
Saudi-Arabien plant in seiner „Vision 2030“ den Ausbau der Gesundheitsinfrastruktur. Bietet das den Medizinprodukteherstellern interessante Wachstumsmöglichkeiten? Und rechnet sich bei gestiegenen Zulassungsanforderungen der Aufwand für die Zulassung?
Erfahren Sie in diesem Artikel, wie Sie die Zulassung in Saudi-Arabien
- vorbereiten,
- durchführen und
- aufrechterhalten.
1. Ein interessanter Markt für Medizinproduktehersteller?
a) Ein großer Markt
Saudi-Arabien hat über 34 Mio. Einwohner. Das entspricht etwa dem Doppelten der Einwohnerzahl von Österreich und der Schweiz. Die Ausgaben für die Gesundheitsversorgung betrugen in Saudi-Arabien im Jahr 2006 nur 3,3 % des BIP. Dieser Anteil war in Österreich etwa dreimal so hoch (10,3 %).
Die hohe Abhängigkeit Saudi-Arabiens vom Erdöl stellt zudem ein Risiko für langfristige Investitionen dar. Andererseits verfolgt das Projekt Vision 2030 das Ziel, genau diese Abhängigkeit zu minimieren.
b) Strategische Investitionen in den Gesundheitsmarkt
Ein Teil dieser Initiative zielt explizit auf die Transformation des Gesundheitssystems. Auch der Gesundheitstourismus soll gefördert werden. Der saudi-arabische Medical Attaché in den USA und Kanada sagt in diesem Kontext:
The government [of Saudi Arabia] is investing a lot of money in healthcare; the budget for just the Ministry of Health is about $10 billion USD. That does not include the National Guard hospitals, Ministry of Defense hospitals and we have 26 universities, most of those have hospitals.
Quelle
c) Fazit
Saudi-Arabien steht laut Global Gender Gap Report weltweit auf einem der letzten Plätze, wenn es um Menschenrechte, Meinungsfreiheit oder Gleichberechtigung geht. Gleichzeitig ist Saudi-Arabien ein Land, das bereits viel Geld für die Gesundheitsversorgung aufwendet, diese Ausgaben weiter steigern wird und daher ein interessanter Markt für Medizinproduktehersteller ist.
Doch Hersteller, die ihre Produkte in diesem Land verkaufen wollen, müssen zuerst die regulatorischen Anforderungen erfüllen, d. h., ihre Produkte in Saudi-Arabien zulassen.
In diesem Artikel erhalten Sie Informationen darüber, wie Sie die Zulassung in Saudi-Arabien vorbereiten, durchführen und aufrechterhalten können. Weiterhin erfahren Sie, welche Vor- und Nachteile es mit sich bringt, dass sich die SFDA an MDR und IVDR orientiert.
2. Zulassung von Medizinprodukten in Saudi-Arabien
a) Saudi Food and Drug Administration
Medizinprodukte dürfen in Saudi-Arabien nur in Verkehr gebracht werden, wenn sie bei der SFDA (Saudi Food and Drug Administration) gemeldet sind und das Medical Devices Law erfüllen. Die Behörde verantwortet die Zulassung und die Überwachung der Medizinprodukte.
b) Neue Herausforderungen und Vorteile für Hersteller aus der EU
Die SFDA fordert, dass alle Produkte, die in Saudi-Arabien verkauft werden, über eine Medical Devices Marketing Authorization (MDMA) zugelassen werden. Wie dies abläuft, erfahren Sie in Kapitel 4 dieses Artikels.
Eine gibt keine Anerkennung bestehender Zulassungen aus anderen Ländern mehr, die zu einer vereinfachten Zulassung in Saudi-Arabien beitragen könnten. Die gute (und gleichzeitig schlechte) Nachricht ist, dass die aktuellen Anforderungen der SFDA sich stark an der MDR (und IVDR) orientieren.
