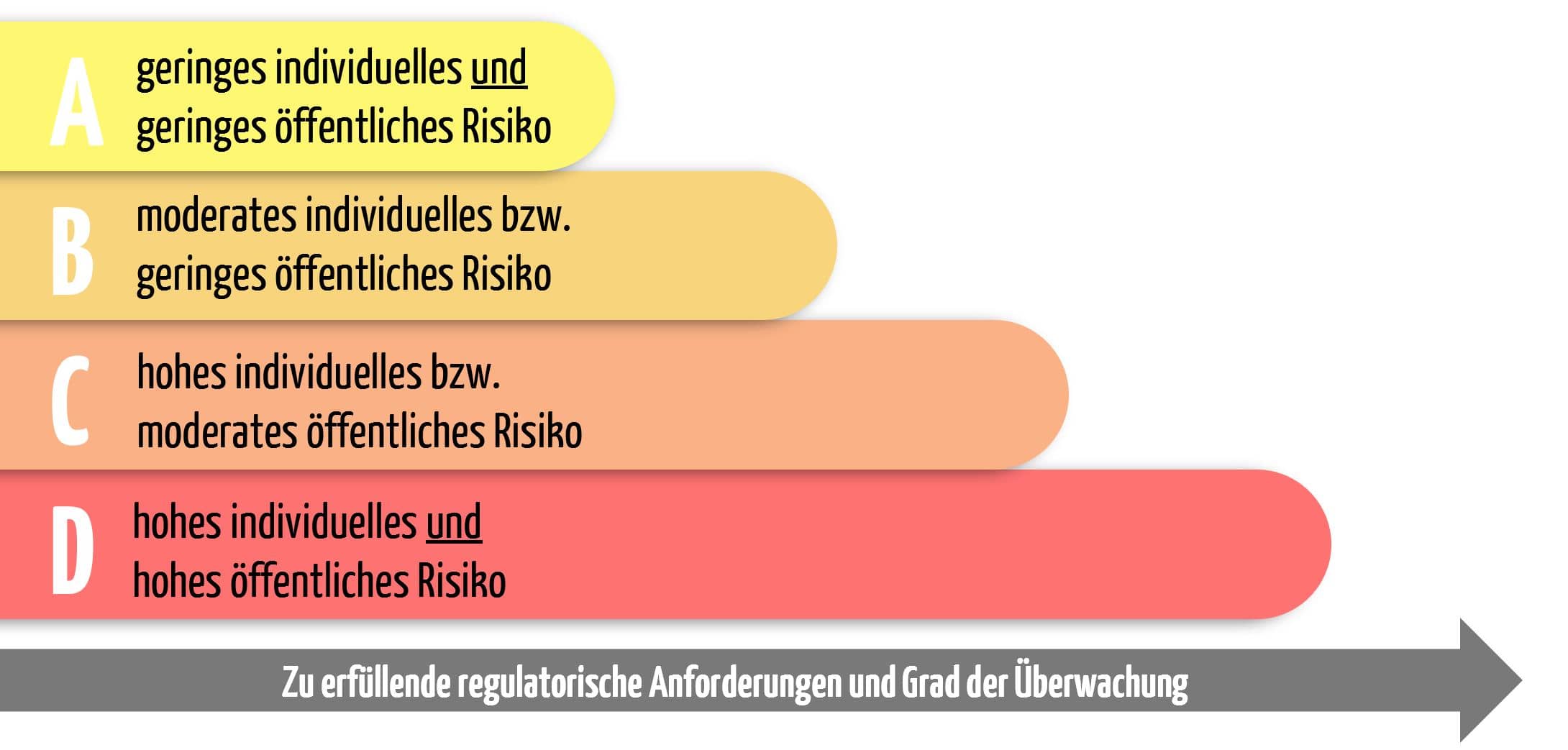
Klasse D – lebensbedrohliche Infektionen – höchstes Risiko
Hohes individuelles und hohes öffentliches Risiko
Von Produkten der Klasse D geht bei einem falschen Ergebnis ein lebensbedrohliches Risiko für mehrere Individuen aus (Regel 1 und 2).
Unter Klasse D fallen Produkte zum/r:
- Nachweis übertragbarer Erreger, die eine lebensbedrohende Krankheit mit einem hohen Verbreitungsrisiko verursachen (Regel 1)
- Beispiel: Detektion hochansteckender und gefährlicher Erreger wie Ebola-V, SARS-V, Lassa-V, Marburg-V
Achtung! Je nach Zweckbestimmung: HIV, Masern, MRSA, MRGN
- Beispiel: Detektion hochansteckender und gefährlicher Erreger wie Ebola-V, SARS-V, Lassa-V, Marburg-V
- Bestimmung des Infektionsgrades einer lebensbedrohenden Krankheit im Rahmen der Überwachung für das Patientenmanagement (Regel 1)
- Beispiel: Infektionsgrad bei Tuberkulosepatienten
- Nachweis übertragbarer Erreger für die Eignung von Blut, Zellen, Geweben oder Organen für Transfusionen oder Transplantationen (Regel 1)
- Beispiel: Tests, die den Status bzgl. HIV, HCV, HBV, HTLV in Blutkonserven detektieren
- Produkte, die zur Blutgruppenbestimmung oder zur Feststellung einer feto-maternalen Blutgruppen-Inkompatibilität oder zur Gewebetypisierung im Rahmen von Transfusions- bzw. Transplantationsmedizin verwendet werden (Regel 2)
- Marker im ABNull-System, im Rhesus-System, im Kell-System, im Kidd-System und im Duffy-System
- Hinweis: Tests zur Bestimmung anderer Marker werden der Klasse C zugeordnet
Klasse C – Produkte mit hohem Risiko
Hohes individuelles bzw. moderates öffentliches Risiko
Von Produkten der Klasse C geht bei falschem Ergebnis ein lebensbedrohliches Risiko für ein Individuum aus (Regel 3).
