
Kombinationsprodukte bestehen aus der Kombination eines Medizinprodukts und eines Arzneimittels. Da hierbei sowohl das Medizinprodukte- als auch das Arzneimittelrecht anwendbar sein kann, gibt es einige Besonderheiten zu beachten.
Erfahren Sie in diesem Beitrag,
- was ein Kombinationsprodukt ist,
- welches Recht anwendbar ist und
- welche besonderen Anforderungen an Kombinationsprodukte gelten.
1. Was sind Kombinationsprodukte?
a) Begriffsbestimmung EU-Recht
Im EU-Recht gibt es den Begriff „Kombinationsprodukt“ nicht offiziell. Zwar wird dieser in mehreren Regelungen umschrieben, jedoch niemals ausdrücklich genutzt.
Die Verordnung EG 1394/2007 (Arzneimittel für neuartige Therapien) zum Beispiel definiert den Begriff „kombiniertes Arzneimittel für neuartige Therapien“ als ein
Arzneimittel für neuartige Therapien, das als festen Bestandteil eines oder mehrere Medizinprodukte enthält.
Die MDR und die Richtlinie 2001/83/EG für Arzneimittel verwenden den Begriff „Kombinationsprodukt“ oder einen ähnlichen Begriff nicht. Die MDR macht dennoch Vorgaben für Produkte, die eine Kombination aus einem Arzneimittel oder einem Wirkstoff und einem Medizinprodukt sind.
In Art.1 Abs. 8 und 9 legt die MDR Regeln fest, wann Hersteller entweder die MDR oder die Richtlinie 2001/83/EG für Arzneimittel anwenden müssen.
(8) Jedes Produkt, das beim Inverkehrbringen oder bei der Inbetriebnahme als integralen Bestandteil einen Stoff enthält, der für sich allein genommen als Arzneimittel im Sinne von Artikel 1 Nummer 2 der Richtlinie 2001/83/EG gelten würde, auch wenn es sich um ein Arzneimittel aus menschlichem Blut oder Blutplasma im Sinne von Artikel 1 Nummer 10 der genannten Richtlinie handelt, dem im Rahmen des Produkts eine unterstützende Funktion zukommt, wird auf der Basis dieser Verordnung bewertet und zugelassen.
Kommt diesem Stoff jedoch eine hauptsächliche und keine unterstützende Funktion im Rahmen des Produkts zu, so gilt für das Gesamtprodukt die Richtlinie 2001/83/EG bzw. die Verordnung (EG) Nr. 726/2004 des Europäischen Parlaments und des Rates. In diesem Fall gelten für die Sicherheit und Leistung des Medizinprodukt-Teils die in Anhang I der vorliegenden Verordnung aufgeführten einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen.
Art. 1 Abs. 8 MDR
(9) Jedes Produkt, das dazu bestimmt ist, ein Arzneimittel im Sinne von Artikel 1 Nummer 2 der Richtlinie 2001/83/EG abzugeben, unterliegt dieser Verordnung unbeschadet der das Arzneimittel betreffenden Bestimmungen dieser Richtlinie und der Verordnung (EG) Nr. 726/2004.
Werden das Produkt, das zur Abgabe eines Arzneimittels bestimmt ist, und das Arzneimittel jedoch so in Verkehr gebracht, dass sie ein einziges untrennbares Gesamtprodukt bilden, das ausschließlich zur Verwendung in dieser Verbindung bestimmt und nicht wiederverwendbar ist, so unterliegt dieses einzige untrennbare Gesamtprodukt der Richtlinie 2001/83/EG bzw. der Verordnung (EG) Nr. 726/2004. In diesem Fall gelten für die Sicherheit und Leistung des Medizinprodukt-Teils des einzigen untrennbaren Gesamtprodukts die in Anhang I der vorliegenden Verordnung aufgeführten einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen.
Art. 1 Abs. 9 MDR
Hieraus kann geschlossen werden, dass die MDR durchaus das Prinzip von kombinierten Produkten kennt, auch wenn sie diese nicht definiert. Außerdem ergibt sich aus den MDR-Regelungen, dass auf diese Produkte primär entweder die MDR oder die RL 2001/83/EG für Arzneimittel anwendbar ist, jedoch nicht beide gleichermaßen.
