
Mit dem Pre-Submission-Programm (kurz „Pre-Sub“) bietet die FDA ein formales Verfahren an, mit dem Hersteller bereits vor der eigentlichen Zulassung ihre Zulassungsstrategie sowie konkrete Fragen klären können. Ein Pre-Sub Request eignet sich unter anderem im Vorfeld von 510(k)s, De Novo Requests oder auch PMAs. Damit können auf beiden Seiten unnötige Kosten und Aufwände vermieden werden.
Das Johner Institut beobachtet, dass viele Hersteller dieses Angebot nicht ausreichend kennen oder/und sich nicht sorgfältig genug auf die FDA Pre-Submission-Meetings vorbereiten.
In diesem Beitrag erfahren Sie, wie Sie mit der FDA in Kontakt treten und wie Sie das Pre-Sub-Programm bestmöglich nutzen können – zum Beispiel, um die Review-Zeit Ihrer Einreichung zu verkürzen.
1. Hintergrund: Das Q-Submission-Programm
Pre-Submissions bieten für Hersteller eine Möglichkeit, im Rahmen des Q-Submission-Programms mit der FDA in Verbindung zu treten und Feedback einzuholen. Mithilfe eines Pre-Submission Requests können im Vorfeld einer Produktzulassung konkrete Fragen besprochen werden.
Die FDA unterscheidet verschiedene Q-Submission-Typen:
- Pre-Submissions
- Submission Issue Requests (SIR)
- Study Risk Determinations (SRD)
- Informal Meetings
- Other Types: Diese Q-Submissions eigenen sich z. B. in Zusammenhang mit
- Breakthrough Devices,
- PMA,
- Klassifizierung von Zubehör oder
- klinischen Studien.
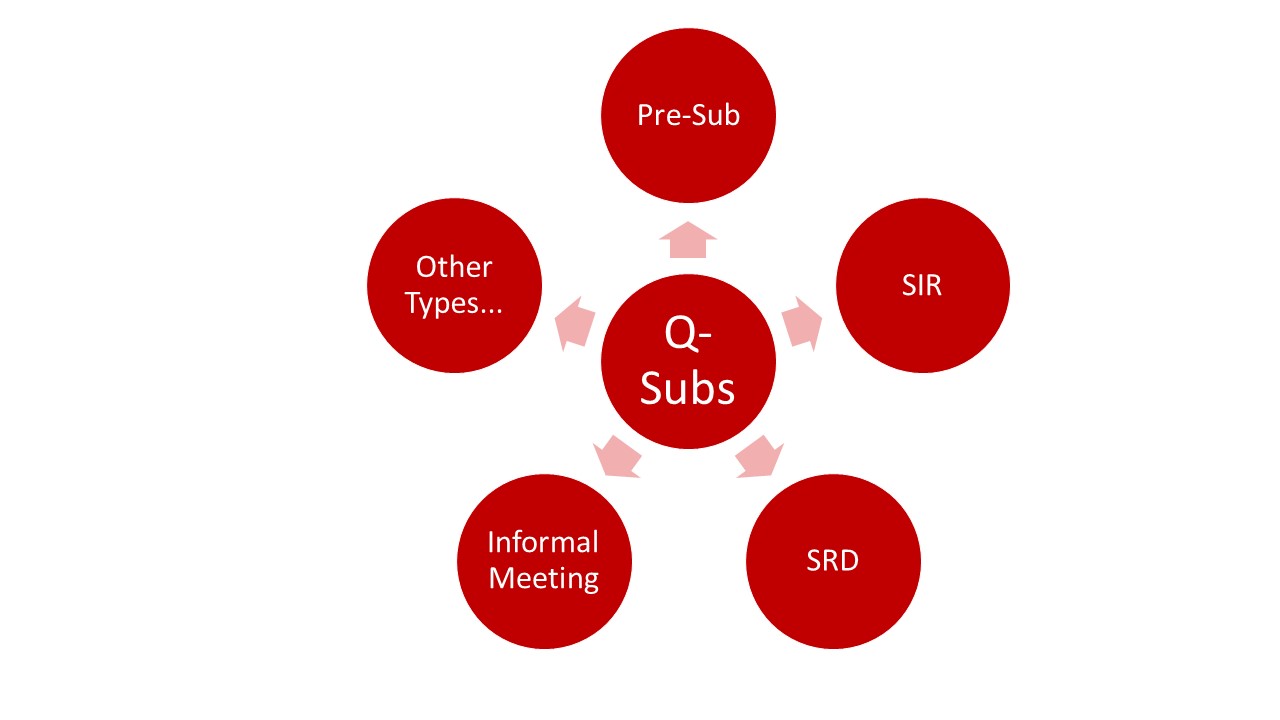
Der folgende Artikel bezieht sich auf die Möglichkeiten der Pre-Submission Requests.
