
Das Breakthrough Devices Program ist ein Zulassungsverfahren für Medizinprodukten, mit dem die FDA schwerstkranken Patienten einen schnelleren Zugang zu neuartigen Medizinprodukten ermöglichen möchte. Die Behörde hat dazu im Dezember 2018 ein „Guidance Document“ veröffentlicht.
Sie erfahren in diesem Artikel,
- wie das „Breakthrough Devices Program“ abläuft und
- welche Voraussetzungen die Hersteller erfüllen müssen, um daran teilnehmen zu dürfen.
1. Ziele des FDA-Programms für Breakthrough Devices

Die FDA will Herstellern einen Weg zu ebnen, damit diese ihre Medizinprodukte schneller in den Markt bekommen können. Dieses Angebot gilt aber nur für neuartige Produkte für Patienten, die entweder an einer lebensbedrohlichen Krankheit leiden oder an einer Krankheit, die sie irreversibel schwächt oder beeinträchtigt.
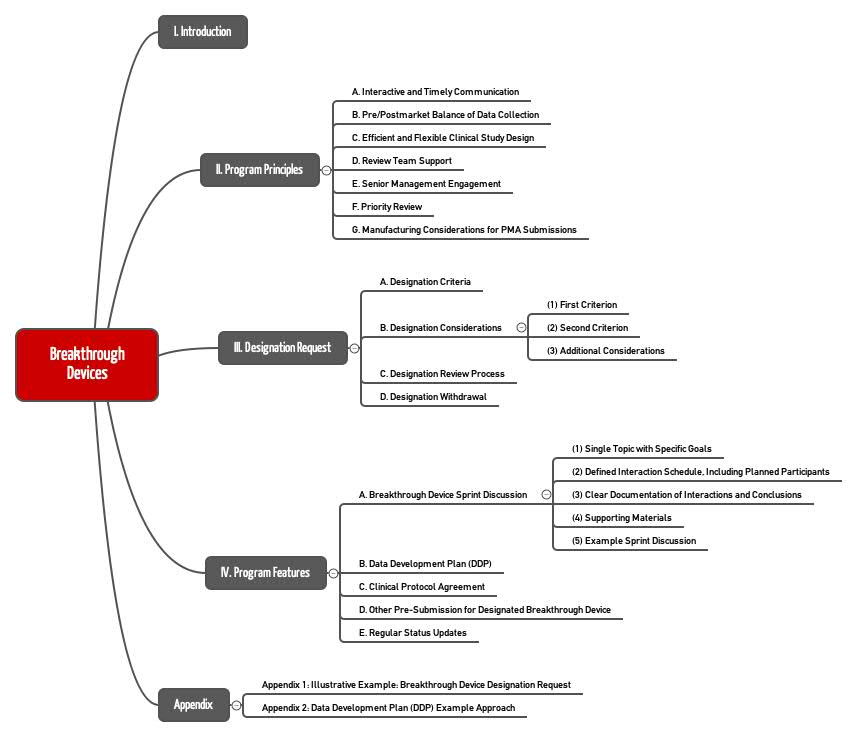
2. „Breakthrough Devices Program“: Die Grundlagen
Das Breakthrough-Programm ist keine neue Form der Zulassung. Es stellt somit keine Alternative zu einer Premarket Notification nach 510(k), zu einem Premarket Approval oder einem De Novo Request dar. Vielmehr schafft die FDA mit dem neuen Programm die Möglichkeit, diese bestehenden Zulassungsverfahren zu beschleunigen.
Lesen Sie hier mehr zu den Zulassungsverfahren Special 510(k) und Abbreviated 510(k).
Das Programm steht für Medizinprodukte und Kombinationsprodukte offen. Es löst die folgenden Programme ab:
- Expedited Access Pathway
- Priority Review
- Innovation Pathway
3. Wie die FDA die Zulassung für Breakthrough Devices beschleunigt
3.1 Der Ansatz
Die FDA stellt den Herstellern, die in dieses Programm aufgenommen wurden, mehrere Verbesserungen in Aussicht:
- Interaktive Kommunikation
Hersteller können direkt mit der FDA interagieren, beispielsweise um den Ablauf klinischer Studien zu klären oder um Zulassungsdokumente zu besprechen. - Höhere Priorität
Die FDA bearbeitet Zulassungen (etwa 510(k), PMAs) mit höherer Priorität. Das Gleiche gilt für Q-Submissions (z. B. Beispiel Pre-Market-Request). Auch Anträge für eine „Investigational Device Exemption“ (IDE) werden mit höherer Priorität bearbeitet. Die IDE ist die Genehmigung, die Produkte für klinische Studien einsetzen zu dürfen. - Flexibilität bei klinischen Studien
Falls keine Sicherheitsbedenken dagegensprechen, gestattet die FDA, einen Teil der Daten erst mit Beginn der Post-Market-Phase zu erheben. Die Behörde ist bereit, angesichts des hohen Nutzens (lebensrettend) temporär höhere Risiken zu akzeptieren. Die FDA gestattet, das Studiendesign während der klinischen Studie und des Zulassungsverfahrens zu adaptieren. - Dedizierte, verfügbare und gut ausgebildete Review Teams
Die FDA stellt ausreichend viel und kompetentes Personal bereit. Die Kompetenz bezieht sich auch auf die Anwendung des Programms für Breakthrough Devices. Selbst das höhere FDA-Management steht bereit. - Qualitätsmanagement
Selbst beim Qualitätsmanagementsystem ist die FDA gegebenenfalls bereit, ein Auge zuzudrücken, indem sie weniger Dokumenten einfordert („FDA may accept less quality system and manufacturing information“) oder nicht auf einer „preapproval inspection“ besteht.
