
Die „Abbreviated 510 (k)“ ist eines der drei 510(k)-Zulassungsverfahren, die die FDA anbietet.
Dieser Artikel verrät Ihnen,
- welche Arbeit Sie sich bei der Zulassung durch die „Abbreviation“ sparen können,
- welche Voraussetzungen Sie dafür erfüllen müssen und
- weshalb der Begriff „abbreviated“ irreführend sein kann.
1. Abbreviated 510(k) und andere Zulassungsverfahren
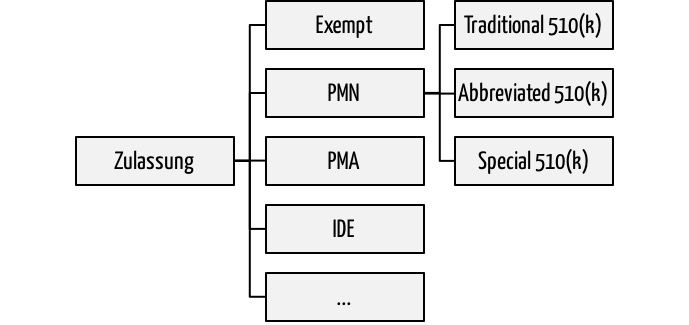
Wollen Hersteller ihre Medizinprodukte in den USA vermarkten, müssen sie eines der Zulassungsverfahren durchlaufen. Zu den am häufigsten angewendeten zählen die Premarket Notifications PMN. Diese sind in Anlehnung an den entsprechenden Artikel im Food Drug and Cosmetic Act besser als 510(k) bekannt. Bei diesen 510(k)-Verfahren unterscheidet die FDA drei:
- Traditional 510(k): Das klassische Zulassungsverfahren bei „me too“ Produkten
- Abbreviated 510(k): Das abgekürzte Zulassungsverfahren unter bestimmten Umständen
- Special 510(k): Vereinfachte Neuzulassung des eigenen Produkts(!) bei Produktänderungen
Lesen Sie hier mehr zur Zulassung nach 510(k) und zur „Special 510(k)„. Beachten Sie v.a. das Guidance Document der FDA „The Abbreviated 510(k) Program“.
2. Abbreviated 510(k): Was Sie im Vergleich zur Traditional 510(k) sparen
Keine verkürzte Zeitdauer
Der Begriff „abbreviated“ lässt vermuten, dass das Zulassungsverfahren kürzer dauern würde. Das stimmt leider nicht ganz. Eine verkürzte Dauer von 30 Tagen gilt bei den „Special 510(k)s“, aber nicht bei den „Abbreviated 510(k)s“. Letztere haben den gleichen zeitlichen Verlauf wie die „Traditional 510(k)“.
Weniger Dokumentation
Auch die Hoffnung, dass die Dokumentation bei einer „Abbreviated 510(k)“ kürzer als bei einer „Traditional 510(k)“ sei, scheint auf den ersten Blick zu trügen. Das Format beider Zulassungsverfahren, d.h. die Kapitelstruktur, unterscheidet sich nicht(!).
Lesen Sie hier mehr zu der Kapitelstruktur, die die FDA bei Traditional 510(k) (und Abbreviated 510(k)) fordert.
Statt ausführlicher Unterlagen dürfen die Hersteller bei einer „Abbreviated 510(k)“ Zusammenfassungen, Verweise auf eingehaltene Normen (Konformitätserklärungen) einreichen. D.h. das Kapitel „9. Declarations of Conformity and Summary Reports“ kann bei einer Abbreviated 510(k) etwas umfangreicher, die Kapitel 14 bis 19 können kürzer ausfallen.
Weil dies weniger Dokumentation bedeutet, die ein FDA Reviewer prüfen muss, kann ein Bescheid doch schneller als bei einer „Traditional 510(k)“ erfolgen. Nur verpflichten möchte sich die Behörde nicht dazu.
Gleiche Kosten
Wer hofft, dass sich der niedrigere Aufwand für die Behörde in niedrigeren Gebühren niederschlägt, wird enttäuscht. Die Gebühren sind für alle 510(k)-Verfahren gleich.
