
Es ist nicht einfach, in Brasilien Medizinprodukte zuzulassen. Das liegt sowohl an der Anzahl und Komplexität der Regularien als auch an der Tatsache, dass Brasilien die meisten Regularien nur auf Portugiesisch veröffentlicht hat.
Dieser Artikel verschafft Ihnen einen Überblick und stellt die Gemeinsamkeiten mit dem europäischen und dem US-amerikanischen System vor. So wird es Ihnen einfacher gelingen, die Regularien zu verstehen und zu erfüllen. Unnötige Rückweisungen durch die brasilianische Behörde und damit einen Verzug der Zulassung können Sie so vermeiden und die Zulassung in Brasilien schneller und einfacher schaffen.
1. Das brasilianische Rechtssystem für Medizinprodukte
ANVISA ist die in Brasilien für Medizinprodukte verantwortliche Behörde. Vergleichbar mit der FDA
- erlässt sie Gesetze zu Medizinprodukten,
- prüft sie deren Zulassung und
- überwacht sie die Konformität der QM-Systeme der Hersteller mit den brasilianischen Anforderungen.
Die Anforderungen der ANVISA weisen viele Ähnlichkeiten mit der europäischen Medizinprodukteverordnung (EU) 2017/745 und den US-amerikanischen Quality System Regulations (21 CFR part 820) auf:
- Hersteller, die über ein QM-System gemäß 21 CFR Part 820 bzw. ISO 13485:2016 verfügen, erfüllen auch weitgehend die brasilianischen Anforderungen an ein QMS.
- So wie die EU und die FDA registrierte lokale „Bevollmächtigte“ bzw. Repräsentanten verlangen, bedarf es eines Brazilian Registration Holders.
- Nach ähnlichen Regeln wie in Europa teilt die Behörde Medizinprodukte in Klassen ein. Diese Klassen bestimmen wiederum den Aufwand und die Dauer für die Zulassung der Produkte. Mehr zur Klassifizierung erfahren Sie weiter unten.
Darüber hinaus gibt es in Brasilien landesspezifische Anforderungen wie die nach einem INMETRO-Zertifikat. Dazu später mehr.
2. Medizinproduktegesetze in Brasilien
Die wichtigsten brasilianischen Gesetze finden Sie in der folgenden Übersicht:
| Produkttyp | Resolution | Inhalt |
|---|---|---|
| Medizinprodukte | RDC 751/2022 | Registrierung, Änderung, Verlängerung und Löschung von Medizinprodukten |
| Medizinprodukte | RDC 270/2019 RDC 423/2020 | Anforderungen an die Registrierung von Klasse-I- und Klasse-II-Produkten |
| In-Vitro-Diagnostika | RDC 830/2023 | Klassifizierungsregeln und Anforderungen zu Registrierung und Kennzeichnung von IVD |
| Alle | RDC 665/2022 | Brazilian Good Manufacturing Practices |
| Alle | Decree No. 8077/2013 | Regelt die Zulassung und Überwachung von Produkten durch die Behörde in Brasilien |
| Alle | Law No. 6360/1976 | Allgemeines Gesetz zur Gesundheitsüberwachung |
3. Klassifizierung gemäß RDC 751/2022
Hersteller, die ein Medizinprodukt in Brasilien zulassen möchten, müssen zunächst die Risikoklasse des Produkts bestimmen. ANVISA unterscheidet dabei die Klassen I (niedriges Risiko) bis IV (hohes Risiko).
Die Klassifizierungsregeln wurden überarbeitet und sind im Anhang I der Resolution RDC 751/2022 enthalten. Neue Regeln adressieren dabei fortschrittliche Technologien, u.a. zu Software als Medizinprodukt sowie Medizinprodukte aus Nanomaterialien. Die Regeln sind weitgehend von der europäischen MDR übernommen.