Zwischenergebnis
Es gibt damit keine spezifische Verordnung, Richtlinie oder ein Gesetz für kombinierte Produkte, die aus einem Arzneimittel oder Wirkstoff und einem Medizinprodukt bestehen. Dennoch lässt sich aus den genannten Regelungen die folgende Definition ableiten:
Ein Kombinationsprodukt liegt vor, wenn die Kombination aus einem Arzneimittel oder Wirkstoff und einem Medizinprodukt ein einziges Produkt bildet.
b) Begriffsbestimmung US-Recht
Im Gegensatz zur EU gibt es in den USA den Begriff Kombinationsprodukt (Combination Product, 21 CFR 3.2(e)) auch offiziell. Dieser bezeichnet Produkte, die eine der folgenden Bedingungen erfüllen:
Kombination aus Arzneimittel und Medizinprodukt und/oder biologischem Produkt,
- (1) hergestellt als eine Einheit (physisch, chemisch oder anderweitig) oder
- (2) gemeinsam verpackt oder
- (3) getrennt verpackt, aber beide Produkte können nur gemeinsam die beabsichtigte Wirkung erzielen, oder
- (4) für die klinische Prüfung bestimmte Kombinationen, die separat verpackt sind und deren gemeinsame Anwendung für die Prüfung erforderlich ist
Beispiele zu den Bedingungen der FDA (den Nummern zugeordnet):
- Autoinjektor mit Adrenalin oder Insulin-Pen (1)
- Chirurgische Sets mit Instrumenten, Abdeckungen und antimikrobiellen Tupfern (2)
- Kardioplegie-Set für eine Herz-Lungen-Maschine (Schläuche zusammen verpackt mit der Kardioplegielösung) (2)
- Produkt für die Foto-Immuntherapie zusammen mit dem Wirkstoff (3)
Kombinationsprodukte im Sinne der FDA sind demnach:
Kombinationen aus Medizinprodukt und Arzneimittel und/oder biologischem Produkt, die entweder physisch verbunden (physically combined), zusammen verpackt (co-packaged) oder querverlinkt gekennzeichnet (cross-labelled) sind.
Festzuhalten ist, dass die FDA den Begriff „Kombinationsprodukte“ sehr viel weiter versteht als die MDR. Von den vier Varianten von „Combination Products” überschneidet sich nur eine mit dem Verständnis der MDR von Kombinationsprodukten, nämlich Variante 1: „hergestellt als eine Einheit (physisch, chemisch oder anderweitig)“.
2. Was sind nach der MDR Kombinationsprodukte NICHT?
Der Begriff „Kombination“ muss im Sinne von „integriert“ oder „zugehörig“ bzw. „unzertrennbar“ verstanden werden. Kombinationsprodukte (Arzneimittel-Geräte-Einheiten) sind demnach nicht:
a) Produkte zur Abgabe von Arzneimitteln
Produkte, die nur dazu bestimmt sind, Arzneimittel(-stoffe) abzugeben und die nicht an ein spezifisches Arzneimittel angepasst wurden, werden als Medizinprodukt behandelt (siehe MDR Artikel 1 (9) Abs. 1). Beispiele für solche Produkte sind Spritzenpumpen für allgemeine Zwecke.
b) Systeme und Behandlungseinheiten
Systeme und Behandlungseinheiten gelten in der EU (anders als in den USA) nicht als Kombinationsprodukte. Es gibt in Art. 22 MDR für Systeme und Behandlungseinheiten spezielle Regelungen.
„Behandlungseinheit“ bezeichnet eine Kombination von zusammen verpackten und in Verkehr gebrachten Produkten, die zur Verwendung für einen spezifischen medizinischen Zweck bestimmt sind;
Quelle: Art. 2 Nr. 10 MDR
„System“ bezeichnet eine Kombination von Produkten, die zusammen verpackt sind oder auch nicht, und die dazu bestimmt sind, verbunden oder kombiniert zu werden, um einen spezifischen medizinischen Zweck zu erfüllen;
Quelle: Art. 2 Nr. 11 MDR
Beinhalten solche Systeme oder Behandlungseinheiten Arzneimittel, gelten für diese Sachgesamtheit die Bestimmungen im Artikel 22 MDR (“Systeme und Behandlungseinheiten”).
Werden Systeme und Behandlungseinheiten zusammen mit Arzneimitteln verpackt geliefert („co-packaged”) oder gemeinsam gelabelt („cross-labelled”), gelten sie bei der FDA als Kombinationsprodukte.
Mehr erfahren Sie im Beitrag über Systeme und Behandlungseinheiten.
3. Anwendbares Recht: Medizinprodukterecht oder Arzneimittelrecht?
Für Kombinationsprodukte gilt jeweils entweder die RL 2001/83/EG für Arzneimittel bzw. die VO 726/2004 oder die MDR für Medizinprodukte vorrangig. Sie werden also entweder vorrangig als Arzneimittel oder vorrangig als Medizinprodukt behandelt, jedoch nicht als beides gleichermaßen. Je nachdem, welches Rechtsgebiet hauptsächlich einschlägig ist, müssen Hersteller das Kombinationsprodukt dann auch weitestgehend wie ein Arzneimittel oder weitestgehend wie ein Medizinprodukt prüfen.
Davon hängt z. B. ab,
- welchen Weg die Hersteller für den Konformitätsnachweis (Medizinprodukt) oder die Zulassung gehen müssen (Arzneimittel),
- wo die erforderlichen Unterlagen eingereicht werden müssen (Benannte Stelle beim Medizinprodukt, Arzneimittelbehörde beim Arzneimittel).