2. Wann Sie ein Pre-Sub durchlaufen sollten und wann nicht
Die FDA erlaubt bei allen Formen der Zulassung, dass Hersteller einen Pre-Submission Request stellen:
- IDE – Investigational Device Exemption
- 510(k) PMN –Premarket Notification (es gibt mehrere 510(k)-Varianten)
- PMA –Premarket Approval
- HDE – Humanitarian Device Exemption
- De Novo Request
- CLIA –Clinical Laboratory Improvement Amendments
- Breakthrough Devices
Einen Pre-Submission Request können Sie einreichen, wenn Sie im Vorfeld der Einreichung konkrete Fragen haben und sich mit der FDA abstimmen möchten. Anhang 2 des zugehörigen Guidance-Dokuments enthält Beispielfragen, an denen sich Hersteller orientieren können. Das Programm ist übrigens freiwillig und kostenlos.
a) Wann die FDA einen Pre-Submission Request empfiehlt
Besonders bei den folgenden Fragestellungen empfiehlt sich ein Pre-Submission Request:
- Wir planen einen De Novo Request. Stimmt die FDA mit der Ansicht überein, dass keine geeigneten Predicate Devices zur Verfügung stehen?
- Hält die FDA das Predicate Device für ausreichend äquivalent?
- Stimmt die FDA mit der Ansicht überein, dass, basierend auf unserer regulatorischen Strategie und den präklinischen Daten, eine klinische Studie nicht notwendig ist?
- Ist die FDA mit dem geplanten Studiendesign einverstanden?
- Hält die FDA die geplanten Usability-Studie für angemessen, um Nachweise zur Sicherheit zu sammeln?
- Ist der Documentation Level bei der Software richtig klassifiziert?
- Wurden Cybersecurity-Management-Plan ausreichend für eine geplante Einreichung? Falls nicht, kann die FDA uns fehlende Informationen mitteilen?
- Hält die FDA, basierend auf den eingereichten Unterlagen, unseren Predetermined Change Control Plan (PCCP) für angemessen?
Besonders bei einer unklaren Zulassungsstrategie, beim Einsatz neuer Technologien oder bei „first-of-kind devices“ sollten Hersteller die Pre-Subs nutzen. Das gilt auch, wenn Regularien nicht ganz klar oder nicht genau anwendbar sind.
b) Wann die FDA einen Pre-Submission Request ablehnt
Die FDA möchte keine Zeit vertun und lehnt Pre-Sub Requests beispielsweise in den folgenden Fällen ab:
- Der Hersteller stellt keine spezifischen Fragen.
- Die Antwort könnte auch ein Reviewer der FDA geben.
- Es geht (nur) um die Klassifizierung des Produkts. Dazu müssen Hersteller einen Antrag gemäß 513(g) stellen.
- Die Anfrage entspricht einem anderen Typ der Q-Submission.