Nutzen Sie die folgende Tabelle als Leitlinie für die Klassifizierung Ihres Produktes:
| EU | Brasilien |
|---|---|
| I | I |
| IIa | II |
| IIb | III |
| III | IV |
Die Verfahren zur Klassifizierung und Registrierung von In-vitro-Diagnostika (IVD) fallen nicht unter diese Resolution und sind weiterhin Bestandteil der RDC 830/2023.
4. Voraussetzungen für die Zulassung von Medizinprodukten in Brasilien
Bevor Hersteller die Zulassungsunterlagen bei der brasilianischen Behörde einreichen können, müssen sie bestimmte Voraussetzungen erfüllen.
1. Voraussetzung: Das B-GMP-Zertifikat
Hersteller müssen die Anforderungen der Brazilian Good Manufacturing Practices (B-GMP) erfüllen. Diese sind stark an die Quality System Regulations (21 CFR 820) der FDA angelehnt.
Hersteller von Produkten der Klassen I und II müssen diese Anforderungen erfüllen, werden jedoch nicht von ANVISA überprüft. Sie sind auch nicht verpflichtet, bei der Zulassung ein B-GMP-Zertifikat einzureichen. Hingegen benötigen Hersteller von Produkten der Risikoklassen III und IV ein B-GMP-Zertifikat für die Produktzulassung.
Bitte beachten Sie: Ein solches Zertifikat kann erst nach einer Inspektion, z.B. durch ANVISA, ausgestellt werden. Es kann lange Vorlaufzeiten von Seiten der Behörde bis zum Audit geben.
Mit der im Oktober 2017 verabschiedeten Resolution RDC-183/2017 hat ANVISA wesentliche Vereinfachungen zur Erlangung des B-GMP-Zertifikats geschaffen:
- ANVISA akzeptiert bei der Beantragung eines Certificado de Boas Práticas de Fabricação (BPF) die Auditberichte des Medical Device Single Audit Program (MDSAP), sofern der Hersteller die brasilianischen Anforderungen erfüllt und während der gesamten Zertifikatsgültigkeit im MDSAP bleibt.
Die Einreichung eines oben genannten Auditberichts kann die Ausstellung des B-GMP-Zertifikats und die Zulassung Ihres Medizinprodukts in Brasilien beschleunigen.
B-GMP-Zertifikate sind standardmäßig zwei Jahre gültig. Für MDSAP-zertifizierte Hersteller wurde die Gültigkeit auf vier Jahre verlängert (s. RDC No. 497/2021). Es wird empfohlen spätestens sechs Monate vor Ablauf des Zertifikats einen Neuantrag durch den Brazilian Registration Holder (BRH) einzureichen.
Die Kosten für ein GMP-Zertifikat machen einen großen Teil der Zulassungskosten aus und können sich auf etwa 25.000 EUR je Gültigkeitszeitraum belaufen.
2. Voraussetzung: Der Brazilian Registration Holder (BRH)
Eine weitere Voraussetzung für die Zulassung in Brasilien ist ein regionaler Vertreter vor Ort, der Brazilian Registration Holder (BRH). Der BRH muss offiziell ernannt werden und ist gesetzlich für die zugelassenen Produkte in Brasilien verantwortlich.
Diese Aufgabe kann ein Firmensitz in Brasilien oder ein Vertriebshändler übernehmen. Auch unabhängige Unternehmen dürfen die Aufgaben des BRH wahrnehmen.
Wählen Sie den BRH mit Bedacht aus, da er Ihre Registrierung hält! Sie sind damit in seiner Abhängigkeit.
Zu den Aufgaben des Brazilian Registration Holders zählen:
- Kommunikation mit ANVISA
- Beantragung des B-GMP-Zertifikats
- Einreichung der Zulassungsunterlagen
- Marktüberwachung (zumindest trägt der BRH die Verantwortung dafür)
- Vigilanz: Der BRH berichtet Rückrufe und Vorkommnisse an ANVISA.
3. Voraussetzung: Das INMETRO-Zertifikat
INMETRO ist das nationale Institut für Messtechnik, Qualität und Technologie. Einige Medizinprodukte benötigen für die Zulassung durch ANVISA ein Zertifikat, welches belegt, dass ein Produkt die zutreffenden brasilianischen Normen und Anforderungen einhält.