Beachten Sie auch den Artikel, der die komplexe Abgrenzung von Arzneimitteln und (stofflichen) Medizinprodukten verständlich erläutert.
a) Regeln aus Art. 1 Abs. 8 und 9 MDR
Welches Rechtsgebiet wann hauptsächlich anwendbar ist, ergibt sich aus Art. 1 Abs. 8 und 9 der MDR. Die Regeln lassen sich folgendermaßen zusammenfassen:
- Alle Produkte, die als integralen Bestandteil ein Arzneimittel zur Unterstützung enthalten, sind Medizinprodukte. Liegt die Hauptwirkung des Produkts jedoch beim Arzneimittel, wird das Gesamtprodukt wie ein Arzneimittel behandelt (Artikel 1 (8) MDR).
- Alle Produkte, die dazu bestimmt sind, ein Arzneimittel abzugeben, sind Medizinprodukte; es sei denn, sie werden als ein einziges untrennbares Gesamtprodukt in Verkehr gebracht, das ausschließlich zur Verwendung in dieser Verbindung bestimmt und nicht wiederverwendbar ist. Dann wird das Gesamtprodukt wie ein Arzneimittel behandelt (Artikel 1 (9) MDR).
Hauptbestandteil ist dabei derjenige Bestandteil, welcher die beabsichtigte Verwendung, Indikation oder Wirkung erzielt. Dieser Bestandteil darf also nicht nur unterstützende Wirkung haben.
Integral ist der Bestandteil, wenn beide Komponenten fest und unzertrennlich miteinander verbunden sind. Weiterhin wird vorausgesetzt, dass die Kombination einem spezifischen Zweck dient.
Kombinierte Produkte müssen immer die Anforderungen aus dem jeweiligen Anhang I beider Rechtsgebiete erfüllen. In der MDR sind dies die grundlegenden Sicherheits- und Leistungsanforderungen, in der RL 2001/83/EG sind dies analytische, toxikologische, pharmakologische und ärztliche oder klinische Vorschriften und Nachweise über Versuche mit Arzneimitteln.
Mögliche Varianten und anwendbares Recht im Detail
Art. 1 Abs. 8 MDR
Medizinprodukt, wenn:
- Arzneimittel integraler Bestandteil des MP
- Unterstützende Funktion Arzneimittel
- Beispiel: Heparin-imprägnierter Katheter (Arznei hat unterstützende Wirkung)
Arzneimittel, wenn:
- Arzneimittel integraler Bestandteil des MP
- Hauptsächliche Funktion: Arzneimittel
- Beispiel: Antiseptisches Pflaster, bei dem die Hauptwirkung das Antiseptikum ist und das Pflaster das Antiseptikum speichert
Art. 1 Abs. 9 MDR
Medizinprodukt, wenn:
- MP dazu bestimmt, Arzneimittel nach Richtlinie 2001/83/EG abzugeben
- Beispiel: Medikamentenpumpen zum Nachfüllen
Arzneimittel, wenn:
- MP dazu bestimmt, Arzneimittel nach Richtlinie 2001/83/EG abzugeben
- Untrennbares Gesamtprodukt
- MP und Arzneimittel ausschließlich zur Verwendung in Kombination bestimmt
- Kombination nicht wiederverwendbar
- Beispiel: Epi-Pen (mit Arzneimittel vorgefüllte Spritze) für den Einmalgebrauch
Im Zweifel gilt die Richtlinie für Arzneimittel
Es gibt Situationen, in denen keine klare Trennung der Wirkungsweise möglich ist und beide Definitionen zutreffen: sowohl Arzneimittel als auch Medizinprodukt.
Beispiel: Als Hauptwirkung eines mit einem Antiseptikum benetzten Wundverbandes kann der Hersteller in der Zweckbestimmung
a) den physischen Schutz der Wunde erklären und/oder
b) die antiseptische Wirkung zur Vermeidung einer Infektion in den Vordergrund stellen.
Im häuslichen Gebrauch steht der physische Schutz im Vordergrund und in der klinischen Anwendung die antiseptische Wirkung wegen der höheren Keimbelastung. Der Wundverband wird für beide Bereiche zugelassen.
In solchen Zweifelsfällen gilt die Richtlinie für Arzneimittel (siehe Art. 2 Abs. 2 2001/83/EG und AMG § 2 Abs. 2 Nr. 7).
b) Berücksichtigung beider Rechtsgebiete beim Konformitätsbewertungsverfahren bzw. der Zulassung
Beim Konformitätsbewertungsverfahren bzw. der Zulassung sind dennoch Vertreter beider Bereiche, also sowohl des Arzneimittel- als auch des Medizinprodukterechts, involviert.
Medizinprodukt vorrangig
Ist primär Medizinprodukterecht anwendbar, muss die Benannte Stelle die zuständige Arzneimittelbehörde einbeziehen und eine wissenschaftliche Bewertung einholen.
