
Das De-Novo-Verfahren, die FDA spricht auch vom „de novo program“ und vom „de novo submission process“, ist eines der Zulassungsverfahren für Medizinprodukte in den USA.
Dieses Verfahren können Hersteller für neuartige Produkte anwenden – wie der Name „de novo“ bereits vermuten lässt.
Hersteller sollten also einen De-Novo-Antrag für ein Produkt stellen, für das es kein vergleichbares Vorgängerprodukt („predicate device“) gibt, das die FDA aber wahrscheinlich in die Klasse I oder Klasse II klassifizieren würde.
1. Hintergrund und Einführung
a) Problemstellung
Für neuartige Produkte dürfen Hersteller das 510(k)-Zulassungsverfahren nicht durchlaufen, denn es fehlt ein vergleichbares Vorgängerprodukt. Für diese neuartigen Produkte hat die FDA auch noch keine Klasse festgelegt, weshalb sie per default in die Klasse III fallen würden (siehe FD&C Act, 513(f)(1)).
Bei neuartigen Produkten bzw. bei Produkten der Klasse III müssten Hersteller eigentlich ein PMA-Zulassungsverfahren durchlaufen. Dieses „Premarket Approval“ (PMA) ist aber ebenso teuer wie arbeits- und zeitaufwändig. Damit wären Hersteller von unkritischen Produkten überfordert.
b) Lösungsansatz
Um diese unverhältnismäßigen Aufwände zu begrenzen, hat die FDA das De-Novo-Verfahren ins Leben gerufen. Es ist ein Verfahren, bei dem die FDA für neuartige Produkte
- die Klasse bestimmt,
- einen neuen Produkt-Code vergibt und
- die zugehörigen „Controls“ festlegt. Beispiele für Controls sind spezielle Labeling-Anforderungen, Design Controls oder Production Controls, also Aspekte eines Qualitätsmanagementsystems. Sie finden hier eine Übersicht über die Controls.
Ein als „de novo“ zugelassenes Produkt kann wiederum als „predicate device“ für andere ähnliche Produkte dienen.
2. Ablauf des De-Novo-Verfahrens
a) Varianten
Es gibt zwei Varianten, mit dem Verfahren zu beginnen (siehe Abb. 1):
- „Klassisches“ de novo
Der Hersteller reicht einen 510(k)-Antrag ein (Premarket Notification, PMN). Die FDA lehnt diesen mit der Begründung ab, dass das Vergleichsprodukt nicht „substantially equivalent“ (NSE) ist. Die FDA kann in der Ablehnung ihre Einschätzung kundtun, dass es sich bei dem zuzulassenden Produkt um einen „De-Novo-Kandidaten“ handeln könnte. Unabhängig von dieser Einschätzung darf der Hersteller dann einen De-Novo-Antrag einreichen, bei dem er sich auf die Ablehnung bezieht. - „direct de novo“
Inzwischen dürfen Hersteller auch direkt einen solchen Antrag stellen. Dies empfiehlt sich, wenn ein Hersteller glaubt, dass es kein passendes Vorgängerprodukt gibt und die FDA das eigene Produkt als Produkt der Klasse I oder II einstufen wird.