Die Prüfung und das zugehörige Zertifikat können von INMETRO selber oder von einer von INMETRO anerkannten Stelle ausgestellt werden. Das INMETRO-Zertifikat ist unbegrenzt gültig, muss jedoch durch regelmäßige Audits aufrechterhalten werden.
Medizinprodukte, die ein INMETRO-Zertifikat benötigen, sind u.a.:
- Elektrische Geräte
- Sterile Injektionsspritzen zum Einmalgebrauch
- Brustimplantate
- Chirurgische und nicht-chirurgische Handschuhe aus Kautschuk
- Kondome
Planen Sie drei bis zwölf Monate ein, um Ihr INMETRO-Zertifikat zu erlangen. Beachten Sie auch, dass für die Aufrechterhaltung des Zertifikats laufende Kosten anfallen.
4. Voraussetzung: Das ANATEL-Zertifikat
ANATEL ist die nationale Telekommunikations-Behörde Brasiliens. Alle Medizinprodukte, die über ein Funkmodul verfügen, benötigen für die Zulassung durch ANVISA ein Zertifikat von ANATEL. Ebenso wie das INMETRO-Zertifikat ist die Notwendigkeit ein ANATEL-Zertifikat einzureichen nicht von der Produktklasse abhängig. Sie benötigen also auch für ein Klasse I Produkt ein ANATEL-Zertifikat, wenn es über ein Funkmodul verfügt.
Aktuell werden nur brasilianische Produkttests von ANATEL anerkannt. Unter Umständen müssen Sie ihr Produkt für den brasilianischen Markt somit erneut prüfen lassen.
Bitte beachten Sie den ausführlichen Artikel zu den INMETRO- und ANATEL-Zertifikaten. Er verrät auch, wie lange diese Prozesse dauern, was diese kosten und welche Fehler zu unnötigen Verzögerungen und Schwierigkeiten führen.
5. Zulassungsprozess
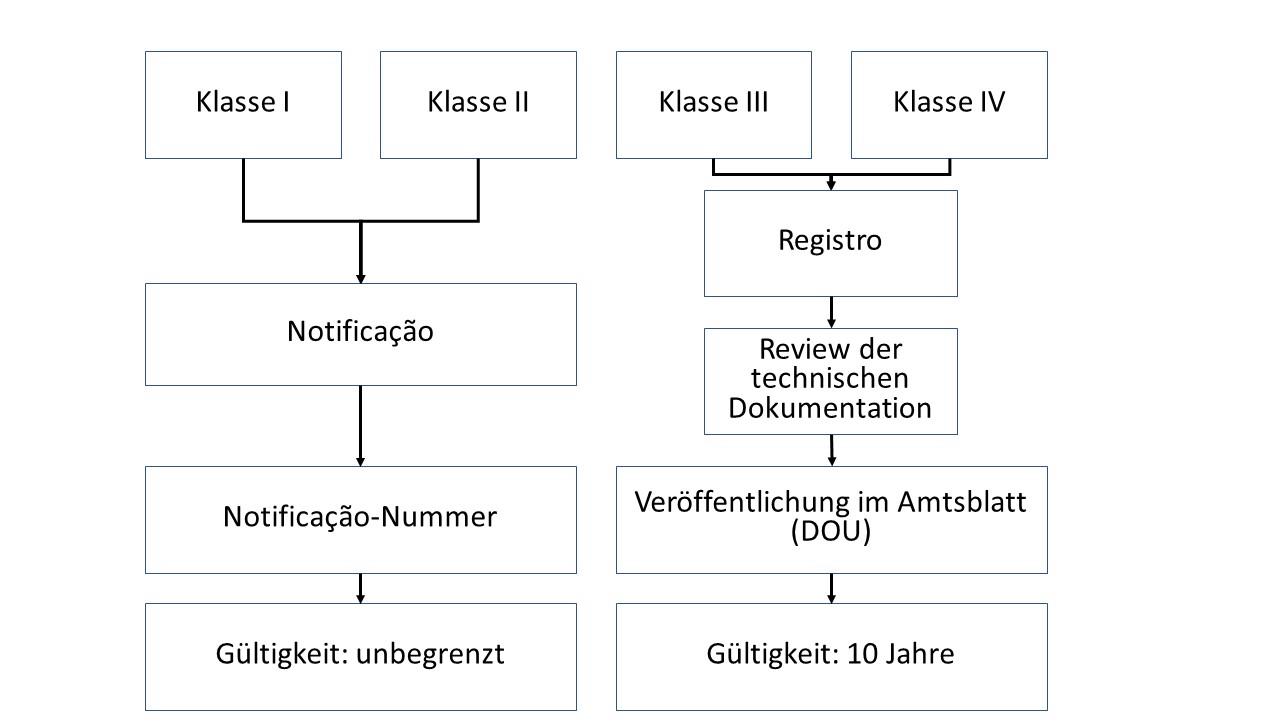
ANVISA unterscheidet zwei Zulassungswege. Diese richten sich nach der Klassifizierung.