Unter Klasse C fallen Produkte für:
- Blutgruppenbestimmung, Gewebetypisierung nicht gelisteter Marker
- Beispiel: HLA Typisierung
- Feststellung des Immunstatus von Frauen auf übertragbare Erreger bei pränatalem Screening (CMV-Tests bei Schwangeren)
- Therapiebegleitende Diagnostika (EGFR (Lungenkrebs), BRAF (Hautkrebs), KRAS (Darmkrebs) zur Ableitung der weiteren Behandlung mit einem dazugehörigen Arzneimittel)
- Krebsvorsorge, -diagnose, Stadieneinteilung (PAP-Test, CIN-Test (Stadieneinteilung Cervixkarzinom))
- Gentests beim Menschen (Carrier-Testing, BRCA1/2 (Brustkrebs-Prädisposition), HLA-DQ2 oder DQ8 (Zöliakie))
- Genetisch bedingte Störungen beim Embryo oder Fötus (NIPT (fötale Trisomien, Mikrodeletionen))
- Untersuchung zu Infektionserregern mit bestehendem Risiko für lebensbedrohliche Situation bei falschem Ergebnis
Klasse C/B – Produkte zur Eigenanwendung („Selbsttests“) und patientennahe Tests
IVD-Produkte zur Eigenanwendung fallen grundsätzlich unter die Risikoklasse C (Regel 4).
Beispiel Selbsttest Klasse C:
- Indikator-Tests
- Probennahme-Sets, sofern den Laien-Anwendern mehr als nur eine einfache Probennahme abverlangt wird
Ausnahmen sind Schwangerschaftstests, Ovulationstests, Tests zur Bestimmung des Cholesterinspiegels oder Tests zum Nachweis von Glukose, Erythrozyten, Leukozyten und Bakterien im Urin. Diese Tests werden der Klasse B zugeordnet (Regel 4).
Patientennahe Tests werden für sich allein, d.h. nach ihrem ausgehenden Risiko, klassifiziert. Das bedeutet, dass patientennahe Tests in Klasse B, C oder auch D eingestuft werden können.
Klasse B – Fall-back-Klasse
Moderates individuelles bzw. geringes öffentliches Risiko
Von Produkten der Klasse B geht kein lebensbedrohliches Risiko bei falschem Ergebnis aus (Regel 6 und 7).
Klasse B ist als eine „Fall-back-Klasse“ zu verstehen.
- Produkte, die nicht unter die Regeln 1 bis 5 fallen, werden der Klasse B zugeordnet.
- Kontrollen ohne einen zugewiesenen quantitativen oder qualitativen Wert werden der Klasse B zugeordnet.
Klasse A – Laborbedarf
Geringes individuelles und geringes öffentliches Risiko
Produkte der Risikoklasse A liefern kein Ergebnis (Regel 5).
Darunter fallen:
- Instrumente, die speziell für die In-vitro-Diagnostik vorgesehen sind, insofern keine integrierte Software zur konkreten Ergebnisberechnung, welche mit einem medizinischen Zweck verbunden ist, beinhaltet ist
- Beispiele: Quantitativer PCR-Thermocycler, Sequenzierer, Massenspektrometer, ELISA-Reader, Durchflusszytometer
- Probenbehältnisse
- Beispiele: Urin-Becher, Spuck-Röhrchen, EDTA-Blutröhrchen, Behälter zur Aufnahme eines Abstrich-Stäbchens
- Erzeugnisse für den allgemeinen Laborbedarf: Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen
- Beispiele: Spezielle Puffer zur Konservierung oder Vorbehandlung von Zellen; spezielle Gefäße mit Beschichtungen, die Störfaktoren für eine bestimmte Untersuchung verhindern
- Allgemeine Nährmedien und histologische Färbungen für spezifische IVD-Untersuchungen
4. Folgen der Klassifizierung
Die Risikoklasse entscheidet über das Konformitätsbewertungsverfahren.