Anhang IX Abschnitt 5.2 der MDR besagt, dass die Benannte Stelle bei einem Medizinprodukt, das ein Arzneimittel nach Art. 1 Nr. 2 der Richtlinie 2001/83/EG für Arzneimittel enthält, die zuständige Arzneimittelbehörde konsultieren muss. Ohne deren positives Votum erhält das Kombinationsprodukt keine Konformitätsbescheinigung.
Hierin liegt eine Abweichung zur MDD/AIMDD. Die MDR legt ausdrücklich fest, dass die Benannte Stelle eine Bescheinigung nicht erteilen darf, wenn die Stellungnahme der Arzneimittelbehörde negativ ausfällt (Anhang IX 5.2 (e) und (f)).
Arzneimittel vorrangig
Gleiches gilt umgekehrt für Kombinationsprodukte, die nach Arzneimittelrecht zugelassen werden. Hier wird eine Benannte Stelle involviert (Art. 117 MDR).
Artikel 117 MDR regelt das Verfahren, wie die Benannte Stelle bei Kombinationsprodukten, die primär als Arzneimittel gelten, einbezogen wird.
- Verwendet der Arzneimittelhersteller ein Medizinprodukt mit EU-Konformitätserklärung des Herstellers, ist das weitere Hinzuziehen einer Benannten Stelle nicht notwendig. Das Produkt erfüllt dann bereits Anhang I MDR. Den Nachweis muss der Arzneimittelhersteller mit dem Zulassungsantrag einreichen.
- Wenn keine Bescheinigung vorliegt und für den Konformitätsnachweis des Medizinprodukts allein eine Benannte Stelle beteiligt werden müsste, muss der Hersteller eine „Notified Body Opinion” von einer Benannten Stelle anfordern, die für solche Produkte benannt wurde. Dabei geht es ebenfalls um den Nachweis, dass der Medizinprodukteteil die Anforderungen im Anhang I der MDR erfüllt.
Ähnlich sind die Regeln für Companion Diagnostics (CDx). Hierzu mehr in unserem Beitrag zum Thema.
4. Regulatorische Anforderungen an Kombinationsprodukte
Aus der Kombination von Medizinprodukt und Arzneimittel ergeben sich neue Gefährdungen bezüglich Interoperabilität und Kompatibilität, die auch im Anhang I der MDR adressiert werden. Bezüglich dieser Gefährdungen gelten weitere Anforderungen.
a) Risikobewertung
Der Hersteller muss nachweisen, dass er den Abs. 12 im Anhang I, Art. II MDR erfüllt.
Anhang I Abs. 12.2 MDR:
Die Wechselwirkung zwischen Arzneimittel und Patient sowie Arzneimittel und Medizinprodukt muss bezüglich der Resorption, Verteilung, Metabolismus, Ausscheidung, lokale Verträglichkeit, Toxizität, Wechselwirkungen mit anderen Medizinprodukten bewertet werden. Es gelten die Anforderungen aus Anhang I der Richtlinie 2001/83/EG.
Die Folgenabschätzung gilt auch für bestimmte Herstellungstechnologien, z. B. die Sterilisation (Anhang I Abs. 11.4). Der Hersteller sollte bewerten, ob die jeweils angewandte Technologie bzw. Methode Auswirkungen auf die Sicherheit und Leistung des Medizinproduktteils hat. In Ergänzung legt die für die Beurteilung des Zulassungsdossiers zuständige Behörde (EMA) und/oder (NCAs) den Schwerpunkt der Beurteilung auf den Arzneimittelteil.
b) Dokumentation
Anhang II Abs. 6.2 a) MDR besagt:
Der Anwender des Medizinprodukts muss wissen, dass das Medizinprodukt ein Arzneimittel beinhaltet. Die Dokumentation enthält die genaue Angabe der Quelle dieses Stoffes sowie die Daten der Tests, die unter Berücksichtigung der Zweckbestimmung des Produkts zur Bewertung der Sicherheit, der Qualität und des Nutzens durchgeführt wurden.
Erforderlich sind damit Angaben auf dem Medizinprodukt zu
- dem Vorhandensein eines Arzneimittels,
- der Quelle des Arzneimittels,
- den Daten der Tests, die seine Sicherheit, seine Qualität und seinen Nutzen belegen.
c) Gebrauchstauglichkeit
Für einzelne Produktklassen hat die FDA spezielle Guidance-Dokumente zum Human Factors Engineering veröffentlicht. So gibt es ein Guidance-Dokument für Kombinationsprodukte.
d) FDA
Die FDA beschreibt ihre Anforderungen an die Kombinationsprodukte zumindest indirekt in ihrem Guidance Document „How to Prepare a Pre-Request for Designation“.
5. Klassifizierung
a) Klassifizierungsregeln
Das Konformitätsbewertungsverfahren bzw. die Zulassung im Arzneimittelrecht richtet sich zunächst danach, ob das Produkt als Arzneimittel oder als Medizinprodukt qualifiziert werden muss.