- Klasse-I- und Klasse-II-Produkte werden an ANVISA gemeldet und erhalten im Anschluss eine Registriernummer (Notificacao).
- Klasse-III- und Klasse-IV-Produkte durchlaufen ein vollständiges Zulassungs- und Prüfungsverfahren (Registro).
6. Anforderungen an die Dokumentation
Die Klasse und damit die Zulassungsverfahren bestimmen auch die Anforderungen an die Dokumentation. Je höher die Klasse ihres Produktes, desto umfangreicher sind die Anforderungen an die Dokumentation.
a) Anforderungen, die für alle Klassen gelten
Unabhängig von der Klasse des Produkts müssen die Hersteller die (Dokumentations-) Anforderungen der RDC 751/2022 erfüllen. Diese orientieren sich an dem ToC-Format des IMDRF und sind in Anhang II der RDC 751/2022 zusammengefasst. Anhang II fordert unter anderem:
- Eine ausführliche Beschreibung des Medizinprodukts, einschließlich der Grundlagen seiner Funktions- und Wirkungsweise, seines Inhalts oder seiner Zusammensetzung
- Indikation, Zweckbestimmung oder Anwendung, für die das Medizinprodukt nach Angaben des Herstellers bestimmt ist
- Vorsichtsmaßnahmen, Einschränkungen, Warnhinweise, besondere Vorsichtsmaßnahmen und Erläuterungen zur Verwendung des Medizinprodukts
- Angaben zu Lagerung und Verpackung
- Kennzeichnungsmuster und Gebrauchsanweisungen
- Beschreibung der Herstellung
- Nachweis über die Erfüllung der Grundlegenden Sicherheits- und Leistungsanforderungen
b) Anforderungen an die Zulassung von Medizinprodukten der Klassen I und II
Produkte der Klassen I und II werden über ein vereinfachtes Verfahren zugelassen. Neben allgemeinen Angaben zu Ihrem Produkt reichen Sie eine Beauftragung des BRH und eine Bestätigung über die Einhaltung der B-GMP-Anforderungen ein. Die Behörde erteilt ohne Prüfung der Technischen Dokumentation eine Registrierungsnummer.
Es ist ANVISA jedoch erlaubt, weitere Dokumente anzufordern.
Bitte beachten Sie: Falls eine Überprüfung durch ANVISA stattfindet, müssen Sie Ihrem BRH die Technische Dokumentation zur Verfügung stellen.