Die Zusammenarbeit ist somit schneller und interaktiver – eher vergleichbar mit einem agilen Ansatz.
3.2 Der Ablauf des Breakthrough Device Programs
Das Programm läuft in zwei Phasen ab. In der ersten Phase muss der Hersteller einen Antrag stellen, in dem er darlegt, weshalb er die o. g. Voraussetzungen erfüllt. Spätestens nach 60 Kalendertagen entscheidet die FDA über den Antrag, wobei sie bei Bedarf nach 30 Tagen zusätzliche Informationen anfragt.

In der zweiten Phase arbeiten die FDA und der Hersteller in Sprints zusammen. Dabei räumt die FDA dem Hersteller nicht nur eine höhere Verfügbarkeit, sondern dem „Projekt“ auch eine höhere Priorität ein als bei anderen Zulassungsverfahren.
Die FDA hat ein ausführliches Guidance Document herausgegeben, das Sie hier herunterladen können.
4. Voraussetzung für die Teilnahme
Damit ein Hersteller sein Produkt mithilfe des Breakthrough-Devices-Programms zulassen darf, muss er mehrere Voraussetzungen erfüllen.
- Das Medizinprodukt bietet eine bessere Behandlung oder Diagnose von lebensbedrohlichen oder irreversibel schädigenden Krankheiten oder Gesundheitszuständen. Beispiele sind Schlaganfall, Herzinfarkt, Krebs, schwere Traumata und ALS.
- Mindestens
eines der folgenden Kriterien muss erfüllt sein:
- Das Gerät stellt einen technologischen Durchbruch dar (Breakthrough Technology). Ein Beispiel wäre ein neuer Gentest, mit dem die Behandlungsoptionen besser abgewogen werden können.
- Es gibt keine zugelassenen Alternativen. So gibt es derzeit – nach Aussage der FDA – noch kein Gerät, das Parkinson direkt diagnostizieren kann.
- Das Produkt bietet im Vergleich zu zugelassenen Alternativen eine signifikante Verbesserung.
- Das Produkt ist im besten Interesse der Patienten. Das wäre der Fall, wenn die Alternativen große Nebenwirkungen haben oder wenn das neue Produkt die diagnostischen Möglichkeiten und die Anwendbarkeit deutlich verbessert.
Wenn Sie unsicher sind, ob Ihr Produkt sich für das „Breakthrough Device Program“ qualifizieren kann, melden Sie sich bitte. Unsere FDA-Experten in Deutschland und den USA können die Chancen rasch abschätzen.
5. Fazit
Einmal mehr zeigt die FDA, dass sie in der Lage ist, Nutzen und Risiken wirklich abzuwägen. Sie hat erkannt, dass man durch ein Verwehren oder Verzögern der Zulassung von Medizinprodukten Patienten ebenso schädigen kann wie durch Medizinprodukte, die nicht ausreichend sicher oder leistungsfähig sind.
Ein solches Programm wäre auch in Europa wünschenswert. Leider beschreitet Europa mit der MDR genau den entgegengesetzten Weg.
Das Johner Institut unterstützt Medizinproduktehersteller weltweit bei der Zulassung.
Es hilft Ihnen bei der Auswahl des passenden Zulassungsverfahrens und unterstützt Ihre Kommunikation mit Behörden wie der FDA.
Melden Sie sich hier, falls Sie Ihr Medizinprodukt möglichst schnell und ohne unnötige Kosten und Aufwände in den USA vermarkten wollen.
Änderungshistorie
- 2024-02-25: Redaktionelle Änderungen, Kapitel neu nummeriert und aufgeteilt
- 2019-01-15: Erste Version des Artikels veröffentlicht


