
Die Medizinprodukte-Sicherheitsplanverordnung (MPSV) trägt den Titel „Verordnung über die Erfassung, Bewertung und Abwehr von Risiken bei Medizinprodukten“. Der Titel dieser deutschen Verordnung macht klar, was sie regelt: Sie legt fest, wer Risiken bei Medizinprodukten erfassen, bewerten und darauf reagieren muss.
Beachten Sie, dass die MPSV mit dem 25.05.21 außer Kraft gesetzt und durch die Medizinprodukte-Anwendermelde- und Informationsverordnung – MPAMIV abgelöst wurde.
1. Ziele der MPSV (Medizinprodukte-Sicherheitsplanverordnung)
Das Ziel der MPSV besteht darin, Risiken für Patienten, Anwender und ggf. Dritte dadurch bestmöglich zu minimieren, dass die Hersteller und Aufsichtsbehörden auf diese Risiken so schnell und angemessen wie möglich reagieren.
2. Adressaten: An wen sich die MPSV wendet
Die Medizinprodukte-Sicherheitsplanverordnung wendet sich an
- Hersteller
- Gewerbliche Nutzer oder Anwender von Medizinprodukten wie Ärzte, Zahnärzte und Kliniken
- Personen oder Organisationen, die berufsmäßig Medizinprodukte an Patienten abgeben wie Apotheker oder Sanitätshäuser
- Vertreiber, Händler
- Prüfer und Sponsoren von klinischen Prüfungen
- Behörden (auf Bundes- und Landesebene)
3. Überblick: Forderungen der MPSV
Die wichtigsten Forderungen der MPSV sind:
- Die oben genannten Adressaten (außer den Behörden) müssen Vorkommnisse und schwerwiegende unerwünschte Ereignisse innerhalb gegebener Fristen und in bestimmter Form den Behörden melden (§§ 3-7).
- Die Hersteller müssen Risiken durch ihre Medizinprodukte schnellstmöglich beseitigen (§§ 14ff).
- Die Behörden müssen überprüfen, ob die Maßnahmen wirkungsvoll sind (§§ 8ff), gegebenenfalls Verbesserungen einfordern oder sonstige Maßnahmen treffen.
- Die Behörden müssen relevante Parteien wie andere Behörden informieren (§§ 19ff).
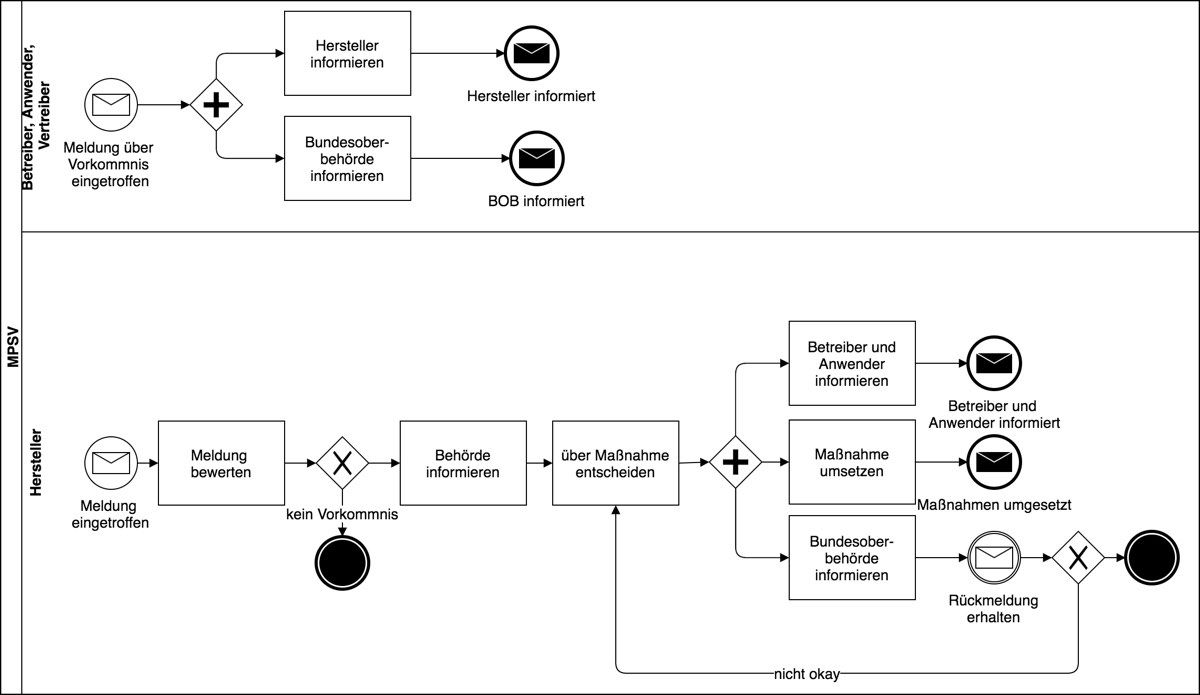
Um diese Forderungen noch besser zu verstehen, ist es notwendig, die Definitionen zu kennen.