Für Produkte der Klassen I und II gilt eine unbegrenzte Zulassung.
c) Anforderungen an die Zulassung von Medizinprodukten der Klassen III und IV
Für Medizinprodukte der Klassen III und IV führt ANVISA eine ausführliche Prüfung der Dokumentation durch. Die Behörde verpflichtet die Hersteller, weite Teile ihrer Technischen Dokumentation einreichen. Dazu zählen unter anderem:
- Produktbeschreibung
- Anforderungen an das Produkt
- Informationen zum Risikomanagement
- Software- bzw. Firmware-Beschreibung
- Klinische Bewertung
- Tests zu Biokompatibilität, elektrischer Sicherheit oder elektromagnetischer Verträglichkeit (soweit zutreffend)
- Nachweise zur Gebrauchstauglichkeit
ANVISA wird die eingereichte Dokumentation prüfen und bei Freigabe im DOU (Diário Oficial da União) veröffentlichen. Sie erhalten kein zusätzliches Zertifikat.
Die Zulassung von Klasse-III- und Klasse-IV-Produkten ist 10 Jahre gültig.
Verliert Ihr GMP-Zertifikat seine Gültigkeit, wird gleichzeitig die Produktregistrierung ungültig! Das GMP-Zertifikat müssen Sie alle zwei Jahre erneuern.
Kapitel VII der RDC 751/2022 definiert nun die inhaltlichen Anforderungen an die zu erstellende Technische Dokumentation. Für die Vorgaben zur Struktur der Technischen Dokumentation im Anhang II der Resolution hat die ANVISA das Table-of-Contents-Format der IMDRF übernommen.
d) Anforderungen an die Zulassung besonderer Medizinprodukte in Brasilien
Für einige Produkte muss zusätzlich ein Wirtschaftsbericht eingereicht werden. Betroffene davon sind unter anderem orthopädische und kardiovaskuläre Produkte.
Die gesamte Liste finden Sie im Beschluss RE n° 3385/2006 und auf der Website von ANVISA. Dort steht auch ein hilfreiches Template für Ihren Wirtschaftsbericht bereit.
Der Wirtschaftsbericht enthält grundlegende Informationen zum Produkt sowie wirtschaftlich relevante Informationen wie Preisgestaltung, Absatzzahlen oder die geschätzte Anzahl behandelter Patienten.
7. Fazit und Zusammenfassung
Die Anforderungen an die Zulassung in Brasilien sind ebenso umfangreich wie jene in Europa oder den USA. Die meisten Unterlagen können die Firmen wiederverwenden, müssen diese allerdings ins Portugiesische übersetzen. Zudem gibt es einige Anforderungen zu erfüllen, die spezifisch für Brasilien sind.
Wie in Europa und den USA bestimmt die Klasse der Medizinprodukte die Dauer und den Aufwand für die Zulassung.
| Klasse I | Klasse II | Klasse III | Klasse IV | |
|---|---|---|---|---|
| Komplexität | niedrig | niedrig | hoch | hoch |
| Dauer | 1 Monat | 1 Monat | ~ 12 Monate | ~ 12 Monate |
| Gültigkeit der Zulassung | unbegrenzt | unbegrenzt | 10 Jahre | 10 Jahre |
| B-GMP-Zertifikat | nicht notwendig | nicht notwendig | Ja. 2 Jahre gültig | Ja. 2 Jahre gültig |
Das Team vom Johner Institut kann Sie bei der Zulassung Ihrer Medizinprodukte in Brasilien unterstützen. Gerne beantwortet es Ihre Fragen, im Rahmen des Micro-Consultings sogar kostenlos.
- 2025-10-13: Überarbeitung der Informationen zum B-GMP Zertifikat und Gültigkeit bei MDSAP-Teilnahme.
- 2024-10-15: Umfassende Überarbeitung zur Einarbeitung neuer Anforderungen durch RDC 751/2022 und RDC 830/2023
- 2023-01-24: ANVISA legt mit der RDC 751/2022 neue Regeln zur Klassifizierung von Medizinprodukten fest und vereint die Regeln für die Meldung, Registrierung, Änderung, Erneuerung und das Löschen von Registrierungen in einer Resolution. Sie löst damit verschiedene Regelwerke ab, u. a. die RDC 185/2001
- 2021-03-21: Hinweis zu Beitrag über ANVISA- und INMETRO-Zertifikate ergänzt
- 2020-10-06: ANVISA eliminiert mit der RDC-423/2020 die Cadastro-Route für Klasse II Produkte: Anpassung der Abb. 1 an das neue System und Aktualisierung des Zulassungsverfahrens in Kapitel 5 und 6.b)



Wie haben ein brasilianisches Haarprotein. Zudem haben wir ANVISA. Was benoetigen wir fuer die Zulassung? Duerfen wir das Produkt in Deutschland verkaufen?