Die SFDA unterstützt Hersteller dadurch, dass sie ihre Anforderungen rund um Medizinprodukte und In-vitro-Diagnostika auf ihrer Website veröffentlicht und (fast) alle Regularien und Gesetze ins Englische übersetzt. Allerdings bereiten die Unübersichtlichkeit und die hohe Anzahl der Veröffentlichungen den Herstellern regelmäßig Probleme.
c) Die wichtigsten Gesetze und Richtlinien
Die wichtigsten Anforderungen hat die saudi-arabische Behörde im „Medical Devices Law“ publiziert. Dieses Gesetz enthält allgemeine Anforderungen an Medizinprodukte, deren Zulassung und Marktüberwachung.
Die Anforderungen dieses übergeordneten Gesetzes werden durch weiterführende Regulations präzisiert. Zudem veröffentlicht die SFDA regelmäßig Richtlinien (Guidelines) zu spezifischen Themen wie Qualitätsmanagementsysteme, UDI oder Software.
Eine Übersicht über die wichtigsten Anforderungen finden Sie in Tabelle 1.
| Dokument | Inhalt |
| Medical Devices Law | Allgemeine Anforderungen an Medizinprodukte, deren Zulassung und Marktüberwachung. Das Gesetz wird durch Regulations (MDS-REQ) und Guidelines (MDS-G) ergänzt. |
| MDS-REQ 1 Requirements for Medical Devices Marketing Authorization | Beschreibt die Anforderungen, die Produkte erfüllen müssen, um eine MDMA zu erlangen (Medical Device Marketing Authorization). Es enthält „Essential Principles of Safety and Performance“ für Medizinprodukte und IVD sowie Regeln zur Risikoklassifizierung. Alle Produkte müssen entsprechend diesen Anforderungen registriert werden. |
| MDS-G008 Guidance on Medical Devices Classification | Anwendungsbeispiele und Erklärungen zur Klassifizierung und zu den Regeln der MDS-REQ 1 |
| MDS-REQ 5 Requirements for Shipments Clearance of Medical Devices at Ports of Entries | Nur Produkte mit gültiger Zulassung werden vom Zoll freigegeben. Das Dokument erklärt, welche weiteren Dokumente beizulegen sind, um Produkte nach Saudi-Arabien einzuführen. |
| MDS-REQ 7 Requirements for Unique Device Identification (UDI) for Medical Devices | Erklärt, wie Medizinprodukte in Saudi-Arabien mit einer UDI versehen werden müssen. Ab September 2023 müssen alle Produkte eine UDI tragen. |
| MDS-REQ 11 Requirements for Post-Market Surveillance of Medical Devices | Enthält Anforderungen zur Marktüberwachung und zur Meldung von Vorkommnissen und Rückrufen an die SFDA |
| MDS-REQ 10 Requirements for Inspections and Quality Management System for Medical Devices | Enthält Anforderungen an das Qualitätsmanagementsystem sowie die Überwachung der Hersteller durch die SFDA. Alle Hersteller benötigen ein Zertifikat nach ISO 13485. |
Die in Tabelle 1 referenzierten Dokumente können Sie auf der Webseite der SFDA herunterladen.
Die SFDA erfindet in puncto Anforderungen an Zulassung, Dokumentation und Klassifizierung das Rad nicht neu: Die Anforderungen weisen viele Parallelen mit der europäischen MDR und IVDR auf. So enthält die MDS-REQ 1 unter anderem die grundlegenden Sicherheits- und Leistungsanforderungen der MDR, ebenso wie identische Anforderungen für die Klassifizierung.
d) Etwas abweichende Definition des Begriffs Medizinprodukt
Die Definition des Begriffs Medizinprodukt weicht in Details von der Definition des Begriffs in Europa ab. Prüfen Sie daher, ob Ihr Produkt unter diese Definition fällt. Der Begriff wird in Artikel 1 des Medical Devices Law wie folgt definiert:
Medical device means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article:
Intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific purpose(s) of:
- Diagnosis, prevention, monitoring, treatment or alleviation of disease,
- Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- Investigation, replacement, modification, or support of the anatomy or of a physiological process,
- Supporting or sustaining life,
- Control of conception
- Disinfection of medical devices,
- Providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived from the human body; and
which does not achieve its primary intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its intended function by such means.”
IVD-Produkte werden nicht eigenständig definiert, sondern sind in der obigen Definition eingeschlossen. Sie unterliegen jedoch separaten Klassifizierungsregeln.