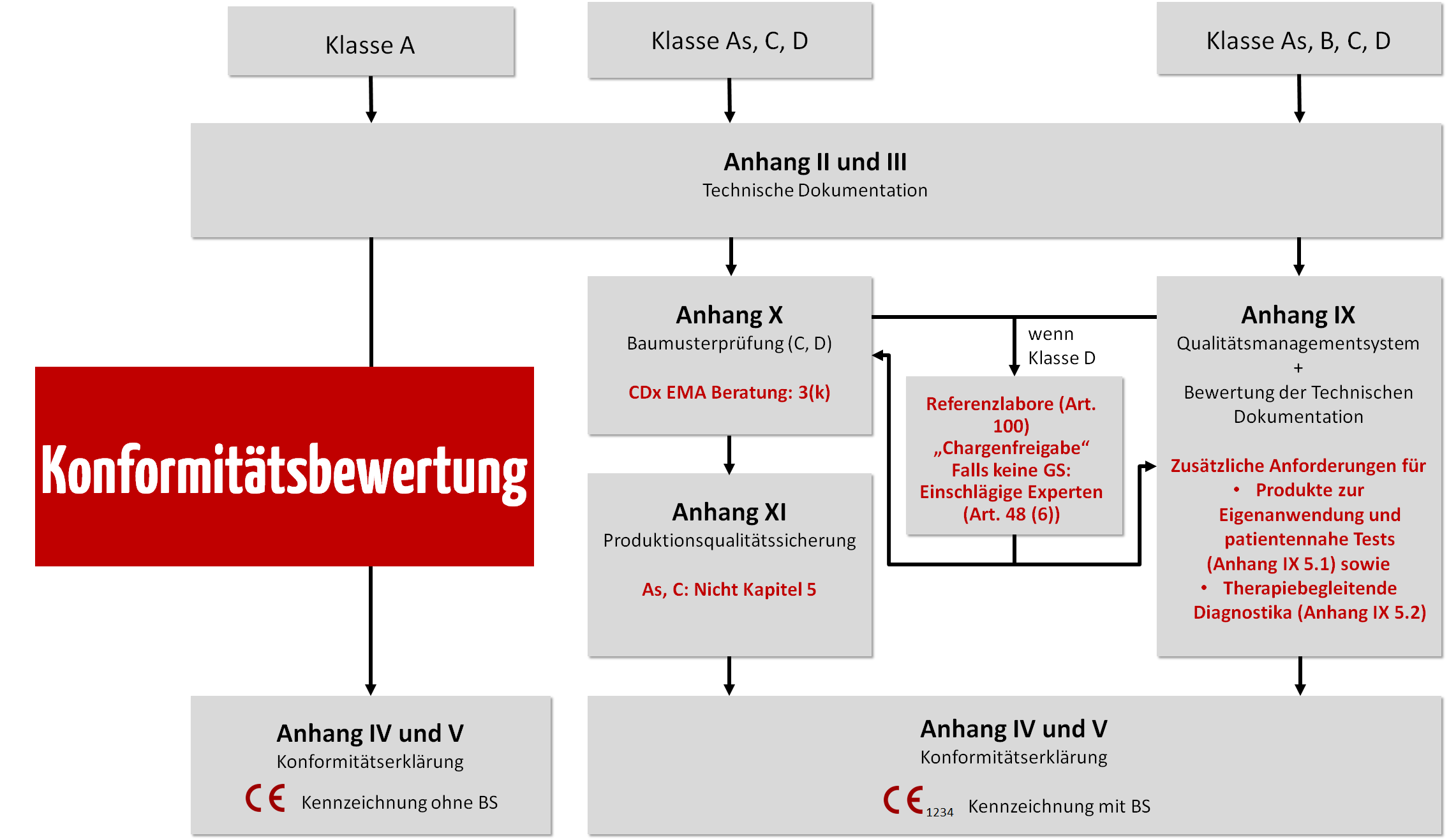
Klasse D
In der höchsten Risikoklasse für IVD wird intensiv auditiert.
Das Referenzlaboratorium prüft jede hergestellte Reagenzien-Charge stichprobenartig. Bei neuartigen IVD dieser Klasse wird zudem das Scrutiny-Verfahren angewendet (Verfahren, bei dem die Benannten Stellen im Rahmen der Konformitätsbewertung ein Expertengremium einbeziehen; Artikel 50 IVDR).
Für Benannte Stellen hat die MDCG das Dokument MDCG 2022-3 „Verification of manufactured class D IVDs by notified bodies“ zu Klasse-D-IVD veröffentlicht. Beachten Sie auch die Common Specifications der EU zu Klasse-D-IVD.
Klasse C
Klasse C Produkte werden wie Klasse B behandelt, jedoch müssen Hersteller nach Art. 81 IVDR von Produkten der Klasse C und D regelmäßig, mindestens einmal jährlich, einen aktualisierten Bericht über die Sicherheit erstellen. Bei speziellen Produkten wie patientennahe Tests (POCT; point-of-care-testing) oder Tests zur Eigenanwendung (Selbsttests) liegt ein besonderer Fokus auf der Gebrauchstauglichkeit. Bei therapiebegleitenden Diagnostika (companion diagnostics, CDx) wird zusätzlich der Arzneimittelbezug unter Einbindung der Europäischen Arzneimittelbehörde (EMA) geprüft.
Klasse B
Ab Klasse B und höher wird das QM-System durch die Benannte Stelle auditiert und die Technische Dokumentation geprüft. Wenn Sie Produktkategorien bilden, werden nur repräsentative Produkte einer Kategorie geprüft.
Mehr Informationen zu Produktgruppen und -kategorien erhalten Sie in unserem Beitrag Produktkategorie, generische Produktgruppe, Medizinproduktegruppe: Bitte nicht verwechseln!
Klasse A
- Bei IVD der Klasse A müssen Hersteller keine Benannte Stelle involvieren, d. h., dass keine Prüfung durch eine Benannte Stelle erfolgt. Allerdings kann die zuständige Landesbehörde Dokumente zur Prüfung einfordern. Hersteller benötigen gemäß IVDR Art. 10 (8) ein vollständiges Qualitätsmanagementsystem (ISO 13485).
