
Mit der „Special 510(k)“, der „Abbreviated 510(k)“ und der „Traditional 510(k)“ unterscheidet die FDA drei 510(k)-Zulassungsverfahren. Mit der „Special 510(k)“ möchte die Behörde bei kleinen Änderungen der Produkte die Zulassung vereinfachen.
Lesen Sie hier, welche Aufwände Sie bei der Zulassung mit einer Special 510(k) sparen können und welche Voraussetzungen Sie dafür erfüllen müssen.
1. Regulatorischer Hintergrund: 510(k) bei Produktänderung
Die sogenannten 510(k)-Zulassungsverfahren nennt man auch Premarket Notification (PMN) Submissions. Der 21 CFR part 807.81 legt fest, wann Hersteller eine PMN einreichen müssen. Das ist auch der Fall, wenn ein Produkt signifikant geändert wurde. Was eine signifikante Änderung ist, beschreit der 807.81 wie folgt:
- A change or modification in the device that could significantly affect the safety or effectiveness of the device, e.g., a significant change or modification in design, material, chemical composition, energy source, or manufacturing process.
- A major change or modification in the intended use of the device.
Zudem hat die FDA ein Guidance Dokument Deciding When to Submit a 510(k) for a Change to an Existing Device veröffentlicht. Dieses nennt Kriterien, wann bei Änderungen am Produkt eine erneute 510(k)-Submission notwendig wird.
2. Voraussetzungen für die Special 510(k)
a) Übersicht
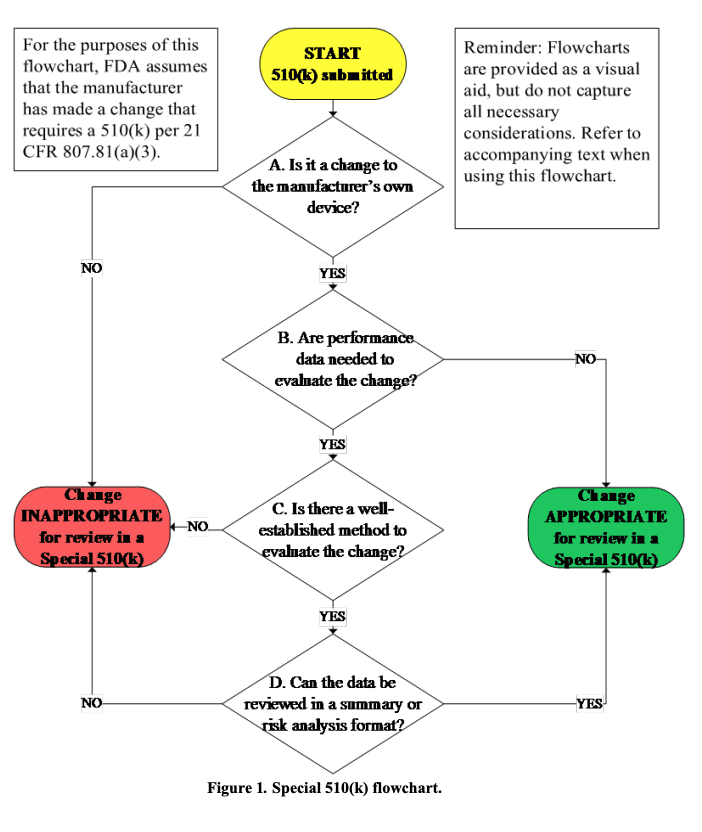
In einen ersten Schritt müssen somit die Hersteller überprüfen, ob bei einer Änderungen des eigenen Produkts überhaupt einer erneuten 510(k)-Submission bedarf. Wenn dies der Fall ist, bietet die FDA mit der Special 510(k) eine Vereinfachung an. Allerdings nur, falls die folgenden Voraussetzungen erfüllt sind:
- Die Änderung betrifft ein Produkt, das der gleiche Hersteller bereits legal vermarktet hat.
- Entweder bedarf es keiner Leistungsdaten („Performance Data“) oder diese können mit „well-established methods“, um diese Änderungen zu bewerten.
- Alle Leistungsdaten für den Nachweis der „substantial equivalence“ (mit dem predicate device(!)) lassen sich in einer Zusammenfassung oder in der Risikoanalyse bewerten.
Diese Überlegungen finden sich auch im Entscheidungsdiagramm, das die FDA im Guidance Document „The Special 510(k) Program“ publiziert hat:
b) Leistungsdaten
Hersteller müssen ihre Produkte nach Änderungen verifizieren und validieren. Allerdings sind nicht alle dieser „Testergebnisse“ notwendig, um die Äquivalenz mit einem „substantial equivalent predicate device“ nachzuweisen.
Als Beispiel führt die FDA ein Produkt an, das der Hersteller zuvor als „Safety in MR Imaging Not Evaluated“ und nun als „Magnetic Resonance (MR) Unsafe“ kennzeichnet. Offensichtlich bedarf es keiner Leistungsdaten, um nachzuweisen, dass das Produkt nicht geeignet für die Anwendung in einem Kernspingerät ist.