3. Inhalt eines Pre-Sub Requests
Die Inhalte eines Pre-Submission Requests hat die FDA genau festgelegt. So möchte sie gewährleisten, dass der Prozess für alle beteiligten Parteien effizient und zielführend ist. Das zugehörige Guidance-Dokument heißt Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program und fordert die folgenden Inhalte:
- Anschreiben (Cover letter)
- Typ des Q-Submission Requests
- Kontaktdaten des Herstellers (Sponsor)
- Name des Medizinprodukts
- Grund für die Frage nach Feedback
- CDRH Premarket Review Submission Cover Sheet (Form FDA 3514)
- Inhaltsverzeichnis
- Genaue Beschreibung des Medizinprodukts einschließlich Zweckbestimmung
- Historie der bisherigen Zulassungen des Produkts oder der bisherigen Kommunikation mit der FDA
- Überblick über die Produktentwicklung inklusive (geplanter) Teststrategie
- Liste der Fragen, zu denen man Antworten wünscht
- Gewünschter Kommunikationskanal (persönlich, schriftlich, telefonisch)
- Drei Terminvorschläge für das Meeting bzw. Telefonat
- Geplante Teilnehmer
4. Typischer Ablauf des Pre-Submission-Verfahrens
Eine Pre-Submission ist ein formalisiertes Verfahren. Die FDA beantwortet die Fragen der Hersteller
- im Rahmen einer Telefonkonferenz (von der FDA bevorzugt),
- schriftlich oder
- während eines persönlichen Treffens („Face to Face Meeting“).
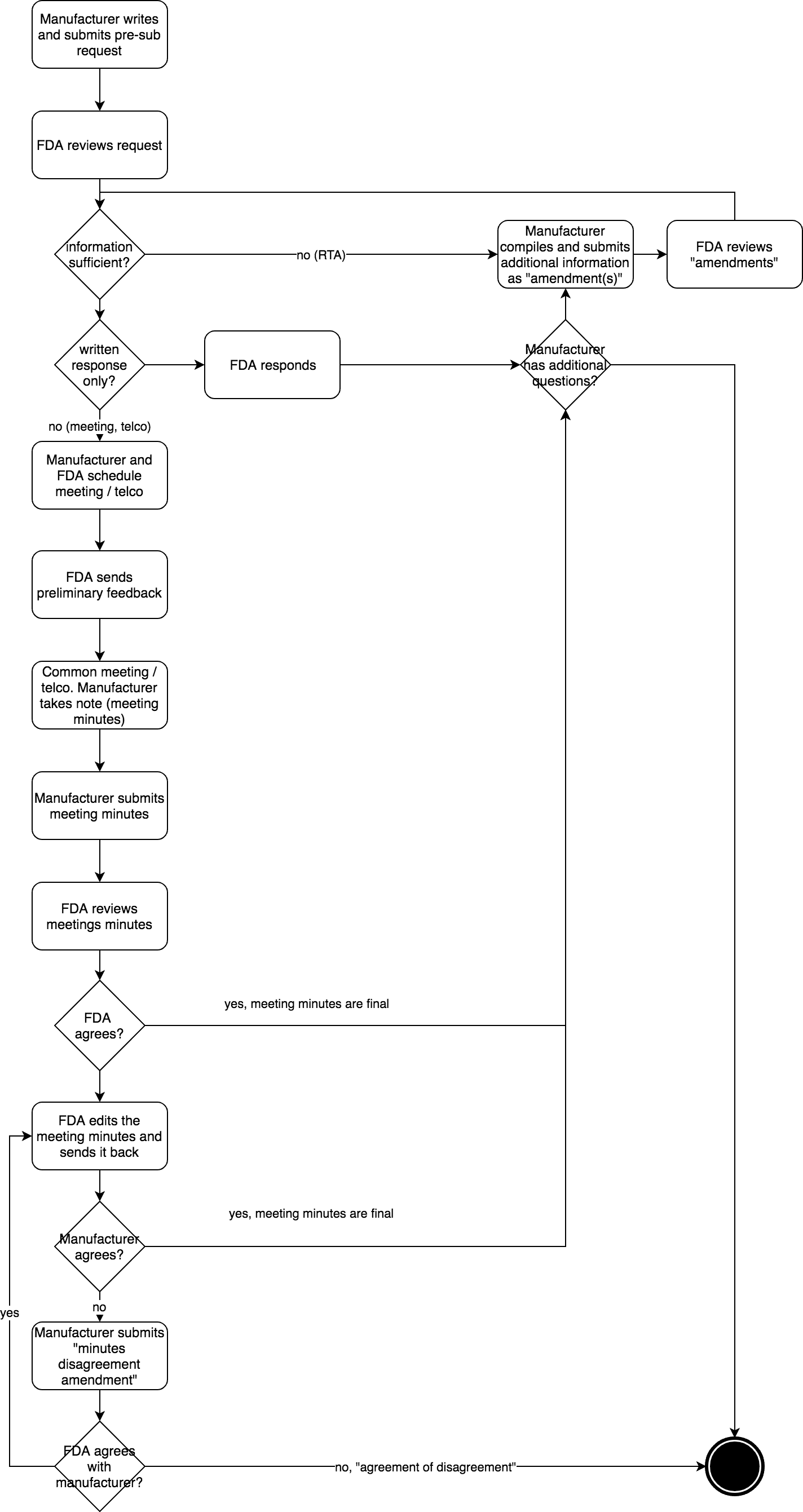
Auch der Ablauf eines Pre-Submission-Verfahrens ist klar festgelegt (s. Abb. 2):
- Der Hersteller verfasst und übermittelt die Pre-Submission an die FDA im eCopy-Format oder im eStar-Format. Diese enthält einen Cover Letter und das CDRH Premarket Review Submission Cover Sheet. Der Cover Letter enthält alle Informationen, die die FDA benötigt, um die Fragen des Herstellers zu beantworten und ggf. ein Meeting zu planen.