Im Vergleich zur MDR fallen in Saudi-Arabien Produkte zur Vorhersage und Prognose von Krankheiten nicht unter die Begriffsbestimmung. Dafür werden Produkte zur Unterstützung der Anatomie oder eines physiologischen Prozesses als Medizinprodukt eingestuft.
Dem gegenüber stehen die Klassifizierungsregeln der MDS-G008, in welcher drei Produktbeispiele genannt werden, deren Zweckbestimmung eine Prognose und Vorhersage ist.
Im Zweifelsfall sollte also über einen Bevollmächtigten (Authorized Representative, AR) mit der SFDA Kontakt aufgenommen werden, um die Einstufung als Medizinprodukt zu klären.
Produkte ohne medizinische Zweckbestimmung, die in Annex XVI der europäischen Medizinprodukterichtlinie aufgelistet sind, müssen auch in Saudi-Arabien gemäß den geltenden Klassifizierungsregeln als Medizinprodukt behandelt werden (MDS-G008, Kapitel 1).
3. Klassifizierung
Produkte werden, abhängig von ihrem Risiko, in die Klassen A bis D eingeordnet. Die Klasse bestimmt das Zulassungsverfahren, das im nächsten Kapitel vorgestellt wird.
Die Klassifizierungsregeln finden sich in der MDS-REQ 1 und sind identisch mit den 22 Regeln der MDR. Entsprechend können Sie Ihre Klassifizierung übertragen (s. Tab. 2).
| Klassifizierung in Saudi-Arabien | Risikolevel | Klassifizierung nach MDR |
| A | Low | I |
| A – Steril | Low–medium | Is |
| A – Messfunktion | Low–medium | Im |
| A – Wiederverwendbare chirurgische Instrumente | Low–medium | Ir |
| B | Low–medium | IIa |
| C | Medium–high | IIb |
| D | High | III |
Die SFDA hat die Richtlinie MDS-G008 zur Interpretation der Klassifizierungsregeln herausgegeben. Das Dokument enthält hilfreiche Erläuterungen und ergänzende Beispiele, wie diese Regeln bei Produkten angewendet werden sollen.
Auch bei der Klassifizierung von In-Vitro-Diagnostika orientiert sich die SFDA an der europäischen Gesetzgebung: Die sieben Klassifizierungsregeln der IVDR finden sich auch in Saudi-Arabien.
| Klassifizierung in Saudi-Arabien | Risikolevel | Klassifizierung nach IVDR |
| A | Low Individual Risk and Low Public Health Risk | A |
| B | Moderate Individual Risk and/or Low Public Health Risk | B |
| C | High Individual Risk and/or Moderate Public Health Risk | C |
| D | High Individual Risk and High Public Health Risk | D |
4. Medizinprodukte erfolgreich zulassen
Die Registrierung bei der SFDA gliedert sich im Wesentlichen in drei Schritte:
- Vorbereitung
- Einreichung der Unterlagen
- Aufrechterhaltung der Zulassung
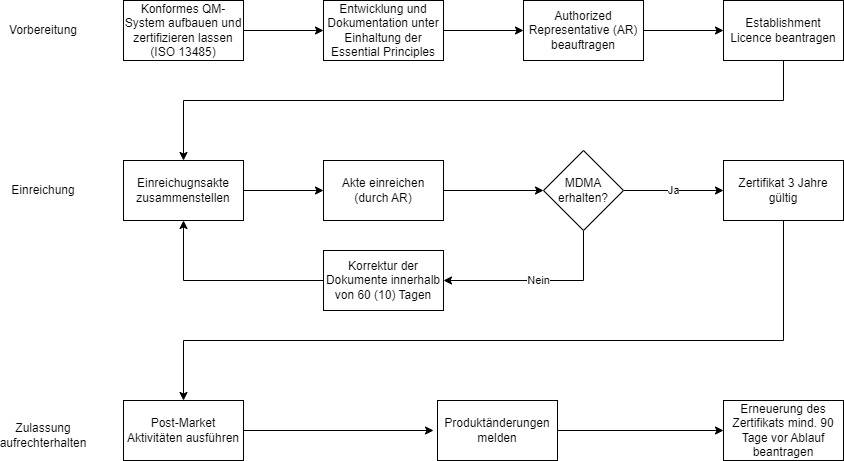
a) Die Schritte zur Vorbereitung der Zulassung
Schritt 1: QM-System errichten
Medizinproduktehersteller müssen über ein QM-System gemäß ISO 13485 verfügen. Das entsprechende Zertifikat und der letzte Auditbericht der Benannten Stellen sind bei der SFDA einzureichen. Dies gilt auch für Hersteller von Klasse-A-Produkten, die in der EU nicht zwingend über ein ISO-13485-Zertifikat verfügen müssen.