3. Wann Sie eine Abbreviated 510(k) nutzen dürfen
Voraussetzungen
Eine Abbreviated 510(k) ist dann erlaubt, wenn eine der folgenden drei Bedingungen erfüllt ist:
- Die FDA hat ein Guidance Document für das entsprechende Produkt oder dessen Technologie veröffentlicht.
- Der Hersteller wendet die „Special Controls“ an. Beispiele dafür sind die Marktbeobachtung (Post-Market Surveillance), die Einhaltung von Entwicklungsrichtlinien oder bestimmte Normen. Welche dieser Special Controls zu befolgen sind, regelt die „Product Classification Database“.
- Die FDA hat relevante Normen anerkannt, die der Hersteller befolgt. Die FDA führt über 400 solcher „recognized standards“ in ihrer Datenbank.
Beispiele
- Beispielsweise listet die FDA für Kernspingeräte 15 „Consensus Standards“. Ein Qualitätsmanagementsystem ist verpflichtend, weitere Special Controls werden nicht explizit genannt. Eine Übersichtsseite nennt jedoch weitere Normen und Guidance Documents.
- Für implantierbare RFID Transponder nennt die FDA die „Special Controls“, für die sie sogar ein eignes Guidance Document publiziert hat.
- Für digitale Mammographie-Systeme listet die FDA vier Normen und verweist auf das zugehörige Guidance Document mit den Special Controls. Zu diesen Controls zählen die Prüfung der elektrischen Sicherheit (nach IEC 60601-1), die Prüfung der Biokompatibilität, das Testen mit Phantomen u.v.m.
Ein europäischer Ansatz
Der Gedanke hinter der Abbreviated 510(k) besteht darin, dass die FDA nicht anhand der Unterlagen des Herstellers selbst beurteilen muss, ob das Produkt den regulatorischen Anforderungen z. B. an die Produktsicherheit genügt. Vielmehr geht die Behörde davon aus, dass dies der Fall ist, wenn der Hersteller die entsprechenden Normen, Guidance Documents bzw. anderen Best Practices erfüllt bzw. befolgt.
Dies entspricht dem Ansatz der europäischen Medizinprodukterichtlinien und Verordnungen, die ebenfalls erlauben, die Konformität zu vermuten, wenn die Hersteller harmonisierte Normen und gemeinsame Spezifikationen (Common Specifications) befolgen.
4. Ablauf des Verfahrens
Konformitätserklärung
Wendet der Hersteller einen „recognized standard“ an, d.h. erklärt er die Konformität mit einer Norm (oder mehreren), so muss er die in Form einer „Declaration of Conformity“ tun, die folgende Angaben enthält:
- Norm(en) (eindeutig identifziert)
- Erklärung, dass die Norm(en) eingehalten wurden (von nicht anwendbaren Anforderungen abgesehen)
- Liste der nicht anwendbaren Anforderungen
- Darlegung aller Abweichungen beim Anwenden der Norm z. B. beim Testen
- Beschreibung aller Unterschiede zwischen dem getesteten Gerät und den Geräten, die in den Markt gebracht werden
- Kontaktdaten des Testlabors
Die FDA betont, dass der Hersteller die Verantwortung für die Konformität trägt und diese nicht auf ein Testlabor abwälzen kann.
Ablehnung
Wenn die FDA eine 510(k)-Premarket-Notification eines Herstellers als nicht für das „Abbreviated“ Programm geeignet hält, bietet die Behörde an, das Verfahren in ein „Traditional“ Verfahren umzuwandeln. Allerdings muss der Hersteller dann davon ausgehen, dass die FDA weitere Dokumente anfordert.
5. Draft Guidance Document „Expansion of the Abbreviated 510(k) 2 Program“
Zielsetzung
Am 12. April hat die FDA einen Entwurf für ein neues Guidance Document veröffentlich. Es trägt den Titel „Expansion of the Abbreviated 510(k) Program: Demonstrating Substantial Equivalence through Performance Criteria”. Das Ziel besteht darin, den Aufwand für 510(k)-Verfahren sowohl für die Hersteller als auch für die Behörde zu verringern.