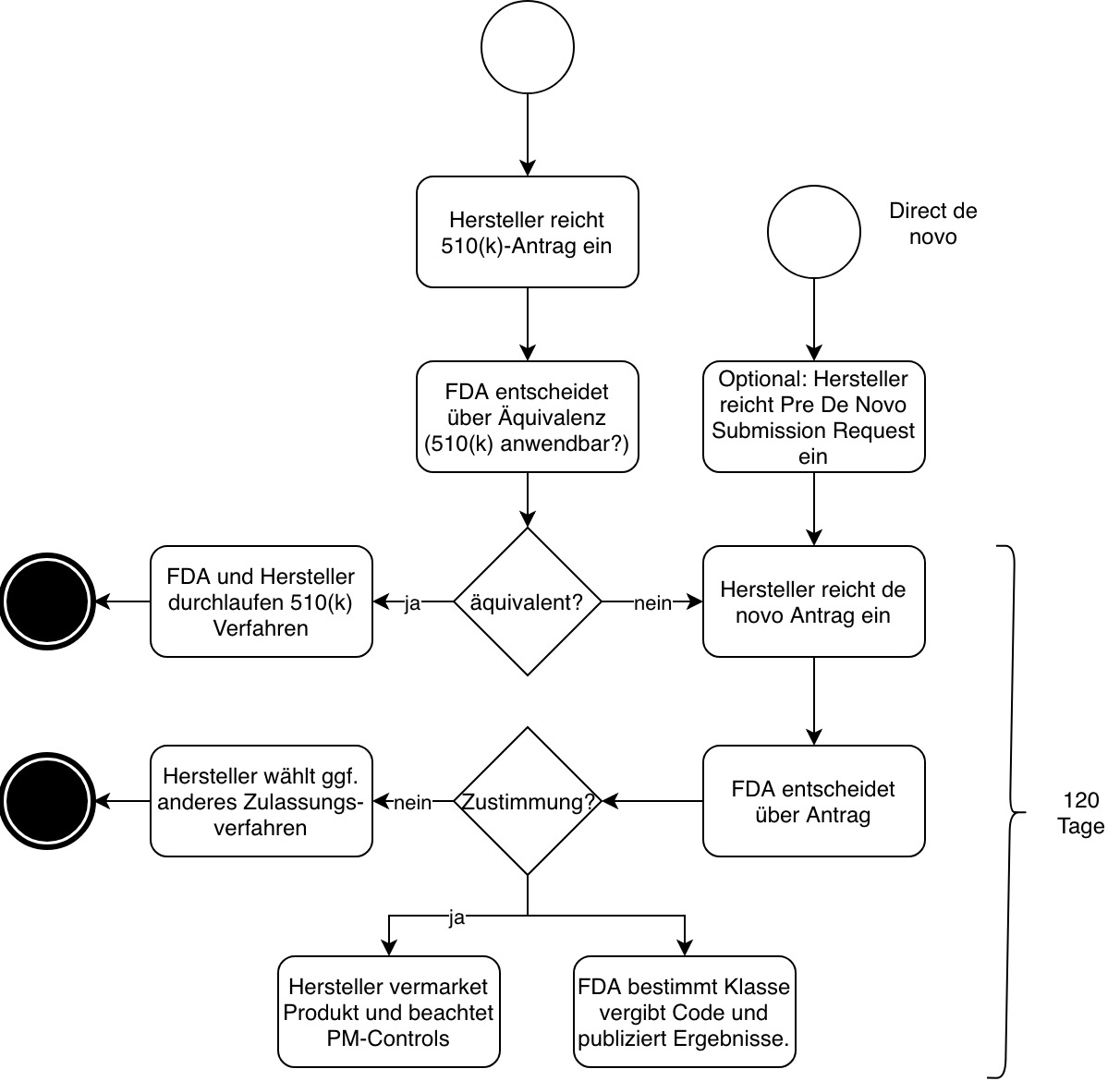
Im zweiten Fall sollten Hersteller zusätzlich eine Pre-Submission beantragen.
Lesen Sie hier mehr über Pre-Submission Requests.
b) Prüfung durch die Behörde und zeitlicher Ablauf
Formale Prüfung
Die formale Prüfung wird mit Verpflichtung zur Nutzung des eSTAR-Einreichungsformats ab dem 01.10.2025 obsolet.
Zuerst prüft die FDA den Antrag anhand einer „Annahme-Checkliste“, die Sie im Anhang A des Guidance-Dokuments Acceptance Review for De Novo Classification Requests finden. Die Behörde wird innerhalb von 15 Tagen entscheiden, ob sie den Antrag überhaupt annimmt.