4. Begriffsdefinitionen der MPSV
Die Medizinprodukte-Sicherheitsplanverordnung definiert die Begriffe Vorkommnis, schwerwiegendes unerwünschtes Ereignis, korrektive Maßnahme und Rückruf.
„Eine Funktionsstörung, ein Ausfall, eine Änderung der Merkmale oder der Leistung oder eine unsachgemäße Kennzeichnung oder Gebrauchsanweisung eines Medizinproduktes, die oder der unmittelbar oder mittelbar zum Tod oder zu einer schwerwiegenden Verschlechterung des Gesundheitszustands eines Patienten, eines Anwenders oder einer anderen Person geführt hat, geführt haben könnte oder führen könnte; als Funktionsstörung gilt auch ein Mangel der Gebrauchstauglichkeit, der eine Fehlanwendung verursacht.“
Im Rahmen von klinischen Prüfungen ist auch der Begriff des schwerwiegenden unerwünschten Ereignisses relevant:
„Jedes in einer genehmigungspflichtigen klinischen Prüfung oder einer genehmigungspflichtigen Leistungsbewertungsprüfung auftretende ungewollte Ereignis, das unmittelbar oder mittelbar zum Tod oder zu einer schwerwiegenden Verschlechterung des Gesundheitszustands eines Probanden, eines Anwenders oder einer anderen Person geführt hat, geführt haben könnte oder führen könnte ohne zu berücksichtigen, ob das Ereignis vom Medizinprodukt verursacht wurde; […]“
Damit würde sowohl ein Vorkommnis als auch ein schwerwiegendes unerwünschtes Ereignis vorliegen, wenn während einer klinischen Prüfung ein Medizinprodukt ausfällt und dies zum Tod eines Patienten hätte führen können.
In diesem Artikel finden Sie mehr zu den Definitionen der Begriffe Rückruf und korrektive Maßnahme.
5. Meldepflicht
Wann gemeldet werden muss
Die Hersteller müssen laut MPSV melden:
- Vorkommnisse
- Rückrufe (als Sonderfall korrektiver Maßnahmen)
- Schwerwiegende unerwünschte Ereignisse
An wen gemeldet werden muss
Die Meldungen gehen an die Bundesoberbehörde (meist das BfArM, ggf. das Paul-Ehrlich-Institut).
In welcher Form gemeldet werden muss
Die Meldung muss schriftlich über ein Formular erfolgen, das das BfArM auf seiner Internetseite bereitstellt. Dieses interaktive Formular enthält einen Button zum Versenden des Inhalts im XML-Format als E-Mail.
D.h. ein Versand per Post ist nicht notwendig.
Wie schnell gemeldet werden muss
| Vorkommnis | Rückruf | Schweres unerwünschtes Ereignis | |
| Hersteller | spätestens nach 30 Tagen, bei Gefahr im Verzug unverzüglich | Spätestens mit Beginn der Maßnahme | Unverzüglich |
| Gewerbliche Nutzer oder Anwender von Medizinprodukten | Unverzüglich | — | — |
| Personen oder Organisationen, die berufsmäßig Medizinprodukte an Patienten abgeben | Unverzüglich | — | — |
| Vertreiber, Händler | Unverzüglich | — | — |
| Prüfer und Sponsoren von klinischen Prüfungen | Wird in Zusammenhang mit schwerwiegendem unerwünschten Ereignis erfolgen | — | Unverzüglich, falls möglicherweise im Zusammenhang mit dem Produkt; sonst vierteljährlich |
6. Verpflichtung, Maßnahmen zu ergreifen
Welche Formen von Maßnahmen es gibt
Die Maßnahmen dienen der Verringerung oder Beseitigung von Risiken. Beispiele für Maßnahmen sind:
- Produkt aus dem Verkehr nehmen
- Produkt für eine Reparatur zum Hersteller zurückrufen
- Produkt durch Techniker vor Ort warten oder Teile austauschen lassen
- Gebrauchsanweisung überarbeiten
- Anwendern Hinweise zur Verwendung des Produkts geben
- Anwender bitten, das Produkt zu vernichten
- Update einspielen
Lesen Sie hier mehr zu Rückrufen (als Sonderfall der Maßnahmen) und weshalb der Begriff so missverständlich ist.