Sehr geehrter Herr Swidan,
herzlichen Dank für Ihre Rückfrage.
Leider ist Ihre Anfrage so umfangreich, dass sie sich nicht in Kürze beantwortet lässt. Folgendes kann ich Ihnen zusammenfassend mitteilen:
Eine Zulassung über ANVISA ermöglicht keinen Verkauf eines Produktes in der EU bzw. in Deutschland.
Handelt es sich bei Ihrem Haarprotein um ein Medizinprodukt im Sinne der europäischen Medizinprodukteverordnung (MDR)? Hierzu formulieren Sie im ersten Schritt die Zweckbestimmung des Produktes und prüfen, ob es unter die MDR fällt. Medizinprodukte dürfen in Europa (und Deutschland) nur vermarktet werden, wenn Sie über eine CE-Kennzeichnung verfügen. Handelt es sich bei Ihrem Produkt um ein Medizinprodukt, muss es ein entsprechendes Konformitätsbewertungsverfahren durchlaufen. Welches Verfahren anwendbar ist, ist von der Klassifizierung des Produktes abhängig und erfordert ggf. die Einbindung einer benannten Stelle.
Lassen Sie mich gerne wissen, wenn wir Sie unterstützen können, z.B. indem wir Ihre Zweckbestimmung und die Anwendbarkeit der MDR für Sie prüfen und mögliche Konformitätsbewertungsverfahren ermitteln.
Herzliche Grüße,
Margret Seidenfaden
Sehr geehrte Frau Seidenfaden,
ich wollte mich über die Zulassung eines brasilianischen Produktes informieren.
Das Produkt ist ein Haarprotein aus Brasilien und ist zugleich ein organisches Produkt aus natürlichen Inhaltsstoffen wie Arganöl, Rosenöl, Macadmia, Zitrone und tropischen Früchten. Es behandelt Haarausfall und pflegt das Haar problemlos.
Hierzu habe ich ein paar Fragen, und ich hoffe, dass Sie mir hierbei weiterhelfen können:
1. Wie wird das oben erklärte Produkt in Deutschland zugelassen? Ziel: Verkauf in Deutschland
2. welche unterlagen werden hierzu benötigt?
3. Was kostet die Zulassung und wie lange dauert diese?
Vielen Dank vorab.
Mit freundliche Grüßen
Osama Swidan
Sehr geehrter Herr Swidan,
herzlichen Dank für die Klarstellung Ihres Anliegens.
Um Ihre Fragen vollumfänglich zu beantworten, werden wir Sie persönlich kontaktieren.
Die Beantwortung nimmt einige Zeit in Anspruch und übersteigt den Rahmen der Möglichkeiten der Kommentare.
Herzliche Grüße,
Margret Seidenfaden
Liebes Johner-Team,
unser Unternehmen hat ein MDSAP Zertifikat, welches die brasilianischen Anforderungen berücksichtigt. Bedeutet das, wenn wir für die Beantragung des B-GMP Zertifikats den MDSAP-Auditbericht einreichen, dass wir alle in RDC 183 – Kapitel II aufgelisteten Dokumente nicht mehr einreichen müssen? Oder müssen trotzdem Dokumente wie das QM-Handbuch, Grundriss der Produktionsstätte, Flussdiagramm vom Herstellprozess, etc. eingereicht werden?
Vielen Dank!
Schöne Grüße
Martina M.
Sehr geehrte Frau Marker,
auch mit MDSAP-Zertifikat reichen Sie unterstützende Dokumentation (wie die von Ihnen genannte) für die Beantragung des B-GMP-Zertifikats ein.