Zuerst bewertet sie den Antrag nach eher formalen Entscheidungskriterien. Dazu zählt auch, ob
- der Hersteller die Gebühr beglichen hat,
- das Produkt überhaupt ein Medizinprodukt ist,
- der Antrag an das richtige Center der FDA gerichtet ist und ob
- für das Produkt überhaupt ein solcher Antrag gestellt werden kann. Falls es z. B. ein Predicate Device gäbe, wäre das nicht der Fall.
Wenn der Antrag diese erste Hürde genommen hat, fährt die FDA mit der Checkliste des Anhangs A fort und prüft:
- Organisatorische Aspekte: Kann sich die FDA in dem Antrag zurechtfinden?
- Vollständigkeit: Sind alle Inhalte vorhanden? Bei Software würde man prüfen, ob der Documentation Level bestimmt und die Dokumentation gemäß des Guidance-Dokuments Content for the Premarket Submissions for Software Contained in Medical Devices vorhanden ist.
- Kombinationsprodukte: Wenn das Produkt Arzneimittel enthält, bedarf es weiterer Unterlagen. Es besteht die Voraussetzung, dass dieses Arzneimittel bereits zugelassen wurde.
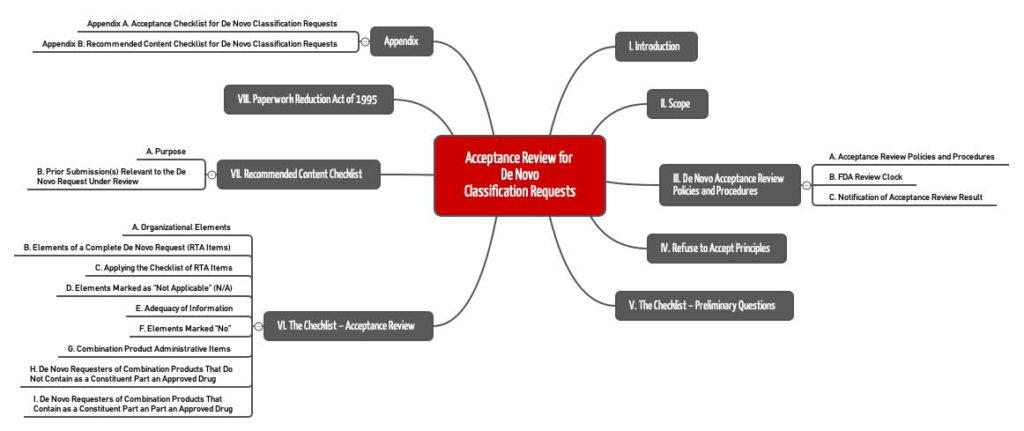
Inhaltliche Prüfung
Für die inhaltliche Prüfung nutzen die FDA-Mitarbeiter dasselbe Guidance-Dokument, jedoch die Checkliste in Anhang B. Diese Checkliste gruppiert Fragen zu folgenden Themen:
- Beschreibung des Geräts inklusive Zubehör, Labeling für Patienten und Anwender usw.
- Begründung, weshalb das De-Novo-Verfahren beantragt wird
- Nennung alternativer Verfahren, Technologien, Produkte
- Zusammenfassung der präklinischen Daten wie Tests und klinische Daten von klinischen Studien
- Aufzählung eingehaltener Normen (insbesondere „consensus standards“)
Zwischen der Einreichung des Antrags und der Entscheidung der Behörde sollen nicht mehr als 120 Tage vergehen, falls der Hersteller keine weiteren Informationen nachreichen muss.