- Die FDA reagiert innerhalb von 15 Tagen und legt fest, ob der Hersteller den richtigen Q-Submission-Typ gewählt hat und ob alle notwendigen Informationen vorliegen. Falls nicht, wird die FDA die fehlenden Informationen anfordern oder die Pre-Submission ablehnen. Dieses Review durch die FDA nennt sich RTA (Refuse to Accept). Die FDA nutzt dazu die Checkliste aus Anhang 1 des Guidance-Dokuments. Der Hersteller reicht ggf. Informationen als „Amendment“ nach. Erst wenn der Hersteller diese fehlenden Informationen geschickt hat, beginnt für die FDA der Ablauf einer Frist von 70 Tagen.
- Beide Parteien vereinbaren das Meetings, welches typischerweise 60 bis 75 Tage nach Erhalt der Pre-Submission stattfindet.
- Die FDA schickt spätestens fünf Tage vor dem Meeting ihre vorläufigen Antworten. Etwaige Präsentationsunterlagen müssen der FDA spätestens zwei Tage vor dem Meeting per E-Mail zugesandt werden.
- Das Meeting findet statt (meist einstündig, mehr Zeit bedarf einer Begründung). Der Hersteller ist verpflichtet, das Meeting zu dokumentieren und die Meetingnotizen an die FDA weiterzuleiten.
- Der Hersteller schickt die Gesprächsnotizen als Amendment der ursprünglichen Pre-Submission im eCopy-Format innerhalb von 15 Tagen an die FDA.
- Die FDA korrigiert ggf. diese Notizen (innerhalb von 30 Tagen) , wenn ihr Verständnis des Geschriebenen nicht mit dem des Herstellers übereinstimmt. Andernfalls akzeptiert sie die Notizen per E-Mail.
- Falls der Hersteller dann innerhalb von 15 Tagen nicht widerspricht, ist das Gesprächsprotokoll „amtlich“. Andernfalls widerspricht er mit einem „Minutes Disagreement Amendment“.
- Nach 75 bis 90 Tagen hat der Hersteller dann das Feedback der FDA, das weitere Anmerkungen und Bedenken der Behörde enthalten kann.
Q-Submissions müssen gemäß dem eCopy-Programm oder per eSTAR (über das PreSTAR-Template) eingereicht werden. Nur wenn die Einreichung den Anforderungen an eCopies entspricht bzw. das PreSTAR vollständig ausgefüllt wurde, beginnt die FDA mit deren Bearbeitung.