Jedoch verringert sich die Anzahl der benötigten Dokumente und Sie werden nicht zusätzlich durch ANVISA auditiert. Weitere Informationen hierzu finden Sie auch in der RDC-687/2022 in Artikel 5.
Beste Grüße,
Margret Seidenfaden
Hallo Frau Seidenfaden,
Wir sind Hersteller für Arzneimittel und Medizinprodukte und haben ein BGMP-Zertifikat und MDSAP-Zertifikat. RDC 16/2013 wird in der oben genannten Tabelle bzgl. GMP-Anforderungen für alle Produkttypen genannt, diese Guideline wird auch in unserem MDSAP-Zertifikat aufgeführt. Nun gibt es aber seit März 2022 die RDC 658 für pharmazeutische Produkte – wissen Sie, ob diese dann nur für den Produkttyp Arzneimittel anwendbar ist?
Liebe Frau Reik,
Herzlichen Dank für dieses interessante Update.
Mit dieser Resolution hatte ich bisher keine Berührungspunkte. Nach einer kurzen Überprüfung komme ich zu dem Schluss, dass diese für Hersteller von Arzneimitteln gilt.
Im Scope der RDC 658/22 (Artikel 1 und 2) steht, dass diese für Hersteller von Arzneimitteln gilt (im Original: fabricação de medicamentos), inklusive experimenteller Arzneimittel (Im Original: Esta Resolução se aplica às empresas que realizam as operações envolvidas na fabricação de medicamentos, incluindo os medicamentos experimentais). Die Resolution legt die Mindestanforderungen an GMP fest, die solche Hersteller berücksichtigen müssen.
Herzliche Grüße,
Margret Seidenfaden
Hallo zusammen,
die RDC No. 16/2013 wurde durch die RDC No. 665/2022 ersetzt.
Dies sollte doch auch in diesem Beitrag betrachtet werden, oder?
Viele Grüße
Laura
Sehr geehrte Frau Kuipers,
vielen Dank für Ihren wertvollen Hinweis! Unser Expertenteam ist bereits an der Aktualisierung des Artikels dran und wird die neue RDC No. 665/2022 entsprechend berücksichtigen.
Herzliche Grüße
Tea Bodrusic
Hallo Frau Seidenfaden,
ich habe eine Frage zur UDI Kennzeichnung in Brasilien. Unsere IIb Produkte sind meines Wissens nach bis 2024 noch von der Übergangsfrist abgedeckt und müssen noch nicht gekennzeichnet werden. Da wir generell umstellen kam die Frage auf, ob es zulässig ist, jetzt einen UDI Code bei Produkten, die nach Brasilien gehen, anzubringen, der die europäischen Anforderungen einhält. Ein Eintrag in die brasilianische Datenbank ist ja derzeit noch nicht möglich
Liebe Grüße
Klaus Steger
Sehr geehrter Herr Steger,
vielen Dank für die spannende Frage! In Vertretung für Frau Seidenfaden hier meine Einschätzung:
Aktuell fordert die ANVISA keine Vergabe einer UDI. Damit ist die UDI, die Sie anbringen wollen, kein regulatorisches Element und sollte entsprechend auch keine meldepflichtige Änderung darstellen. Dafür spricht auch die Aussage der ANVISA in Anhang II, Section II, Artikel 12, wonach eine Änderung der UDI-DI an sich nicht gemeldet werden muss. Vergleichen wir nun die brasilianischen Anforderungen an die UDI mit denen aus der MDR, so ergeben sich ohnehin keine signifikanten Unterscheide, wodurch Sie zukünftig wohl auch die „EU-UDI“ für Brasilien nutzen können.
Herzliche Grüße
Christopher Seib
Guten Tag,
ich habe eine Frage zum BRH. Wenn wir einen Händler als BRH wählen, dürfen wir nur über diesen in Brasilien vertreiben oder können wir auch weitere Händler in Brasilien beliefern?