- Bei Instrumenten und Produkten, die auch Maschinen sind, gelten die EMV-Richtlinie und die Maschinenverordnung.
- Bei sterilen Produkten, z. B. Probenahmegefäßen, gelten entsprechende Normen wie die EN 556-Reihe.
Achtung: Für die Prüfung der Sterilaspekte benötigen auch Klasse-A-Hersteller eine Benannte Stelle. Jedoch beschränkt sich deren Prüfung ausschließlich auf diesen Bereich.
Grundlegende Sicherheits- und Leistungsanforderungen
Unabhängig von der Klassifizierung müssen Hersteller die anwendbaren grundlegenden Sicherheits- und Leistungsanforderungen des Anhangs I der IVDR erfüllen. Somit müssen bei allen Klassen, auch bei Klasse A, die Nachweise zur Einhaltung des Stands der Technik dokumentiert sein:
- Risikomanagement
- Gebrauchstauglichkeit
- Verifizierung und Validierung
Bei Produkten, die Software sind oder Software enthalten, sind weitere Nachweise erforderlich:
- Software-Lebenszyklus
- IT-Sicherheit
5. Wie Sie geschickt klassifizieren
Bei IVD kann es leicht zu einer falschen oder zumindest unglücklichen Einstufung in die Risikoklassen kommen. Meist landet das Produkt in diesem Fall in einer zu hohen Klasse und wird damit intensiver geprüft als nötig.
Das passiert vor allem dann, wenn sich Hersteller auf die möglichen Anwendungen des Produkts fokussieren und nicht auf die festgelegte Zweckbestimmung (also nur die vorgesehene Anwendung). Relevant ist jedoch, wofür das Produkt gedacht ist, und nicht, wofür es abweichend davon zum Einsatz kommen kann.
Durch gezielte Maßnahmen und Argumente lassen sich die häufigsten Klassifizierungsfehler vermeiden. Wir stellen Ihnen im Folgenden drei typische Probleme bei der Klassifizierung nach IVDR und passende Lösungsmöglichkeiten vor.

Meistern Sie Ihr IVDR-Zulassungsprojekt mit Leichtigkeit
Lernen Sie mithilfe des Auditgarants, wie Sie Ihr IVD sicher qualifizieren, wie die Klassifizierung von IVD unter der IVDR funktioniert, wie Sie einen Plan zur Umsetzung der regulatorischen Anforderungen erstellen und schlussendlich die Konformität Ihrer Akte prüfen können.
a) Systeme in Produkte aufteilen
Problem:
Mit dem Klassifizierungssystem der IVDR werden zahlreiche Produkte, die aus verschiedenen Teilen bestehen, in eine höhere Klasse eingeordnet. Ein Analysesystem rutscht beispielsweise aufgrund einer einzigen kritischen Komponente in die höchste Klasse. Für die meisten Teile des Systems ist dies jedoch unnötig.
Beispiel:
Sie kombinieren ein IVD-Analysegerät mit einem Roboter für die Probenaufbereitung und mit einer Software für die Ergebnisinterpretation als System. Das System ist in einer technischen Dokumentation als Ganzes beschrieben und wird als Gesamtlösung in Verkehr gebracht. Dann ist die Klassifizierung für das gesamte System abhängig vom untersuchten Biomarker und der klinischen Aussage, die mit dem System getroffen wird.
Wenn mit dem beschriebenen Analysesystem Antikörpernachweise zur Feststellung eines Allergiestatus vorgenommen werden, kann dies eine Zuordnung zu Klasse B über Regel 6 nach sich ziehen. Soll gleichzeitig der Immunstatus schwangerer Frauen in Bezug auf eine Cytomegalovirus-Infektion (CMV) mit dem System nachgewiesen werden, liegt die Eingruppierung bei Klasse C. Falls mit dem System auch die Kompatibilität von Blutkonserven für potenzielle Empfänger getestet werden soll, fällt es in Klasse D.
Lösung:
Eine geschickte Abgrenzung ist der Schlüssel zur differenziellen Klassifizierung. Dies führt zu einem schlankeren und schnelleren Zulassungsprozess. Der Preis ist, dass Sie für jedes der kombinierten Einzelprodukte eine technische Dokumentation erstellen müssen.
Sie können dieses System in einzelne Produkte aufteilen und deren Schnittstellen so beschreiben, dass diese in Kombination ihren jeweiligen Zweck erfüllen. Jede Systemkomponente erhält dann eine eigene Zweckbestimmung und eine eigene technische Dokumentation. Damit kann zumindest das Analysegerät in Klasse A fallen und das Reagenzprodukt je nach Zweckbestimmung in eine höhere Klasse.
b) Mit geschickter Software-Architektur die Klassifizierung optimieren
Problem:
Die klinischen Aussagen in Ergebnisberichten gehen weit über die formulierte Zweckbestimmung hinaus. Dies katapultiert das Produkt indirekt in eine höhere Klasse oder schafft Sonderfälle, welche die Benannte Stelle zusätzlich bewerten muss.
Beispiel:
Eine IVD-Software mit der Zweckbestimmung „zur Feststellung der Pathogenität genetischer Varianten“ fällt in Klasse C, da es hier meist um Krebsdiagnostik oder Gentests (Feststellung von Erbkrankheiten) geht. Wird beim Ergebnisbericht neben der Feststellung der Pathogenität der Variante zudem ein Arzneimittel als Therapie vorgeschlagen, eventuell sogar mit Angaben zur Sicherheit des Arzneimittels im Kontext der gefundenen Variante(n), handelt es sich um ein therapiebegleitendes Diagnostikum.