Ist in erster Linie die MDR anwendbar, fallen die Kombinationsprodukte gemäß Regel 14 in die Risikoklasse III.
Alle Produkte, zu deren Bestandteilen ein Stoff gehört, der für sich allein genommen als Arzneimittel im Sinne des Artikels 1 Nummer 2 der Richtlinie 2001/83/EG gelten kann, auch wenn es sich um ein Arzneimittel aus menschlichem Blut oder Blutplasma im Sinne des Artikels 1 Nummer 10 der genannten Richtlinie handelt und dem im Rahmen des Medizinprodukts eine unterstützende Funktion zukommt, werden der Klasse III zugeordnet.
7.1. Regel 14
An Klasse-III-Produkte werden nach der MDR hohe Anforderungen gestellt. Dies bedeutet
- hohen Aufwand in der Dokumentation,
- hohen Aufwand in der Überwachung und
- weniger Auswahl bei den Benannten Stellen.
b) Regulatorische Strategie passend zur Klassifizierung (Beispiele)
Um ein Kombinationsprodukt bestmöglich durch das Konformitätsbewertungsverfahren bzw. das Zulassungsverfahren zu bringen, sollten Hersteller strategisch vorgehen. Sie können beispielsweise die folgenden Strategien wählen:
Beispiel Regulatorische Strategie 1

Der Zweck des Produkts ist die Messung eines Enzymspiegels über die Haut. Der Sensor besteht aus dem Sensorkopf (1) mit einer Messkammer und einer Messzelle (2) mit einem Stoffgemisch, die im Austausch mit der Gewebeflüssigkeit das Messsignal erzeugt. Das Messsignal wird auf der Anzeige (3) in einen Messwert umgerechnet und angezeigt.
Regulatorische Strategie
| Kombinierte Produktteile | Das Stoffgemisch (2) dient zur Erstellung einer ärztlichen Diagnose und muss gemäß Definition Art. 1 Abs.1 (b) 2001/83/EG als Arzneimittel qualifiziert werden. Der Sensorkopf (2) und die Anzeige (3) sind ein Gerät bzw. Apparat zum Zweck der Diagnose sowie Überwachung einer Krankheit und sind das Medizinprodukt. |
| Integraler Bestandteil | Das Arzneimittel ist ein integraler Bestandteil des Medizinprodukts und somit ist die Gesamtheit ein integriertes Produkt (Kombinationsprodukt). |
| Hauptwirkung | Hauptwirkung ist die Berechnung des Enzymspiegels über das durch das Stoffgemisch erzeugte Messsignal. |
| Unterstützende Wirkung | Die von der Messgröße erzeugte Druckänderung wird von dem Stoffgemisch verstärkt, wodurch die Messung genauer und stabiler wird. |
| Konformitätsbewertung | Maßgeblich ist die MDR, da die Hauptwirkung bei dem Medizinprodukt liegt. Wegen Anhang I Abs. 12.2 gelten weiterhin die Anforderungen aus Anhang I der 2001/83/EG. |
| Klassifikation | Klasse III wegen Regel 14 |
| Behörden | Benannte Stelle (Berechtigung Klasse III) für das Medizinprodukt |
Beispiel Regulatorische Strategie 2
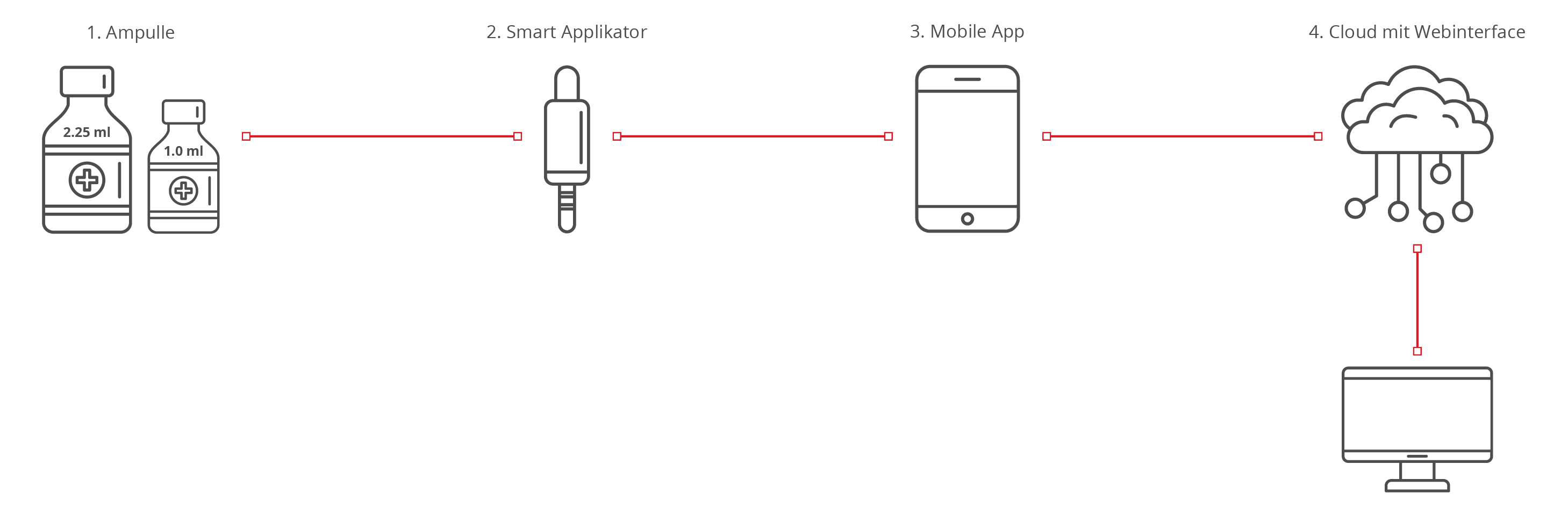
Das Produkt dient zur medikamentösen Behandlung einer Krankheit. Das Medikament wird von Hersteller A vordosiert in Ampullen (1) geliefert. Die Verabreichung erfolgt mit dem Smart Applikator (2), der das Medikament kontrolliert und vollständig abgibt und die Daten des Vorgangs an eine mobile App (3) übermittelt. Die mobile App (3) zeichnet die Zeitreihen auf, die ein Arzt remote über die Cloud (4) auswerten kann. Hersteller für den Smart Applikator und die Softwareprodukte ist Hersteller B. Hersteller B verpackt die Teile (2) und (1) gemeinsam. Der Anwender kann die Ampullen (1) in einer Apotheke nachkaufen.
Regulatorische Strategie