Hersteller dürfen im Laufe des Verfahrens Unterlagen ergänzen. Diese „Amendments“ tragen die „Q-Nummer“ und den Post-Fix „/A001“, „/A002“ usw. Stellt der Hersteller zusätzliche Fragen, erfolgt dies in Form von „Supplements“ mit dem Post-Fix „/S001“.
Wenn der Hersteller allerdings das Gerät (v. a. dessen Zweckbestimmung) substanziell ändert oder wenn das Q-Submission-Verfahren selbst geändert wird (z. B. von Pre-Sub auf „Determination Meeting“), vergibt die FDA eine neue Q-Nummer.
5. Fehler, die Sie bei Pre-Submission-Meetings vermeiden sollten
Das Johner Institut beobachtet bei den Pre-Submissions immer wieder Fehler durch die Hersteller. Diese sollten Sie unbedingt vermeiden, um sich mit den Pre-Subs nicht mehr zu schaden als zu nutzen:
a) Fehlende Klarheit über die eigenen Ziele
Zu Beginn eines jeden Prozesses sollten die Ziele absolut klar sein. Bei einem Pre-Submission Request bedeutet das, dass der Hersteller seine Fragen absolut präzise formuliert. Nur dann kann die FDA genauso präzise antworten. Und nur dann ist auch klar, welche Informationen notwendig sind, um die Antworten geben zu können.
Die FDA schätzt es nicht, wenn sich Hersteller iterativ an die eigentlichen Fragestellung „heranarbeiten“ und scheibchenweise Daten liefern. Vielmehr möchte sie ein Thema innerhalb einer Iteration abarbeiten.
Die FDA erlaubt allerdings, weitere Fragen innerhalb des gleichen Verfahrens (identische Q-Nummer) zu ergänzen.
b) Unklare Strategie
Es ist unerlässlich, dass der Hersteller konsequent eine klare und einfach verständliche Argumentationslinie verfolget. Sonst überlässt er es der FDA, sich einen Reim zu machen, und das geht nicht immer gut.
Der Hersteller sollte vor dem Treffen wissen, welche Tests und klinischen Studien er durchführen will und weshalb er glaubt, dass diese ausreichen. Andernfalls lässt er sich von der FDA zu weiteren Tests „überreden“, was enorme Auswirkungen auf den Zeitplan und die Kosten hat.
Zu einer klugen Strategie zählt auch die richtige Form der Interaktion mit der FDA. Das Johner Institut empfiehlt bei De-Novo-Verfahren und bei PMAs persönliche Gespräche (in Washington), in vielen anderen Fällen Telefonkonferenzen.
c) Schlechte Vorbereitung und Abstimmung
Der wahrscheinlich häufigste Fehler besteht in einer unzureichenden Vorbereitung.
Es darf nicht vorkommen, dass beim Pre-Submission-Meeting Informationen oder gar die Experten des Herstellers fehlen und dann nicht alle Fragen beantwortet werden können (z. B. die Kliniker-Fragen zu geplanten Studien oder die Entwickler-Fragen zur Technologie). Noch schlimmer ist es, wenn sich die Experten im Meeting widersprechen.
Ein Meeting muss geübt werden – fast wie ein Theaterstück, bei dem alle Akteure jederzeit wissen, was zu sagen oder auch nicht zu sagen ist.
Wenn die Hersteller den größten Teil des Meetings mit eigenen Präsentationen „verbraten“, dürfen sie sich nicht wundern, wenn am Schluss keine Zeit mehr für die Antworten der FDA bleibt. Die Agenda ist somit ein wichtiges Element der Vorbereitung.