Schritt 2: Technische Dokumentation erstellen
Bei der Erstellung der konformen Dokumentation haben europäische Hersteller einen Vorteil: Die Anforderungen an die technische Dokumentation sind in Anhang 3 (Medizinprodukte) und Anhang 4 (IVD) der MDS REQ1 enthalten und orientieren sich an den Anhängen 2 der MDR bzw. IVDR. Die SFDA schlüsselt noch detaillierter auf, welche Informationen sie von den Herstellern erwartet.
Die Beschreibung der SFDA ist stichpunktartig im Dokument MDS-REQ 1 festgehalten. Dies ist zum einen übersichtlicher als in der MDR, zum anderen lässt es weniger Spielraum für Interpretation.
Die Zweckbestimmung muss gemäß SFDA mindestens folgende Informationen enthalten:
- Hauptindikationen des Produkts
- Zu behandelnde Krankheit / Behinderung
- Zu behandelnde Patientenpopulation
- Kontraindikationen
Auch die „Information zur Auslegung und Herstellung“ sind ein schönes Beispiel. Sie umfassen in der MDR lediglich drei Unterpunkte. Während Hersteller sich hier überlegen müssen, welche Informationen unter der MDR wohl gefordert sind, um „die Auslegungsphasen, die das Produkt durchlaufen hat, zu verstehen“, gibt die SFDA in acht Unterpunkten vor, welche Dokumente und Information sie bei der Auslegung und Herstellung konkret erwartet.
Schritt 3: Authorized Representative etablieren
Hersteller ohne Sitz in Saudi-Arabien benötigen einen Bevollmächtigten im Land, einen Authorized Representative (AR). Dieser muss sich bei der SFDA registrieren. Die Aufgaben des AR können der Importeur, der Distributor oder eine unabhängige Stelle übernehmen.
Die Aufgaben des AR sind vielseitig: Er ist beispielsweise für die Übermittlung der Einreichungsunterlagen und die Kommunikation mit der SFDA verantwortlich. Ebenso muss er dafür Sorge tragen, dass das Produkt die Anforderungen der SFDA einhält und beispielsweise mit der entsprechenden Kennzeichnung versehen ist. Die Zulassung von Medizinprodukten darf nur der AR durchführen.
Ihr Authorized Representative muss auch ein Qualitätsmanagementsystem aufrechterhalten.
Schritt 4: Medical Device Establishment License beantragen
Alle Hersteller von Medizinprodukten müssen vor der Zulassung von Produkten eine Establishment License bei der SFDA beantragen. Die Lizenz ist fünf Jahre gültig und wird elektronisch über das GHAD-System beantragt. Für Hersteller außerhalb Saudi-Arabiens übernimmt der Authorized Representative diese Tätigkeit.
Die Establishment License wird nicht nur für Hersteller gefordert, sondern auch für Importeure und Distributoren.
b) Die Schritte zur Einreichung
Alle Produkte benötigen eine Medical Device Marketing Authorization (MDMA) und werden, unabhängig von ihrer Klassifizierung, über das TFA-Verfahren zugelassen. Im ersten Schritt stellen Sie die Einreichungsakte zusammen und lassen diese durch Ihren AR bei der SFDA einreichen.
Schritt 1: Einreichungsakte zusammenstellen
Bei der Einreichung unterscheidet die SFDA
- eine vereinfachte Einreichung für Klasse-A-Produkte und
- eine reguläre Einreichung für alle übrigen Produkte.