Wann welche Maßnahme ergriffen werden muss
Über die Form und den Zeitpunkt, zu dem die Maßnahme ergriffen wird, entscheidet der Hersteller basierend auf dem Risiko. Allerdings ermächtigt die Medizinprodukte-Sicherheitsplanverordnung die Bundesoberbehörde, zu entscheiden, ob diese Maßnahmen angemessen sind. Sie kann dann auch selbst (Notfall-)Maßnahmen ergreifen.
Entscheidet sich der Hersteller zu einem Rückruf, muss er nicht nur die Behörde, sondern auch die Betreiber und Anwender informieren.
Treten bei einer klinischen Prüfung Probleme auf, entscheiden Sponsor und Prüfer über die notwendigen Maßnahmen und informieren die Behörde. Auch hier gibt die MPSV die Art der Maßnahmen nicht vor.
7. Änderungen an der MPSV
Mit dem 01.01.2017 trat die geänderte Fassung der MPSV in Kraft. Zu den wesentlichen Änderungen zählen:
- Die Definition des Begriffs „Vorkommnis“ wurde geändert und dahingehend erweitert, dass auch ein Mangel an Gebrauchstauglichkeit betrachtet wird. Eine Gegenüberstellung der Versionen finden Sie hier.
- Vorkommnisse und schwerwiegende unerwünschte Ereignisse dürfen zusammen gemeldet werden.
- Meldungen müssen jetzt elektronisch erfolgen.
- Die Meldungen an die Behörde über Maßnahmen müssen jetzt enthalten:
- Empfehlungen für Maßnahmen an die Betreiber und Anwender
- Auszug aus der Risikoanalyse
- Begründung der Maßnahmen
Am 26.05.2021 wurde die MPSV außer Kraft gesetzt. Sie wird durch die MPAIMV abgelöst.



Hallo liebes Johner-Team,
wird die MPSV durch das MPDG zum 26.05.2021 für Produkte, welche unter die MDR fallen ersetzt, oder wird die MPSV auch nach diesem Datum weiter bestehen bleiben? Falls letzteres, sind Änderungen der MPSV geplant?
Mit freundlichen Grüßen
B. Winter
Sehr geehrter Herr Winter,
die MPSV wird noch nach dem 26.05.21 weiter existieren, schon weil die IVD weiter geregelt bleiben müssen. Von geplanten Änderungen am MPSV ist mir nichts bekannt.
Die MDR-Produkte werden ab dem 26.05.21 durch die Vorgaben des MPDG geregelt. Die MPSV bezieht sich nur auf das MPG und wird damit für MDR-Produkte obsolet.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
wird die MPSV nicht am 26.05.2021 von der MPAMIV abgelöst und gilt das dann nicht für alle Produkte?
Schafft man es überhaupt bis dahin die MPAMIV fertig zu haben?
Mit freundlichen Grüßen
T. C. Stratemann
Sehr geehrte/r Herr/Frau Stratemann,
danke für Ihre Frage! Sie machen mich damit auf einen Beitrag aufmerksam, der dringend der Überarbeitung bedürfte!
Es ist, wie Sie sagen, die MPSV wird durch die MPAMIV abgelöst. Einen entsprechenden Beitrag hatten wir verfasst.
Allerdings gibt es noch das MPG und damit die MPSV für IVD.
Ob man es schafft, die MPAMIV im Mai in Kraft zu setzen, weiß ich auch nicht. Dass so etwas zwei Monate vor der geplanten Inkraftsetzung der MDR noch immer nicht der Fall ist, finde ich selbst als befremdlich.
Nochmals vielen Dank für Ihren wichtigen Hinweis!
Viele Grüße, Christian Johner
Ich kann das MPSV auf den offiziellen Seiten im Internet nicht mehr finden. Gilt das jetzt noch für IVDs oder gilt schon die MPAIMV?
Wir sind ein Labor, also Anwender/ Eigenhersteller von IVDs und nach IVDR dann ggf Betreiber von LDTs.
Sehr geehrte Frau Bausewein,
danke für Ihre Frage!
Für In-vitro-Diagnostika gilt bis einschließlich 25. Mai 2022 die MPSV. Diese Einschätzung finden Sie auf dieser Seite.
Die MPSV finden Sie noch hier.
Viele Grüße, Christian Johner