
Mit dem Safer Technologies Program (STeP) möchte die FDA Herstellern innovativer Medizinprodukte einen neuen Zulassungsweg bahnen. Damit verfolgt die Behörde das Ziel, dass Patienten schneller von diesen Innovationen profitieren.
Lesen Sie in diesem Artikel, welche Vereinfachungen und Hilfestellungen die FDA für Sie als Hersteller vorsieht und welche Voraussetzungen Sie erfüllen müssen, damit Sie Ihre Produkte mit Hilfe des ‚Safer Technologies Programs‘ schneller und einfacher in den amerikanischen Markt zu bringen.
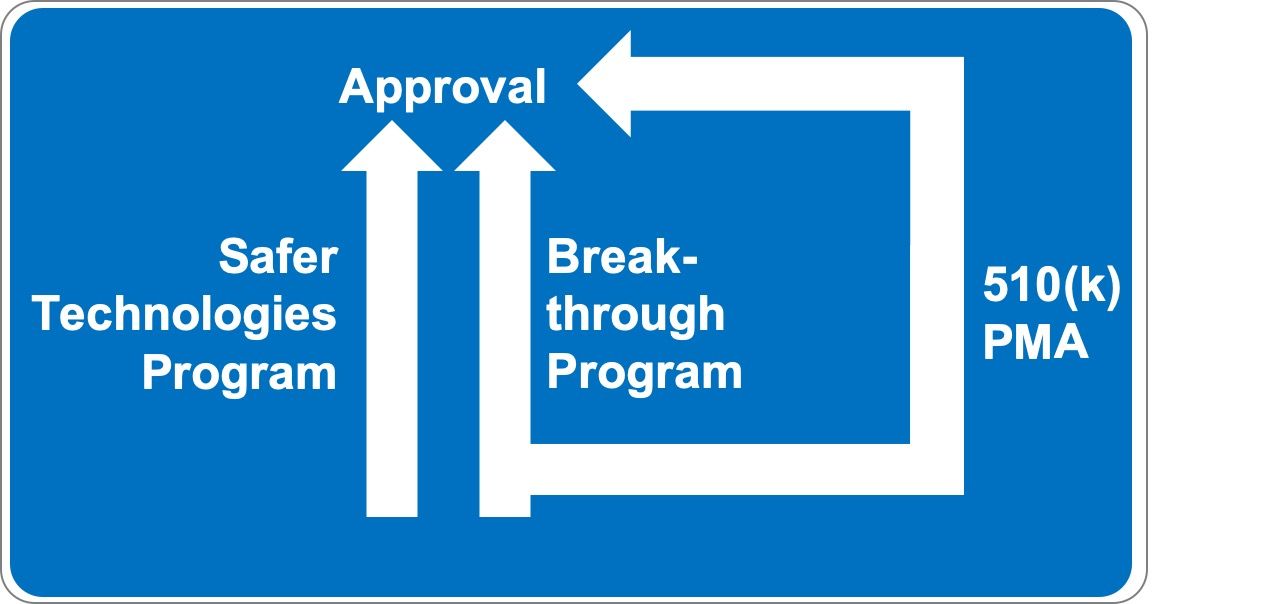
1. Was ist das Safer Technologies Program for Medical Devices?
a) Alternativen
Die FDA plant mit dem Safer Technologies Program (STeP) ein weiteres Zulassungsverfahren für Medizinprodukte. Es bildet unter gewissen Voraussetzungen eine Alternative zu den klassischen Zulassungsverfahren insbesondere
- Premarket Approval (PMA)
- De Novo Classification
- Premarket Notification (PMN), besser bekannt als 510(k) Verfahren
- Breakthrough Devices Program
b) Zielsetzung
Die FDA möchte Patienten schneller Zugang zu neuen Technologien und Produkten verschaffen. Mit dem „Breakthrough Devices Progam“ hat sich bereits ein beschleunigtes Zulassungsverfahren ins Leben gerufen. Mit dem „Safer Technologies Program“ möchte sie etwas Vergleichbares ins Leben rufen.
Allerdings ist das Safer Technologies Program im Gegensatz zum Breakthrough Program für Produkte gedacht, die weniger schwerwiegende Krankheiten und Verletzungen diagnostizieren oder behandeln sollen, beispielsweise keine lebensbedrohlichen oder irreversiblen Erkrankungen.
c) Prinzipien des Safer Technologies Programs
Das Safer Technologies Program zeichnet sich durch folgende Prinzipien aus:
- Die Kommunikation erfolgt interaktiv und zeitnah.
- Die FDA involviert Teams an Experten, darunter auch hochrangige „Directors“.
- Die Anträge werden mit hoher Priorität bearbeitet.
- Auch hier trifft die Behörde die Entscheidungen basierend auf dem Nutzen-Risiko-Verhältnis. Das sind Entscheidungen wie, ob das Produkt überhaupt in das STeP aufgenommen wird oder welche klinischen Daten vor und nach der Inverkehrbringung erhoben werden müssen.
- Klinische Studien sollen effizient und flexibel geplant, umgeplant und durchgeführt werden können.
Das Verfahren erinnert an agile Konzepte. Die FDA spricht sogar von „sprint discussions“. Dazu später mehr.
2. Ablauf des Safer Technology Programs STeP
a) Antrag
Der Hersteller beantragt die Teilnahme am Safer Technologies Program über eine sogenante Q-Submission. Dieser Antrag sollte folgende Informationen enthalten:
- Hintergrundinformationen
- Beschreibung des Geräts inkl. Funktionsweise, Bilder, Zeichnungen
- Erwartete Verbesserung der Sicherheit
- Zweckbestimmung
- Historie bisheriger Interaktionen mit der Behörde z.B. andere Anträge
- Begründung, weshalb das Produkt ins STeP aufgenommen werden darf. Das schließt insbesondere das Verfahren ein, das sonst durchlaufen werden müsste, und eine Begründung, weshalb das Gerät nicht ins Breakthrough Program aufgenommen werden kann.