Dies stellt einen Sonderfall bei der Prüfung der technischen Dokumentation dar. Die europäische Arzneimittelbehörde EMA muss involviert werden.
Lösung:
Technologie-Plattformen können als modulares System aufgesetzt und die Module jeweils als eigenständige Software beschrieben werden. Das zieht zwar die Erstellung mehrerer technischer Dokumentationen nach sich, die Plattform selbst muss jedoch unter Umständen gar nicht als Medizinprodukt qualifiziert sein. Für diese Lösung sind Software-Entwickler gefragt, welche die Software-Architektur so aufsetzen, dass die Module unabhängig voneinander agieren.
Wie dieses Beispiel zeigt, erfüllt Software neben ihrem medizinischen Zweck meist auch zahlreiche nichtmedizinische Funktionen. Die Klassifizierung von IVD-Software ist daher äußerst komplex. Wir haben dieses Thema in einem separaten Fachartikel IVD-Software richtig klassifizieren genauer beleuchtet.
c) Reagenzien-Kits in Komponenten aufteilen
Problem:
Viele Hersteller bieten sich ähnelnde Reagenzien-Kits an. Obwohl sich die einzelnen Komponenten dabei überschneiden, werden sie bei jedem einzelnen Kit neu beschrieben. Viele Teile der technischen Dokumentationen weisen redundante Inhalte auf.
Manche Hersteller bieten außerdem Kits mit Multiplex-Nachweis an. In diesem Fall gilt die höchste Klasse je nach Zweckbestimmung des kritischsten Parameters. Als Konsequenz rutscht das ganze Kit aufgrund der Zweckbestimmung in die höheren Klassen B, C oder D.
Lösung:
Die meisten Reagenzien-Kits sind von vornherein komponentenbasiert: Puffer, Enzyme, Analyt-spezifische Reagenzien wie Primer, Sonden oder Antikörper, Kontrollen und ggf. Kalibratoren wirken zusammen. Die Kits können zerlegt werden. Kit 1 enthält dann beispielsweise die generischen Komponenten. Der generische Teil muss nur einmal beschrieben werden, da er stets gleich ist. Dieses Kit fällt dann in Klasse A. Kit 2 umfasst die Analyt-spezifischen Komponenten und würde in eine höhere Risikoklasse fallen.
Dadurch sind zwar mehr technische Dokumentationen nötig, diese werden aber schlanker und können wie ein Baukastensystem erweitert werden.
6. Quellen, die bei der Klassifizierung helfen
Als Entscheidungshilfe dafür, welcher Klasse Sie Ihr Produkt zuordnen müssen, können Sie neben der IVDR auch auf weitere Quellen zurückgreifen. Hierzu zählen:
- Guidance document – In vitro diagnostic medical devices – Borderline and Classification issues. A guide for manufacturers and notified bodies – MEDDEV 2.14/1 rev.2
- MDCG 2020-16 rev.4 „Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746”; das Team NB hat eine Stellungnahme zur Klassifizierung von SARS-CoV-2-Tests veröffentlicht
- Manual on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices Version 4: Das Borderline Manual der EU-Kommission ist noch nicht vollständig und hilft nur bedingt bei der Klassifizierung. Bis es eine vollständige Version gibt, kann das Vorgängerdokument Manual on borderline and classification in the Community Regulatory framework for medical devices als State-of-the-Art angesehen und zur Klassifizierung herangezogen werden.
- Principles of In Vitro Diagnostic (IVD) Medical Devices Classification IMDRF/IVD WG/N64
7. Fazit
Die IVDR teilt die IVD in vier verschiedene Risikoklassen (Klasse A, B, C und D) ein. Gehen Hersteller nicht richtig vor, werden ihre Produkte einer unnötig hohen Klasse zugeordnet. Dies kann wie im Artikel beschrieben weitreichende Folgen haben.
Durch eine präzise beschriebene Zweckbestimmung und eine kluge Abgrenzung der Komponenten kann eine zu hohe Klassifizierung von IVD-Produkten vermieden und nebenbei noch die Technische Dokumentation verschlankt werden.
Wenden Sie sich bei Fragen zur Zulassungsstrategie oder zur Klassifizierung gern jederzeit an das Johner Institut. Nutzen Sie dafür das Formular oder schreiben Sie einfach eine E-Mail. Mit dem kostenfreien IVD-Klassifikator des Johner Instituts können Sie außerdem die Risiko-Klassifizierung Ihres IVD-Produkts schnell und einfach abschätzen.