Umgekehrt können erneute EMV-Prüfungen notwendig werden, wenn der Hersteller neue Nutzungsumgebungen vorsieht z.B. der Einsatz in einem OP, bei dem HF-Chirurgiegeräte zum Einsatz kommen.
c) „Well-established Methods“
Falls Leistungsdaten notwendig sind, wünscht sich die FDA „well-established methods“ zum Nachweis. Zu diesen Methoden zählen Methoden, die
- die FDA bereits in vorangegangenen 510(k) Zulassungsverfahren akzeptierte,
- in „consensus standards“ der FDA beschrieben sind,
- die FDA in den eigenen Guidance Documents nennt oder
- sonst in der wissenschaftlichen Literatur den Stand der Technik reflektieren.
Beispielsweise dürften EMV-Prüfungen gemäß IEC 60601-1 zu diesen „well-established methods“ zählen.
d) Bewertung der Daten in Zusammenfassung oder Risikoanalyse
Zusätzlich erwartet die FDA, dass sich die Ergebnisse dieser Prüfungen in einer kurzen Zusammenfassung wiedergeben lassen. Das hängt möglicherweise damit zusammen, dass die FDA es sonst nicht schaffen würde, den Antrag in der kurzen Zeit zu bewerten.
Die FDA beschreibt auch, wie sie sich diese Zusammenfassungen wünscht: In einer „Risikotabelle“:
| Änderung | Risiko | Methode(n) für V&V | Akzeptanz-kriterien | Ergebnisse (Zusammen-fassung) |
| Material-änderung auf XY | Allergische Reaktion | Biokompatbilitäts-prüfung wie in Guidance Document XY beschreiben und/oder Cytoxitätstest konform mit ISO 10993-5 | Reaktitätsgrad = 0 (wie bei ursprünglichem Produkt) | Grad = 0. Artikel ist nicht zytotoxisch |
e) Voraussetzungen vor September 2019
Hinweis: Vor September 2019 existierten höhere Voraussetzungen für eine Special 510(k). Die FDA erlaubte sie nur unter diesen Umständen:
- „The modification does not affect the intended use of the device or alter the fundamental scientific technology of the device“.
- Der Hersteller hat mit einer Risikoanalyse sowie mit Verifizierungs- und Validierungsaktivitäten sichergestellt, dass die „Design Outputs“ (noch immer) die „Design Input Requirements“ erfüllen.
3. Special 510(k): Worin die Vereinfachungen bestehen
a) Umfang der einzureichenden Unterlagen
Mit der Special 510(k) können die Hersteller auf die Einreichung eines Teils der Unterlagen verzichten:
| Dokument | Traditional 510(k) | Special 510(k) |
|---|---|---|
| Coversheet | X | X (Hinweis, dass Special 510(k) |
Basisinformationen gemäß 21 CFR part 807.87 z.B.
| X | X |
| Verweis auf bestehende 510(k) Clearance | X | |
„Declaration of Conformity“, die bestätigt, dass
| X | |
| Spezifikation, Beschreibung des physischen Produkts | X | |
Performance-Daten z.B. Beschreibung der Methodik und Ergebnisse von.
| X | |
„Software-Akte“, deren Inhalt vom „level of concern“ abhängt, z.B. mit
| X | |
| Nachweis der Äquivalenz mit „predicate device“ | X |
b) Reaktionszeit der FDA
Zudem beabsichtigt die FDA die Special 510(k) Einreichungen innerhalb von 30 Tagen zu bearbeiten.
Lesen Sie hier mehr zum Thema Abbreviated 510(k)
4. Aktuelles
September 2019: Die FDA hat das Guidance Document in der finalen Version publiziert.



Kann man sich bei einer Special 510(k) auf ein Fremdprodukt beziehen oder werden immer die Modifikationen des eigenen Produktes betrachtet?
Herzlichen Dank
Antje Katzer
Die Modifikationen beziehen sich auf das eigene Produkt. Eine Modifikation eines anderen Produkts hat keine Auswirkung auf die Zulassung des eigenen.
Hallo!
Vielen Dank für die Info. Ich habe aber folgende Fragen:
Kann man wirklich bei einer Special 510(k) auf dem Nachweis der Äquivalenz mit „predicate device“ verzichten?
Reicht es mit einer unterschriebenen Aussage, dass Design Controls implementiert sind, oder muss man Dokumente zu den Risiko Analyse und V&V einreichen?
Wo liegt man die Änderung an dem Gerät fest?
Herzlichen Dank,
Ara Alcala
Die Special 510(k) nutzen Sie nur bei Änderungen zu einem bereits über ein 510(k)-Verfahren zugelassenen Produkt. Für dieses wiederum war ein Predicate Device Voraussetzung (s. Ablaufdiagramm).