| Kombinierte Produktteile | Die Ampulle (1) mit dem Medikament ist das Arzneimittel. Die Ampulle ist die Primärverpackung. Der Smart Applikator (2) ist das Medizinprodukt (kein Zubehör, da Zubehöre an Medizinprodukte und nicht an Arzneiprodukte gebunden sind). |
| Integraler Bestandteil | Ampulle (1) ist kein integral fest verbauter Bestandteil des Smart Applikators (2) und somit liegt keine Kombination im Sinne des Artikels vor. |
| Hauptwirkung | nicht relevant |
| Unterstützende Wirkung | nicht relevant |
| Artikel 1 Abs. 9 der MDR | Der Smart Applikator (2) ist dazu bestimmt, das Medikament abzugeben, und unterliegt damit der MDR. |
| Konformitätsbewertung | Hersteller A: Ampulle mit Arznei nach 2001/83/EG Hersteller B: Komponenten 2, 3, 4 nach MDR (siehe Artikel 1 Abs. 9) Für die zusammen verpackte Gesamtheit muss Hersteller B eine Konformitätserklärung nach Artikel 22 ausstellen. Die Produktteile müssen für die gemeinsame Verwendung gegenseitig gekennzeichnet werden. |
| Klassifikation | Smart Applikator (2) und Software (3, 4) können getrennt klassifiziert werden. |
| Behörden | Hersteller A: mit EMA/NCA für die Ampulle mit Arzneimittel Hersteller B: mit Benannter Stelle für das Medizinprodukt |
6. Zusammenfassung
Mit dem Begriff „Kombinationsprodukt“ sind solche Medizinprodukte gemeint, die ein Arzneimittel enthalten. Diese Produkte müssen im EU-Recht entweder schwerpunktmäßig als Medizinprodukt oder schwerpunktmäßig als Arzneimittel betrachtet werden. Die Regeln hierfür ergeben sich aus Art. 1 Abs. 8 und 9 MDR.
a) Arzneimittel vorrangig – 2001/83/EG ist maßgeblich
Für solche Produkte, die vorrangig als Arzneimittel betrachtet werden, integriert die MDR über den Artikel 117 ihre Anforderungen aus Anhang I in die Richtlinie für Arzneimittel. Diese Neuerung hat einen Einfluss auf Hersteller von solchen kombinierten Arzneimitteln. Für die Einreichung ist zusätzlich ein Nachweis nötig, dass der Hersteller ein bereits CE-gekennzeichnetes Medizinprodukt mit Bescheinigung einer Benannten Stelle verwendet. Andernfalls ist die Stellungnahme einer Benannten Stelle nötig.
Ist der Medizinproduktteil wegen des Übergangs zur MDR von einer Höherklassifizierung betroffen (das Einbeziehen einer Benannten Stelle wird notwendig), müssen Arzneimittelhersteller bis zum Ablauf der Übergangsfristen für Klasse-I-Produkte beim Hersteller des Medizinprodukts die entsprechenden Bescheinigungen nachfordern.
b) Medizinprodukt vorrangig – MDR ist maßgeblich
Für Hersteller von kombinierten Produkten, die vorrangig als Medizinprodukt behandelt werden und bei denen die MDR maßgeblich ist, gelten weitere Anforderungen bezüglich der Wechselwirkung aus Anhang I der MDR. Für die Arzneimittelkomponente gelten zusätzlich die Anforderungen aus Anhang I der Richtlinie 2001/83/EG. Diese Produkte werden per se in Klasse III eingeordnet. In der Regel wird ein wissenschaftliches Gutachten der zuständigen Behörde (EMA) und/oder (NCAs) erforderlich sein, um die Risiken der Kombination abzuschätzen.
c) Getrennt verpackt oder kreuzreferenziert
Hersteller, die Arzneimittel und Medizinprodukte zusammen oder getrennt verpacken und für die eine gemeinsame Verwendung vorgesehen ist, müssen die Anforderungen der MDR zusammen mit Artikel 22 MDR erfüllen.