Versionshistorie:
- 2025-10-06: Unter 6) 4. Version des Manuals on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices verlinkt
- 2025-04-10: Unter 6) 4. Revision des MDCG 2020-16 sowie die Stellungnahme vom Team NB verlinkt
- 2024-10-10: Unter 1c) Hinweis zu MDCG 2024-11 ergänzt
- 2024-03-08: Kapitel zur Qualifizierung von IVD ergänzt; Schaubild zu Konformitätsbewertungsverfahren aktualisiert
- 2024-01-25: Link zu EU-Referenzlaboratorien ergänzt
- 2023-12-04: Vollständige Überarbeitung des Artikels
- 2023-02-28: Anpassungen bezüglich gültiger IVDR; Link zum Guidance-Dokument MDCG 2020-16 rev.2 ergänzt; Ergänzung des Borderline Manual 2022
- 2022-07-11: Link zu Klasse-D-Common-Specifications ergänzt
- 2022-03-04: Unter 2d) Hinweis zu MDCG 2022-3 ergänzt
- 2022-01-28: Link zum Guidance-Dokument MDCG 2020-16 rev.1 ergänzt



Vielen Dank für den tollen Artikel. Die zu wählende Strategie ist offensichtlich ausschlaggebend. Dabei spielt die Aufteilung der Produkte und somit des jeweiligen Intended Use der einzelnen Produkte eine bedeutende Rolle. Können Sie eine Aussage dazu machen, ob es aus Sicht anderer Märkte (z.B. FDA) Aspekte gibt, die den in diesem IVDR-geprägtem Artikel beschriebenen Prinzipien entgegenstehen bzw. wo man an Grenzen stößt?
VG und Dank vorab,
H. Stiefel
Lieber Herr Stiefel,
vielen Dank für Ihre positive Reaktion auf den Artikel, das freut uns sehr! Zu Ihrer Frage der Anwendbarkeit des Konzeptes in anderen Märkten: Ja es macht durchaus einen Unterschied in anderen Märkten. So wäre eine Zulassung in den USA vielmehr abhängig von einem bereits registrierten Produkt eines Wettbewerbers (equivalent device für ein 510k-Verfahren). Wenn es ein solches gibt, dann sind Sie deutlich schneller bei der Zulassung, wenn Sie den Intended Purpose und den Intended Use möglichst identisch zum Vergleichsprodukt beschreiben. Ähnlich gilt dies auch für China.
Besten Dank für die spannende Frage und herzliche Grüße,
Sebastian Grömminger
Lieber Herr Grömminger,
vIelen Dank für die schnelle Rückmeldung und die Hinweise bzgl. 510k etc., die vermutlich auch erst mit einer Zerlegung in einzelne Produkte so richtig in die Praxis umzusetzen ist.
Ein Fragezeichen bleibt bei mir jedoch und das geht eher in die Richtung die Strategie der Aufteilung der Produkte und ob mit der Aufteilung ansich Probleme auftauchen können. Um ihr Beispiel aufzugreifen:
„Sie kombinieren ein IVD-Analysegerät mit einem Roboter für die Probenaufbereitung und mit einer Software für die Ergebnisinterpretation als System. Das System ist in einer technischen Dokumentation als Ganzes beschrieben und wird als Gesamtlösung in Verkehr gebracht. Dann ist die Klassifizierung für das gesamte System abhängig vom untersuchten Biomarker und der klinischen Aussage, die mit dem System getroffen wird.
..
Lösung:
Eine geschickte Abgrenzung ist der Schlüssel zur differenziellen Klassifizierung. Dies führt zu einem schlankeren und schnelleren Zulassungsszenario. Der Preis ist, dass Sie für jedes der kombinierten Einzelprodukte eine technische Dokumentation erstellen müssen.“
Meine Frage ist eigentlich, ob Ihre Lösung (die ich übrigens für richtig halte) in anderen Märkten an ihre Grenzen stößt?
VG,
H. Stiefel
Lieber Herr Stiefel, ja, der Ansatz kann in anderen Märkten an Grenzen stoßen, da Sie getrennte Produkte unter Umständen nur durch ein komplettes Neuzulassungsverfahren in den jeweiligen Markt bekommen. Aber auch dafür gibt es eine Lösung. Sie müssen ohnehin für andere Märkte eigene Dossiers erstellen. So kann es für die Zulassung in USA zum Beispiel Sinn machen, die für Europa bereits getrennten Produkte im 510k-Verfahren als System zu beschreiben, um die Äquivalenz zum Vergleichsprodukt herzustellen. Am Ende sind Sie mit abgegrenzten Produkten immer flexibler, denn das Verbinden von Produkten ist einfacher als deren Abgrenzung von einander. Ich hoffe, das beantwortet Ihre Frage. Im Einzelfall muss man ohnehin Vor-Und Nachteile der Abgrenzung abwägen, um die beste Strategie zu finden, insbesondere wenn man mehrere Märkte bedienen möchte.
Herzliche Grüße,
Sebastian Grömminger
Lieber Herr Grömminger,
vielen Dank – das beantwortet meine Frage und bestätigt mein Gefühl.
Viele Grüße,
H. Stiefel
Sehr geehrter Herr Grömmiger,
Sie schreiben, dass unter Punkt „2C)“, dass Probenentnahme-Sets unter die Kategorie C fallen, da Sie zur den „Produkten zur Eigenanwendung“. Ist das wirklich so?
Laut der Beschreibung in „MANUAL ON BORDERLINE AND CLASSIFICATION IN THE COMMUNITY REGULATORY FRAMEWORK FOR MEDICAL DEVICES“ (V1.22 05-2019) definieren die Autoren reine „Probenentnahme-Sets“, die dem Kunden kein direkten Ergebnis anzeigen (Vgl. zu Schnelltest) explizit nicht als „Produkt zur Eigenanwendung“, da die Analyse durch Dritte (Labor, Arzt etc.) durchgeführt wird. Das Manual bezieht in diesem Fall zwar auf die IVDD, aber ich kenne noch kein gültiges Borderline Manual für die IVDR. Des Weiteren ist die Definition der „Produkte zur Eigenanwendung“ zwischen IVDR und dem Borderline Manual nahezu identisch. Damit gehe ich davon aus, dass das Borderline Manual noch Anwendung findet.