Dieser Aufwand entsteht v.a. durch den Nachweis, dass das neu zuzulassende Produkt genauso sicher und leistungsfähig ist wie ein Vergleichsprodukt, das „Predicate Device“. Die FDA möchte zunehmend den Nachweis dadurch ermöglichen, dass die Hersteller die Äquivalenz nicht mehr durch direkten Vergleich und direkte Messungen nachweisen. Vielmehr soll es Ihnen gestattet sein, auf Normen, „Special Controls“ und Guidance Dokumente zu verweisen.
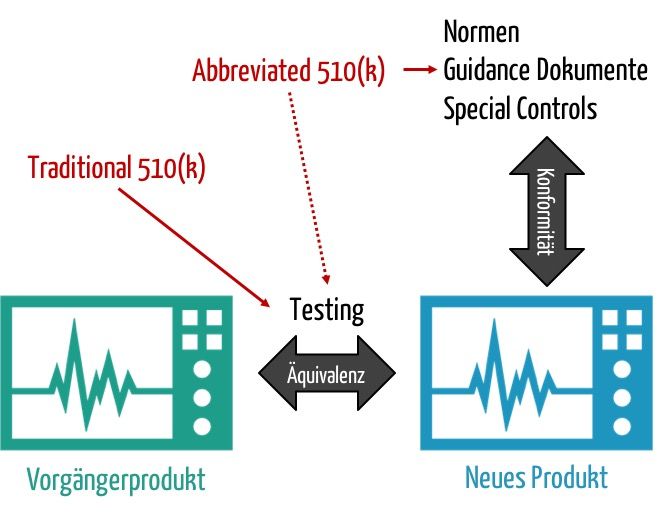
Die FDA beabsichtigt somit, dass noch mehr Zulassungen über das „Abbreviated Programm“ möglich werden. Die Behörde plant aber keine Änderung am Format der Unterlagen.
Voraussetzungen
Als Voraussetzung, dass die Hersteller an diesem erweiterten „abbreviated“ Programm teilnehmen können, sieht die FDA (weiterhin):
- Das neue Produkt hat eine Zweckbestimmung und technische Charakteristiken, die keine neuen Fragen bezüglich Sicherheit und Wirksamkeit aufwerfen, die über die des Äquivalenzprodukts hinausgehen.
- Die Leistungskriterien entsprechen denen des bzw. der legal vermarkteten Produkte des gleichen Typs.
- Das Produkt erfüllt auch diese Leistungskriterien.
Produkte, die unter dieses erweiterte Zulassungsverfahren fallen
Um herauszufinden, ob ein Medizinprodukt im Rahmen dieses erweiterten Programms zugelassen werden darf, bietet die FDA Unterstützung im Rahmen der Pre-Submission Meetings an.
Künftig möchte die FDA eine Liste an Produkten publizieren, die sich für dieses erweiterte Programm eignen. Sie möchte auch die zugehörigen Voraussetzungen (Normen, Special Controls, Guidance Documents) für jedes dieser Produkte benennen.
6. Fazit und Zusammenfassung
Speziell Hersteller, die Normen befolgen, sollten ein „Abbreviated 510(k)“ Verfahren anstreben. Sie ersparen sich dadurch den Aufwand, viele Dokumente nochmals für die FDA zusammenstellen und Nachweise führen zu müssen, die beispielsweise durch eine Prüfung durch ein Testhaus bereits geführt wurden.
Eine Voraussetzung ist, dass die Normen von der FDA als „recognized standard“ gelistet sind. Alternativ bzw. zusätzlich müssen die Hersteller die „Guidance Documents“ der FDA beachten.
Speziell europäische Hersteller, die bereits nach harmonisierten Normen arbeiten, bietet sich das Verfahren an. Selbst wenn die FDA den Weg der „Abbreviated 510(k) nicht anerkennt, ist nichts verloren. Das Verfahren lässt sich als „Traditional 510(k)“ weiterführen.
Das neue Guidance Document zur Abbreviated 510(k) lässt erkennen, dass die FDA diesem Zulassungsverfahren zunehmend mehr Bedeutung schenken will. Das ist aus Sicht der Hersteller sehr zu begrüßen.