Ebenso ist es ein Zeichen guter Vorbereitung, wenn der Hersteller eine Person organisiert, deren ausschließliche Aufgabe darin besteht, das Treffen zu protokollieren. Ton- oder gar Videoaufnahmen sind verboten.
d) Nicht zeitnah agieren
Manche Hersteller scheinen nach den Pre-Submission-Meetings so erleichtert oder erschöpft zu sein, dass sie es versäumen, die Fristen zu wahren. Zudem ist es sehr wichtig, das Treffen zu dokumentieren, solange alles noch frisch im Gedächtnis ist – auch bei der FDA.
e) Formale Fehler
Die FDA möchte bei Pre-Submission Requests nur bestimmte Fragestellungen beantworten. Klassifizierungen zählt sie nicht dazu. Das ist das Ziel eines Antrags gemäß 513(g). Fragen, die ein Reviewer direkt beantworten kann, haben in einer Pre-Submission ebenfalls nichts verloren.
Hersteller sollten sich der verschiedenen Typen von Q-Submissions sowie der anderen Verfahren bewusst sein, z. B. der nicht-formale Anfragen und des 3rd-Party-Review-Programms. Letzteres ist aber nicht für alle Medizinprodukte und Zulassungsverfahren möglich.
6. Fazit
Die FDA bietet Herstellern mit dem Pre-Submission-Programm ein effektives und effizientes Verfahren, um ein schnelles und dennoch verbindliches Feedback auf Fragen rund um die Zulassung von Medizinprodukten zu erhalten.
Aus gutem Grund empfiehlt die Behörde selbst das Verfahren. Hersteller sollten es auch nutzen, um Zulassungsrisiken und Zeitverzug zu minimieren und unnötige Aufwände insbesondere bei Studien (auch bei solchen ohne IDE bzw. außerhalb der USA) zu vermeiden.
Natürlich sollten die Hersteller nur dann einen Pre-Submission Request stellen, wenn sie tatsächlich Fragen haben. Sonst kann das Verfahren die Zulassung sogar verzögern.
Das ganze Verfahren muss exakt vorbereitet sein. Auch wenn wir hier „pro patria“ sprechen: Erwägen Sie, sich bei der Vorbereitung und Durchführung von Pre-Submission-Meetings von einem Berater unterstützen zu lassen. Das ist keine große Sache, hilft aber, den Prozess zu beschleunigen und sicherer zu machen. Melden Sie sich gerne.
Änderungshistorie:
- 2024-10-16: Anpassung an die aktuelle Ausgabe des Q-Sub Guidances von 2023
- 2021-10-08: Anpassung an die aktuelle Ausgabe des Q-Sub Guidances von 2021
- 2020-11-06: Überarbeitung der Artikelstruktur, Einfügen aktueller Referenzen, Aktualisierungen basierend auf dem Guidance-Dokument von Mai 2019



Guten Morgen Herr Johner,
könnte es sein, dass unter Abschnitt 2 b) Punkt 3 ein 513(g) statt eines 503(k) gemeint ist?
Viele Grüße aus München
Simon Spiegel
Sehr geehrter Herr Spiegel,
vielen Dank für Ihren Hinweis. Es ist in der Tat ein 513(g) gemeint. Es gibt auch ein 503(g) („Requests for Designation“) für Kombinationsprodukte. Diese werden ebenfalls gesondert angefragt und nicht über eine Q-Submission.
Herzliche Grüße
Luca Salvatore
Grüezi Herr Salvatore
Besten Dank für den wertvollen Artikel. Wir hatten vor kurzem unser Pre-Submission Meeting mit der FDA und wurden darauf hingewiesen, dass wir die Meeting Minutes nicht per E-Mail an den Lead Reviewer senden sollen, sondern (ähnlich wie die ursprüngliche Submission) per Post an die FDA.
Zudem hatten wir nach dem Erhalt des FDA Feedbacks (in unserem Fall 10 Tage vor dem Meeting) die Meeting Agenda und Slides vorgängig an den Lead Reviewer emailen.
Beste Grüsse,
A. Meyer
Sehr geehrter Herr Meyer,
vielen Dank für Ihre Hinweise. Der Artikel hatte in der Tat noch den alten Stand des Q-Sub Guidances als Grundlage. Ich habe diesen nun an das aktuelle Guidance von 2021 angepasst. Dies deckt sich somit mit den von Ihnen geschilderten Erfahrungen.
Herzliche Grüße
Luca Salvatore