Wenn Sie Fragen zu Kombinationsprodukten haben oder Hilfe beim Konformitätsbewertungsverfahren benötigen, nehmen Sie gern Kontakt zu den Expertinnen und Experten des Johner Instituts auf.
Wir bedanken uns herzlich beim TÜV SÜD für wichtigen Input zu diesem Beitrag.
Änderungshistorie
- 2025-11-13: Abschnitt 4.d) eingefügt (FDA Guidance)
- 2023-09-16: Kapitel 4.c) mit Anforderungen der FDA an die Gebrauchstauglichkeit ergänzt
- 2022-03-01: Erste Version veröffentlicht



Hallo Herr Klessascheck,
Sie nennen im Abschnitt 1a die Verordnung 726/2004/EG als Arzneimittel für neuartige Therapien, die den Begriff “kombiniertes Arzneimittel für neuartige Therapien“ definiere. Den Begriff „kombiniertes Arzneimittel für neuartige Therapien“ (combined advanced therapy medicinal products (cATMP)) ist jedoch in der Verordnung EC 1394/2007 über Arzneimittel für neuartige Therapien und zur Änderung der Richtlinie 2001/83/EG und der Verordnung (EG) Nr. 726/2004 definiert (dort Art. 2(1d)).
Zudem möchte ich darauf hinweisen, dass Arzneimittel für neuartige Therapien (ATMP) nicht gleichzusetzen sind mit den Arzneimitteln gem. Definition Art. 1(2) der Directive 2001/83/EC. Sie werden in dieser in Art. 1(4a) definiert, und für ATMPs gilt die MDR gem. MDR Art. 1(6c) nicht.
Viele Grüße
M. Diercks
Lieber Herr Diercks,
vielen Dank für den wichtigen Hinweis. Ich habe das gleich geändert. Vielen Dank auch für Ihre Klasrtellung, dass die MDR nicht für Arzneimittel für neuartige Therapien im Sinne der Verordnung (EG) Nr. 1394/2007 anwendbar ist.
Herzliche Grüsse, Mario Klessascheck
Sehr geehrter Herr Klessascheck,
Hat ein Händler die Pflicht, von einem Medizinprodukt die Konformitätserklärung einzufordern, wenn dieses in einer Fertigarzneimittelpackung für die Applikation des Arzneimittels beigelegt ist, aber nicht untrennbar verbunden?
Beispielsweise eine Dosierspritze zur Applikation eines Saftes, der sich aber nicht in der Spritze, sondern in einer Glasflasche befindet? Analog gilt die Frage für Dosierbecher oder -löffel, die mit dem AM zusammen verpackt sind. Das gesamte Produkt würde ja als Arzneimittel eingestuft werden.
Beste Grüße
Lieber Herr Weseloh,
Die Pflichten des Händlers gemäß der MDR sind in Artikel 14, insbesondere in Absatz (2), festgelegt. In diesem Zusammenhang besteht zunächst die Verpflichtung des Händlers, die korrekte Kennzeichnung zu überprüfen. Zusätzlich dazu sieht Absatz (6) vor, dass der Händler einer Behörde erforderliche Informationen und Dokumente zum Konformitätsnachweis zur Verfügung stellen muss. Hieraus ergibt sich, dass der Hersteller die Konformitätserklärung auf Anfrage vom Händler bereitstellen muss. Diese Verantwortung kann auch auf den Bevollmächtigten übertragen werden. Daher sollten Sie mit dem Hersteller eine Vereinbarung treffen, um sicherzustellen, dass Sie im Bedarfsfall Zugang zur Konformitätserklärung erhalten.
Hilft Ihnen das weiter? Sonst haken Sie gern noch einmal nach.
Herzliche Grüsse, Mario Klessascheck
Lieber Herr Klessascheck,
vielen Dank für die Antwort. Viele Hersteller kooperieren inzwischen auch ohne große Probleme. Dennoch bleibt mir die Frage, ob die Kontrollpflicht des Händlers auch für Medizinprodukte gilt, die in einer Fertigarzneimittelpackung beigelegt sind (Beispielsweise Dosierlöffel oder Kanülen). Der Hersteller übernimmt ja in diesem Fall eigentlich die Verantwortung für das gesamte Arzneimittel nach Arzneimittelgesetz. Haben Sie solch einen speziellen Fall schon einmal im Institut diskutiert?