Schritt 1a: Vereinfachte Dokumentation
Produkte mit einem geringen Risiko (reine Klasse-A-Produkte) können mit einer vereinfachten technischen Dokumentation eingereicht werden.
In dem Fall sind folgende Dokumente erforderlich:
- Produktbeschreibung und Spezifikationen
- Informationen zum Hersteller
- Checkliste zu den Essential Principles
- Nachweisdokumente zur Einhaltung der Essential Principles
- Risikomanagement-Akte
- PMS-Plan und PMS-Report
Es muss immer die vollständige Dokumentation gemäß Anhang 3 bzw. 4, MDS-REQ 1 erstellt werden. Die SFDA behält sich vor, zusätzliche Dokumentation anzufordern, und gewährt für die Nachreichung eine Frist von nur 10 Tagen.
Ausgenommen von diesem vereinfachten Verfahren sind IVD-Produkte und Klasse A-Produkte der folgenden Kategorien:
- As: Steril
- Am: mit Messfunktion
- Ar: wiederverwendbare chirurgische Instrumente
- Neuartige Produkte
Schritt 1b: Vollständige Dokumentation
Diese und alle weiteren Produkte müssen über das vollständige TFA-Verfahren (Technical File Assessment) zugelassen werden.
Eine bereits bestehende Zulassung, z. B. ein Home-Country Approval, ist keine Voraussetzung für das TFA-Verfahren.
Wie der Name vermuten lässt, beruht das TFA-Verfahren auf einer Bewertung der Technischen Dokumentation. Das Ziel des TFA-Verfahrens ist es, anhand der Technischen Dokumentation zu prüfen, ob die „Essential Principles of Safety and Performance“ eingehalten wurden. Diese sind in Anhang 1 der MDS-REQ 1 enthalten. Sie dürften Ihnen beim Lesen bekannt vorkommen, denn weite Teile wurden aus Anhang 1 der MDR übernommen (inkl. Nummerierung).
Für die Prüfung bei der SFDA müssen Sie nun Ihre komplette Dokumentation gemäß Anhang 3 (für Medizinprodukte) und Anhang 4 (für IVD) aus der MDS-REQ 1 einreichen. Dies ist vergleichbar mit der technischen Akte, die Sie Ihrer Benannten Stelle zur Prüfung einreichen; sie enthält z. B.:
- Produktbeschreibung
- Informationen zum Hersteller
- Informationen zum Design und zur Herstellung des Produkts
- Checkliste der „Essential Principles of Safety and Performance”
- Risikomanagement-Akte
- Durchgeführte Verifizierungs- und Validierungstätigkeiten
- PMS-Plan
- PMS-Report (Klasse A) bzw. PSUR (Klasse B, C und D)
Die Verantwortung für die Übermittlung der Unterlagen an die SFDA liegt bei Ihrem Authorized Representative. Die SFDA prüft anschließend innerhalb von etwa drei bis vier Monaten Ihre Unterlagen und stellt nach erfolgreicher Prüfung die schriftliche Marketing Authorization aus.
Wenn der AR Rückfragen der SFDA nicht selbst beantworten kann, wird er sich an den Hersteller wenden.
Hier müssen die Hersteller am Ball bleiben: Erhält die SFDA innerhalb von 60 Tagen keine Antwort auf Rückfragen, wird die Einreichung gelöscht. Dies gilt auch, wenn die Rückfragen der SFDA innerhalb von drei Zyklen nicht ausreichend beantwortet wurden. Die Gebühr erstattet die Behörde nicht zurück.
War die Einreichung erfolgreich, erhalten Sie ein Zertifikat, das drei Jahre lang gültig ist.
c) Zulassung aufrechterhalten
Mit erfolgreicher Zulassung enden die Verpflichtungen des Herstellers nicht. Sie müssen unter anderem die Anforderungen an die Post-Market Surveillance und Meldungen von Änderungen berücksichtigen.
- Anforderungen an die PMS sind in MDS-REQ 11 enthalten. Dort geregelt ist z. B., welche Informationen an die SFDA gemeldet werden müssen und welche Zeiten zu berücksichtigen sind.
- Änderungen am Produkt müssen je nach Umfang nach 10 bzw. 30 Tagen über den AR an die SFDA gemeldet werden.