c) Einzureichende Dokumentation
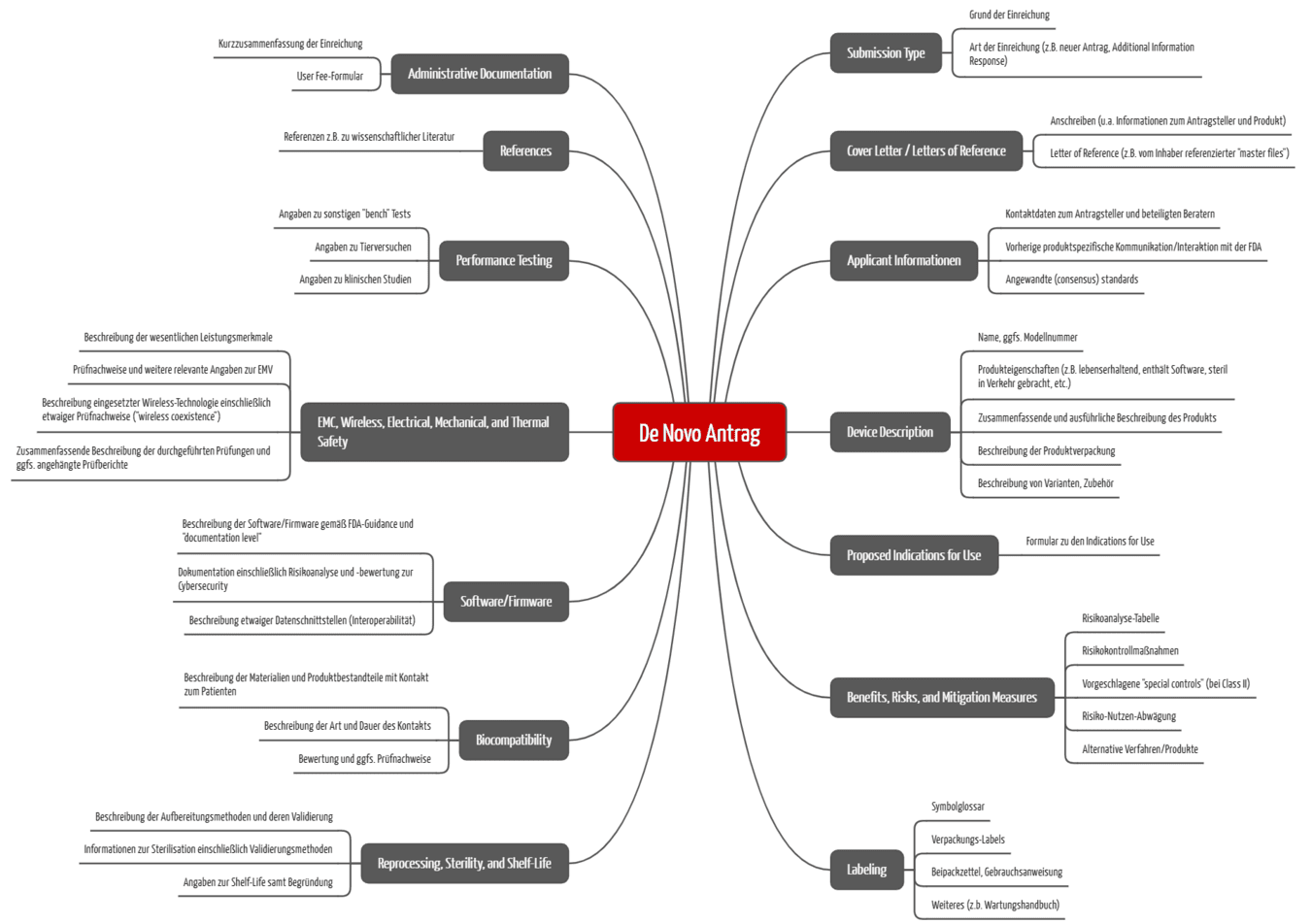
Die FDA verlangt für beide der unter a) genannten Varianten ähnliche Informationen. Sie hat eine Struktur erarbeitet, wie die Informationen vorliegen sollen. Für Anträge ab dem 1. Oktober 2025 erfolgt die Einreichung im eSTAR-Format. Die FDA beschreibt die Struktur und die Inhalte im Guidance-Dokument Electronic Submission Template for Medical Device De Novo Requests. Sie fordert u. a.:
- Administrative Informationen (Hersteller/Antragsteller, Produktname, Art der Einreichung etc.)