Die Autoren schreiben:
„[…]These kits are in vitro diagnostic medical devices by means of applying article 1 (2) b IVDD which states that: ‚Specimen receptacles are considered to be in vitro diagnostic medical devices. ‘Specimen receptacles’ are those devices, whether vacuum-type or not, specifically intended by their manufacturers for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination.‘
The question arises whether these specimen receptacles could be considered as a ‘device for
self-testing‘ in accordance with article 1(2)d IVDD according to which ‘device for ’self-testing‘ means any device intended by the manufacturer to be able to be used by laypersons in a home environment;
In this determination, the notion ‘used’ is essential. Firstly, it is necessary to examine the
instructions for use. Where the instructions for use require an action to be taken by the end-user of the device in question, the notion ‘used’ is fulfilled. In addition, the definition of ‘self-testing’ provides guidance on the action to be taken i.e. “testing”. Thus where a specimen receptacle is simply used by the patient to contain a specimen it
remains a specimen receptacle. To become a ‘device for self-testing’, either the filling of the receptacle with a specimen should result directly in a result being given or the patient should need to do something directly to the specimen prior to its despatch in order to fulfil the concept of ‘used’“
-> Damit komme ich zu dem Schluss, dass „Probenentnahme-Sets“ keine „Produkte zur „Eigenanwendung“ sind und demnach nicht in Klasse C fallen. Die Frage sind die Probenentnahme-Sets dann einfache „Specimen receptacles“ (Rule 5) und fallen denmach in Klasse A oder fallen diese in die Klasse B (Rule 6)?
Vielen Dank für die Auskunft.
Lieber Herr Salis, danke dass Sie diesen Punkt aufbringen. Auch wir warten sehnsüchtig auf ein neues Borderline-Manual oder eine MDCG-Leitlinie zu diesem Thema.
Die Fragestellung ist so komplex, dass sie sich für einen eigenen Artikel eignen würde. Aufgrund der unklaren Situation unter IVDR ist ein Probennahmeset für Laien unter sehr konservativer Betrachtung der IVDR aktuell in Klasse C. Nun ergeben sich aber weitere Möglichkeiten durch Produktabgrenzung und genauere Betrachtung. Wenn Das Probennahme-Set „nur“ eine Zusammenstellung eines Probennahme-Systems ist (z.B. swab oder Lanzette zusammen mit einem IVD-Probenbehältnis) und alle Komponenten CE-markierte Produkte oder anderweitig legale Produkte wie zum Beispiel Verpackungsmaterialien gemäß Verpackungsanweisung P650 für Biomaterialien sind, können Sie sogar Artikel 22 der MDR anwenden und das Probennahme-Set als „Behandlungseinheit“ deklarieren. Das erspart viele regulatorische Anforderungen. Mehr zu Artikel 22 im Allgemeinen finden Sie hier: https://www.johner-institut.de/blog/regulatory-affairs/systeme-und-behandlungseinheiten/. Wenn es sich um einen Direct-to-Consumer-Test handelt, sehe ich das Klasse C als durchaus sinnvoll an, denn die Laien werden ggf. mit dem Ergebnis der Untersuchung alleine gelassen was zu Risiken führen kann, die man unter ärztlicher Behandlung ausschließen könnte. Der Ergebnisbericht wird sozusagen zur Benutzerschnittstelle für die gesamte Untersuchung und sollte daher auch den Gebrauchstauglichkeits-Anforderungen gemäß IEC 62366 gerecht werden.
Als Fazit kann ich sagen, Sie haben Recht: So pauschal kann man ein Probennahme-Set für Laien nicht in Klasse C fallen lassen. Niedrigere Klassen sind prinzipiell möglich und sogar eine Behandlungseinheit gemäß MDR Art. 22, sofern ein MDR-konformes Medizinprodukt enthalten ist. Eine Differenzielle Betrachtung ist jedoch erforderlich, um eine Argumentationskette für die Klassifizierung aufbauen zu können und wie so oft bedarf es einer ordentlich formulierten Zweckbestimmung.
Mit besten Grüßen,
Sebastian Grömminger
Sehr geehrter Herr Dr. Grömminger,
im Artikel steht:
„Beispiele für Produkte, die getrennt klassifiziert werden:
[…]
Zubehör
An einem Mikroskop, das speziell für die Pathologie vorgesehen ist, kann als Zubehör eine Digitalkamera angeschlossen werden, damit gleichzeitig ein zweiter Pathologe auf das histologische Schnittbild sehen kann. Alle Teile werden gemeinsam klassifiziert.“
Hier passt die Aussage ncht zur Überschrift. Wie ist es richtig? Können Sie meine Verwirrung verstehen und auflösen?
Sehr geehrter Herr Schäfer, vielen Dank für Ihren Kommentar. Sie haben recht, das war ein Fehler im Text. Das Zubehör wird getrennt klassifiziert. Das habe ich umgehend korrigiert.