Ein Antrag auf Erneuerung des Zertifikats muss spätestens 90 Tage vor Ablauf eingereicht werden. Bei einem Wechsel des Authorized Representative haben Sie die Möglichkeit, einen Lizenz-Transfer durchzuführen.
5. Fazit und Kosten
Die SFDA hat umfangreiche Gesetze und Leitlinien zu Medizinprodukten erlassen. Dabei orientiert sie sich stark an der europäischen Gesetzgebung. Ob sie sich damit einen Gefallen getan hat, bleibt abzuwarten.
Zulassungsweg, Dauer und Aufwand für die Registrierung richten sich nach der Klasse des Produkts. Zusätzliche Kosten entstehen durch die jährliche Gebühr für Ihren Repräsentanten.
| Klasse A* | Klasse Am, Ar, As | Klasse B | Klasse C | Klasse D | |
| SFDA-Gebühr [SR] | 15.000 | 15.000 | 19.000 | 21.000 | 23.000 |
| Prüfzeiten bei SFDA | wenige Wochen | ~ 3–4 Monate | ~ 3–4 Monate | ~ 3–4 Monate | ~ 3–4 Monate |
| Gültigkeit | 3 Jahre | 3 Jahre | 3 Jahre | 3 Jahre | 3 Jahre |
* ausgenommen Klassen As, Ar, Am
Das Team vom Johner Institut unterstützt Sie bei der Zulassung Ihrer Medizinprodukte in Saudi-Arabien. Gerne beantwortet es Ihre Fragen, im Rahmen des Micro-Consultings sogar kostenfrei.
Änderungshistorie
- 2025-02-26: Überarbeitung von Kapitel 4b
- 2023-06-26: Grundlegende Überarbeitung zum Einpflegen der geänderten Gesetzgebung und Anforderungen unter dem Medical Devices Law
- 2020-10-07: Einfügen der Anmerkungen in den Kapiteln 4.4 und 6 sowie Hinzufügen der Spalte für Klasse Am, Ar, As in Kapitel 8



Hallo Frau Seidenfaden ich hab ein Frage, aber nicht zu Saudi Arabien sondern zu UAE. Können sie uns da auch weiter helfen? Unsere Distri. möchte ein nach MDR 2A Gerät an private Krankenhäuser verkaufen und teilt uns mit, das für private Krankenhäuser MD ohne Registrierung verwenden dürfen. Laut Aussage von dem Distri braucht es nur eine Registrierung für den Verkauf an (staatlichen) MOH KHs. Das können wir fast nicht glauben. Wir glauben, das sehr wohl eine generelle Produktregistrierung von Nöten ist und dies hier nicht gemeint ist, sondern eine Registrierung in einer Art Medizinprodukteregister, aus dem dann die Kliniken ihrer Produkte auswählen können. Können Sie uns hier weiterhelfen und uns die Situation in UAE hierzu kurz schildern. VG Stefan Kieslinger
Lieber Herr Kieslinger,
vielen Dank für Ihre Anfrage. Ein solches Vorgehen ist uns und unserem Partner vor Ort nicht bekannt.
Aus unserer Erfahrung und den regulatorischen Vorgaben muss ein Produkt immer bei MOHAP eingereicht werden. Dabei werden grundlegend zwei Möglichkeiten unterschieden: Der Classification Pathway, welcher für Produkte für professionelle Nutzer angewendet wird und der Registration Pathway, über welchen Produkte für Laien bzw. den Heimgebraucht registriert werden.
Der Classification Pathway ist relativ unkompliziert. Hier erhalten Sie am Ende einen „Classification Letter“ der es Ihnen erlaubt das Produkt ohne eine Registrierung über den Registration Pathway zu verkaufen. Möglicherweise meint Ihr Distributor diesen Pfad.
Ich hoffe diese Information hilft Ihnen weiter. Morgen veröffentlichen wir außerdem einen Fachartikel zum Thema UAE, in welchem Sie die Anforderungen detaillierter ausgearbeitet finden.
Melden Sie sich gerne, wenn es weitere Rückfragen gibt oder wir Sie unterstützen können.
Herzliche Grüße,
Margret Seidenfaden