- Referenz zu etwaiger Kommunikation mit der FDA und FDA-Historie (z. B. IDEs, Pre-Submissions) vor der Einreichung
- Angewandte Recognized Consensus Standards
- Name und ausführliche Beschreibung des Produkts einschließlich Zweckbestimmung bzw. Indications for Use
- Vorschlag für die Klassifizierung mit Begründung
- Zusammenfassende Beschreibung des Nutzens, zur Risikoanalyse sowie Beschreibung der risikominimierenden Maßnahmen
- Begründung, weshalb das Produkt sicher ist und die Zweckbestimmung erreicht unter Nutzung wissenschaftlicher Nachweise (Risiko-Nutzen Diskussion). Typischerweise sind dafür klinische Daten zum Produkt notwendig.
- Bei Produkten, die vermutlich in die Klasse II fallen, einen Vorschlag möglicher „Special Controls“, um die Risiken zu minimieren
- Labeling, v. a. Gebrauchsanweisung
- Angaben zur Aufbereitung und deren Validierung
- Angaben zur Sterilisation und deren Validierung
- Angaben zum „shelf life“
- Bewertung zur Biokompatibilität
- Besondere Angaben und Nachweise zur Software/Firmware und Cybersecurity sowie Interoperabilität (falls zutreffend)
- Testergebnisse (z. B. Labortests, Softwaretests, Tierversuche, klinische Prüfungen)
- Referenzen (z. B. zu wissenschaftlichen Artikeln)
- Antworten zu Nachfragen der FDA („Additional Information requests“)
Die FDA verlangt den Antrag im eSTAR-Format mit vorgegebener Kapitelstruktur (s. Abb. 3).
3. Regulatorischer Hintergrund
Der Food, Drug & Cosmetic Act beschreibt das De-Novo-Verfahren in Artikel 513(f) im Abschnitt (2). Er nennt das Verfahren auch „Evaluation of Automatic Class III Designation“.
Dieser Artikel macht klar, dass dieses Verfahren nur für Produkte gestattet ist, die noch nicht klassifiziert sind.
4. Kosten für ein De-Novo-Verfahren
Ein De Novo Request ist nicht preisgünstig (siehe Preisliste): Ab dem Fiskaljahr 2025 ruft die FDA über 162.000 USD auf. Für kleine Firmen reduziert die FDA die Gebühr auf etwas über 40.000 USD.
5. Fazit
Für neuartige und wenig kritische Produkte ist das De-Novo-Verfahren das Verfahren der Wahl. Auch hier ist die sorgfältige Vorbereitung das A und O. Die Hersteller müssen nachvollziehbar begründen,
- weshalb es kein vergleichbares Produkt gibt,
- weshalb sie eine bestimmte Klasse und entsprechende „Controls“ vorschlagen,
- welche Risiken bestehen und
- wie und weshalb diese beherrscht werden, sodass der Nutzen das Risiko überwiegt.
Eine klare Zweckbestimmung, eine strukturierte Beschreibung des Produkts und Testergebnisse stellen eine Voraussetzung für diese Argumentation dar.
Unterstützung
Das Johner Institut hilft Herstellern schnell und unbürokratisch:
- Entscheiden, ob ein De-novo-Antrag eingereicht werden soll
- Überprüfen, ob der Antrag vollständig ist, um unnötigen Zeitverlust oder gar eine Ablehnung zu vermeiden
- Entscheiden, ob ein Pre-Sub-Antrag oder eine 510(k)-Einreichung die bessere Strategie darstellt
- Fehlende Informationen ergänzen und Anträge verbessern
- An Pre-Submission-Meetings teilnehmen
- Die Kommunikation mit der FDA übernehmen
Damit gelingt es Herstellern, Zulassungsrisiken und damit unnötige Kosten und Verzögerungen zu minimieren und ihre Produkte schnell und sicher in den US-Markt zu bringen.
Steht bei Ihnen eine Zulassung Ihres Produkts in den USA an? Dann nehmen Sie gleich Kontakt mit uns auf.
Änderungshistorie
- 2024-10-11: Überarbeitung aufgrund des ab 2025 geforderten eSTAR-Formats