Mit besten Grüßen,
Sebastian Grömminger
Gewebeproben werden täglich tausendfach in Kunststoffbehältern je nach Präparatgröße mit einem Nennvolumen von wenigen ml bis mehreren Litern in Formaldehydlösung konserviert und zur medizinischen Diagnostik z.B. in die Pathologie transportiert. Nach der Diagnose und einer darauf folgenden Aufbewahrungsfrist können die Flüssigkeiten und Präparate entsorgt werden. Viele Pathologien bereiten die verbleibenden Kunststoffbehälter mittels manueller und häufig maschineller Reinigung auf und bringen sie wieder in Umlauf zur Aufnahme der nächsten Proben. Ist dieser Prozess IVDR-konform oder müssten die Behälter entsorgt bzw. dürften sie nur von einer zertifizierten Stelle aufbereitet und von ihr wieder in Umlauf gebracht werden?
Lieber Herr Weger,
vielen Dank für Ihre spannende Frage.
Sie beschreiben, dass die Behälter zum Probentransport zur anschließenden medizinischen Diagnostik eingesetzt werden. Somit sind nach Art. 2 der IVDR (2) die Probenbehältnisse In-vitro-Diagnostika und müssen die Anforderungen der IVDR erfüllen. Ob und unter welchen Umständen ein Probenbehältnis wiederverwendet werden kann, ist abhängig von dessen Zweckbestimmung. Wenn zutreffend, wird vom Hersteller weiterhin festgelegt, wie eine Aufbereitung zu erfolgen hat. Beides können Sie der entsprechenden Gebrauchsanweisung des Herstellers entnehmen. Gemäß Anhang I, Kapitel III, 20.4.1. n) vi) ist dieser bei wiederverwendbaren Produkten dazu verpflichtet, Angaben über geeignete Aufbereitungsverfahren zu machen.
Erfolgt eine Verwendung außerhalb der Zweckbestimmung und/oder abweichend von der Gebrauchsanweisung, liegt ein Off-label Use vor. Es kann nicht sichergestellt werden, dass die Probenbehältnisse sicher und wirksam sind und nicht den klinischen Zustand des Patienten gefährden, wie es im Anhang I der IVDR gefordert ist.
Herzliche Grüße,
Juliane Havlicek
Sehr geehrtes Johner Team,
unter welche IVDR Klasse, fallen Produkte aus der IVD Annex II List B (Kontrollen) und Produkte aus Other not for self testing (Reagenz, Calibrator und Control)
Vielen Dank im Voraus!
Markus
Lieber Markus,
um Ihre Frage zu beantworten, helfen zunächst die Durchführungsvorschriften in Anhang VIII der IVDR weiter. Diese besagen in Punkt 1.5 „Kalibratoren zur Verwendung mit einem Produkt werden derselben Klasse zugerechnet wie das Produkt“ und in Punkt 1.6 „Kontrollmaterialien mit quantitativen oder qualitativen Werten, die für einen spezifischen oder mehrere Analyten bestimmt sind, werden derselben Klasse zugerechnet wie das Produkt“. Die Klassifizierung von Kalibratoren und Kontrollmaterialien ist demnach vom dem Produkt und dessen Zweckbestimmung abhängig, mit welchem diese zusammen verwendet werden.
Die Klassifizierung von Reagenzien ist ebenfalls abhängig von deren Zweckbestimmung. Handelt es sich beispielsweise bei den Reagenzien um allgemeinen Laborbedarf, wie z.B. Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen, allgemeine Nährmedien oder histologische Färbungen (die vom Hersteller dafür vorgesehen sind, in Zusammenhang mit einer spezifischen diagnostischen Untersuchung eingesetzt zu werden), werden diese nach Regel 5 des Anhangs VIII IVDR der Klasse A zugeordnet.
Herzliche Grüße,
Juliane Havlicek
Sehr geehrtes Johner Team,
benötigen IVDR Hersteller der Klasse A, ein QM System nach ISO 13485 und muss dieses auch zwingend zertifiziert sein? Ich lese es muss ein QM nach ISO vorhanden sein, aber nicht ob dieses zwingend auch ein Zertifikat vorhanen sein muss.
Vielen Dank im Voraus.
Lieber Markus,
Sie haben Recht. Artikel 8 der IVDR besagt, dass bei Anwendung von harmonisierten Normen eine Konformitätsvermutung besteht. Dies gilt auch explizit für QM-Anforderungen. In Artikel 10(8) der IVDR wiederum steht: „Das Qualitätsmanagementsystem umfasst alle Teile und Elemente der Organisation eines Herstellers, die mit der Qualität der Prozesse, Verfahren und Produkte befasst sind.“ Dabei trifft die IVDR keine Unterscheidung zwischen den unterschiedlichen Risikoklassen. Aufgrund der Konformitätsvermutung bei Anwendung der ISO 13485 für das QM-System müssen alle Hersteller alle für sie anwendbaren Anforderungen der ISO 13485 erfüllen. Falls Sie das nicht tun, benötigen sie eine sehr gute Begründung, warum sie dennoch die Anforderungen aus der IVDR bzgl. des QM-Systems erfüllen. Aus diesem Grund ist für IVD-Hersteller von Klasse A-Produkten ein QM-System nach ISO 13485 erforderlich. Eine Zertifizierung des QM-Systems ist jedoch nicht notwendig.
Herzliche Grüße,
Juliane Havlicek