Viele Grüße
Uwe Ledworuski
Lieber Herr Ledworuski,
in Brasilien benötigen Sie einen BRH. Dieser autorisiert weitere Händler, über welche Sie ihr Produkt vertreiben.
Wenn Sie einen Händler als BRH einsetzen, kann es natürlich passieren, dass dieser keine weiteren Händler benennen möchte.
Sprechen Sie die Thematik am besten vorab mit Ihrem gewünschten BRH ab und lassen Sie sich vertraglich zusichern, dass weitere Händler benannt werden dürfen. Können Sie sich nicht einigen, haben Sie die Möglichkeit einen unabhängigen BRH zu benennen.
Herzliche Grüße,
Margret Seidenfaden
Hallo Frau Seidenfaden,
vielen Dank für die vielen Antworten zu der Brasilienzulassung von Medizinprodukten. Stimmt es, dass nur derjenige als Hersteller in Brasilien gilt, der die letzten Schritte (z.B. Verpackung und Sterilisation) durchgeführt hat? Dann müsste ja oft der Dienstleister, der dies für den eigentlichen Hersteller durchführt, als Hersteller gelistet sein. Das kann ich mir gerade nicht vorstellen.
Vielen Dank für Ihr Feedback und schöne Grüße
Liebe Frau Klein,
vielen Dank für Ihre Anfrage.
Ähnlich wie in der EU ist auch in Brasilien derjenige der Hersteller, unter dessen Namen das Produkt in Verkehr gebracht wird. Die Definition finden Sie in der RDC 751/2022 Artikel 4, XIX. Dort steht, dass jede juristische Person, die für die Herstellung, Verpackung, Kennzeichnung,… eines Produktes verantwortlich ist, mit dem Ziel diese unter eigenem Namen in Verkehr zu bringen, als Hersteller gilt. Es ist möglich diese Tätigkeiten auszulagern. Der Auftragnehmer wird dadurch nicht zum Hersteller.
Herzliche Grüße,
Margret Seidenfaden
Ganz lieben Dank für Ihre schnelle und kompetente Antwort!
Guten Tag,
leider sind die hier aufgeführten Informationen nicht mehr aktuell, da die ANVISA im Dezember 2023 eine neue Resolution zum Thema Risikoklassifizierung, Zulassungen und Kennzeichnungspflichten publiziert hat (RDC No. 830).
Liebe Frau Bergs,
ein wichtiger Hinweis, vielen Dank!
Das soll so natürlich nicht sein. Ich werde die Informationen im Artikel prüfen und aktualisieren.
Nochmals vielen Dank für Ihre Aufmerksamkeit und Ihre Unterstützung dabei, unsere Inhalte auf dem neuesten Stand zu halten.
Herzliche Grüße,
Margret Seidenfaden
Guten Tag,
in welcher Verordung wird nun der Aufbau der TD konkret bescheiben (vergleichbar mit ANNEX 2 der MDR)
Viele Grüße
Max Schwanau
Lieber Herr Schwanau,
vielen Dank für Ihre Rückfrage.
Die Anforderungen an die technische Dokumentation werden in Kapitel VII der RDC 751/2022 beschrieben. Die Struktur findet sich in Anhang II dieser RDC. Weiterhin verweist die ANVISA auf das ToC-Format des IMDRF. So detaillierte Anforderungen, wie sie in Anhang 2 der MDR beschrieben sind, gibt es nicht.
Herzliche Grüße,
Margret Seidenfaden
Hallo Frau Seidenfaden,
ist es eigentlich möglich mehrere BRH’s zu benennen?
Lieber Herr Faber,
vielen Dank für Ihre Rückfrage.
In Brasilien ist es möglich mehrere BRHs zu benennen. Sie können entweder verschiedene BRHs für verschiedene Produkte oder auch für ein Produkt mehrere BRHs benennen. Bitte beachten Sie, dass über jeden BRH eine eigene Produktregistrierung erfolgen muss.
Herzliche Grüße,
Margret Seidenfaden