Herzliche Grüße
Jochen Weseloh
Lieber Herr Weseloh,
Die Lesart wäre so, dass Sie lediglich die Kennzeichnung für das Gesamtprodukt bewerten müssen. Sie müssten nur tiefergehend prüfen, wenn Sie einen Grund zur Annahme haben, dass das Produkt nicht konform ist. Was begründete Annahmen sein könnten, haben wir intern noch nicht besprochen und mir fallen auch leider keine Beispiele ein.
Herzlichst, Mario Klessascheck
Hallo Herr Klessascheck,
vielen Dank für diesen Beitrag bzgl. Kombinationsprodukten!
Inwieweit ist Artikel 22 MDR (basierend auf den momentanen „Kombinationsprodukte“ Regularien, Guidelines etc.) sinnig / im Einklang mit diesen („Kombinationsprodukte“ Regularien, Guidelines etc.).
Als ein Beispiel: Artikel 22, Punkt 4
(4) Enthält das System oder die Behandlungseinheit Produkte, die keine CE-Kennzeichnung tragen, oder ist die gewählte Kombination von Produkten nicht mit deren ursprünglicher Zweckbestimmung vereinbar oder wurde die Sterilisation nicht gemäß den Anweisungen des Herstellers durchgeführt, so wird das System oder die Behandlungseinheit als eigenständiges Produkt behandelt und dem einschlägigen Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen. Die natürliche oder juristische Person ist den für die Hersteller geltenden Pflichten unterworfen.
Für das Beispiel eines „co packaged“ Kombinationsproduktes (Medizinprodukt mit CE Kennzeichnung + Arzneimittel ohne CE Kennzeichnung) würde die Anwendbarkeit von Punkt 4, Artikel 22 bedeuten, dass das „co packaged“ Kombinationsprodukt einem Konformitätsbewertungsverfahren nach Artikel 52 MDR unterzogen werden müsste?
Viele Grüße
Konstantin Rischuk
Lieber Herr Rischuk,
Vielen Dank für Ihre interessante Frage.
Solange das Arzneimittel für sich genommen verkehrsfähig ist (Arzneimittel tragen keine CE Kennzeichnung), und Sie es mit einem Medizinprodukt verpacken, zum Bsp. Dosierlöffel mit einem Hustensaft, dann müssen Sie für die Kombination keine erneute Konformitätsbewertung durchführen. Im Artikel 22 steht im Absatz (1): „Produkte mit einer CE-Kennzeichnung mit folgenden anderen Medizin- oder sonstigen Produkten […]“. Das Arzneimittel wäre dann ein sonstiges Produkt, siehe auch Absatz (1) c).
Falls ich Ihre Frage falsch verstanden habe oder meine Antwort nicht hilfreich war, fassen Sie gern nach.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
vielen Dank für ihr Feedback. Ich sehe es auch so.
Eine andere Frage… für das Beispiel eines „co packaged“ Komb.produktes, ist Punkt 1, Artikel 22
(1) Natürliche oder juristische Personen, die Produkte mit einer CE-Kennzeichnung mit folgenden anderen Medizin- oder sonstigen Produkten in einer mit der Zweckbestimmung der Medizin- oder sonstigen Produkte vereinbaren Weise und innerhalb der vom Hersteller vorgesehenen Anwendungsbeschränkungen kombinieren, um sie in Form eines Systems oder einer Behandlungseinheit in Verkehr zu bringen, müssen eine Erklärung abgeben
so zu verstehen, dass das Unternehmen (entweder Medizinproduktehersteller oder Arzneimittelhersteller oder ein anderes Unternehmen) welches als „Hersteller“ (= Inverkehrbringer) auf der Verpackung des „co packaged“ Komb.produktes genannt ist, das von Artikel 22 geforderte Statement ausstellen muss?
Viele Grüße
Konstantin Rischuk
Lieber Herr Rischuk,
Ja das verstehen Sie richtig. Diejenige Person, die die Behandlungseinheit in Verkehr bringt, muss die Artikel 22 Erklärung ausstellen.
Liebe Grüsse, Mario Klessascheck
Sehr geehrtes Johner-Team,
wie sieht es bei Kombinationsprodukten aus, die nach MDR Artikel 1(9) zweiter Subparagraph nach nach Dir 2001/83/EC zugelassen werden und Medizinprodukteseitig den Annex I der MDR erfüllen müssen, müssen die Hersteller einen PRRC benennen und eine SRN haben, oder wird das vollständig über die Dir 2001/83/EC abgedeckt?
Danke.
Freundliche Grüße,
Martina
Liebe Frau Wotsch,
in dem Fall benötigen Sie kein PRRC. Die führende Richtlinie für das Produkte ist die Richtlinie 2001/83/EC. Aus der MDR müssen Sie für den Device Teile nur den Anhang I anwenden.
Liebe Grüsse, Mario Klessascheck