- Genaue Beschreibung, wie das Gerät das Nutzen-Risiko-Verhältnis verbessert. Das sollte einer Faktoren sein, die weiter unten im Abschnitt „Verbesserungen“ genannt sind.
b) Kommunikation und Interaktion mit der Behörde
Wird er zugelassen, bietet die FDA mehrere Optionen der Interaktion an:
- Sprint Discussions
Diese Diskussionen haben zum Ziel, eine bestimmte Fragestellung innerhalb eines gegebenen Zeitrahmens zu beantworten. Sie möchte häufiger und zeitnäher die Meetings und Antworten auf Anfragen des Herstellers anbieten. - Data Development Plan
Der Hersteller reicht diesen Plan ein, in dem er darlegt, welche klinischen und nicht klinischen Daten er wann mit welchen Verfahren erheben will. Die FDA bietet an, diesen Plan ähnlich den Sprint Discussions zu bewerten. - Sonstige „Pre-Submissions“
Falls die Hersteller mehr als ein Thema diskutieren möchte, wie das bei den beiden o.g. Varianten möglich wäre, erwägt die FDA zusätzliche Pre-Submissions anzubieten. Das hängt aber von der Verfügbarkeit der Behördenmitarbeiter ab. - Status Updates
Auch außerhalb der genannten Varianten steht die FDA den Hersteller regelmäßig (sie nennt das Intervall „zweimonatlich“ als Beispiel) zur Verfügung. Diese dürfen aber nicht dazu genutzt werden, um offizielles Feedback einzuholen.
3. Was bringt Ihnen das STeP der FDA?
Für Hersteller erscheint das STeP aus mehreren Gründen attraktiv:
- Schnellere Zulassung
Die Zulassung kann schneller erfolgen. Die Behörde behandelt den Antrag mit hoher Priorität. Sie möchte innerhalb von 60 Tagen entschieden. - Weniger Aufwände durch gezielte Handlungsleitung
Der Hersteller erfährt schneller und während des Verfahrens die Ansichten und Bedenken der Behörde und kann während des Verfahrens darauf reagieren.
Die Behörde plant, innerhalb von 30 Tagen auf Anfragen zu antworten.
Beides wird das Verfahren beschleunigen und dazu beitragen, unnötige Aufwände zu vermeiden.
4. An wen wendet sich das STeP?
Hersteller können unter den folgenden Voraussetzungen eine Teilnahme am Safer Technologies Program in Betracht ziehen:
a) Formale und organisatorische Voraussetzungen
- Der Antrag erfolgt frühestens 60 Tage nach dem 19. September 2019
- Das Produkt ist nicht für Patienten mit schweren, insbesondere lebensbedrohlichen
oder potentiell irrreversiblen Krankheiten und Verletzungen vorgesehen. - Das Produkt müsste normalerweise einer PMA, einem De Novo oder einem 510(k), aber
nicht einem Breakthrogh Verfahren unterzogen werden. - Der Hersteller muss bereit sein, sehr kurzfristig auf Anfragen der FDA zu reagieren
und eng mit der Behörde zusammenzuarbeiten. - Er muss die Behörde sehr früh d.h. bereits während der Entwicklung in seine Pläne
einweihen.
b) Voraussetzungen an das Produkt
Das Produkt muss dahingehend innovativ sein, dass es im Vergleich zu seinen Alternativen das Nutzen-Risiko-Verhältnis verbessert, indem es
- Ernste Nebenwirkungen verringert
- (Geräte-)Fehler verringert
- Nutzungsfehler verringert, die zu Gefährdungen führen können, oder
- Die Sicherheit eines anderen Geräts oder eines anderen Verfahrens erhöht. Damit meint die FDA Zubehör.
Die FDA spricht zwar von einer Verbesserung des Nutzen-Risiko-Verhältnisses, sie benennt aber vor allem Verbesserungen durch die Reduktion von Risiken und nicht die Verbesserung des Nutzens. Das erklärt auch den Namen des Programms: Safer Technologies Program.
Das STeP wendet sich weniger an geänderte Produkte.
5. Fazit
Die FDA setzt ihren Weg fort, Patienten über agilere Zulassungsverfahren und „risk-based“ schneller Zugang zu neuartigen Medizinprodukten zu verschaffen. Das Breakthrough Medical Devices Program und das Precert Program sind weitere Zeugnisse dieser Bemühungen.
Der Name Safer Technologies Program STeP ist tatsächlich Programm und Ziel: Es geht um sicherere Technologien und damit um eine Verbesserung des Nutzen-Risiko-Verhältnisses durch eine Reduktion der Risiken.
Die Alternativen wie das SteP und Breakthrough Program stärkt die FDA wahrscheinlich auch deshalb, um das von ihr selbst kritisierte 510(k)-Programm zunehmend abzulösen. Weshalb dieses Programm kritikfähig ist, zeigt auch John Oliver sehr pointiert auf.
Unterstützung
Wünschen Sie Unterstützung bei der Zulassung Ihres Produkt in den USA? Unsere FDA-Spezialisten helfen schnell und gerne, damit Sie Ihr Produkt schnell und ohne unnötige Aufwände und teure Umwege in den US-Markt bekommen. Melden Sie sich einfach bei uns.


