
Regulatory Science wird meist etwas holprig als Regulierungswissenschaften übersetzt. Im deutschsprachigen Bereich wird die Regulatory Science in systematischer Weise kaum betrieben.
Genau dieser Missstand fällt den Medizinprodukteherstellern auf die Füße und schadet sowohl dem Gesundheitswesen als auch dem Standort. Höchste Zeit, dies zu ändern!
1. Was Regulatory Science ist
a) Definition von Regulatory Science
„Regulatory Science“ ist eine Wissenschaft. Sie entwickelt die (technischen) Grundlagen, Prozesse, Methoden und Werkzeuge,
- um darauf aufbauend regulatorische Anforderungen zu formulieren, die die Sicherheit, Leistungsfähigkeit und Wirksamkeit von Medizinprodukten gewährleisten, und
- um ökonomische und andere Auswirkungen dieser regulatorischen Anforderungen auf die Gesundheitssysteme und die Wirtschaft zu verstehen und zu antizipieren.
Eine kurze und etwas vereinfachende Definition könnte lauten:
b) Abgrenzung zu Regulatory Affairs
Die Regulatory Science kümmert sich im Gegensatz zu Regulatory Affairs nicht darum,
- wie Regularien (Gesetze, Verordnungen, Richtlinien) juristisch korrekt formuliert und verkündet werden,
- wie diese Vorschriften überwacht (z.B. auditiert) und durchgesetzt werden oder
- welche Unterlagen Hersteller in welcher Form bereitstellen müssen.
Umgekehrt ist es nicht die Aufgabe von Regulatory Affairs, die Sinnhaftigkeit und Wirksamkeit der Regularien wissenschaftlich zu bewerten.
Lesen Sie hier mehr zum Thema Regulatory Affairs.
2. Weshalb wir die Regulatory Science benötigen
a) Entweder erfolgt die Regulierung im kompletten Blindflug …
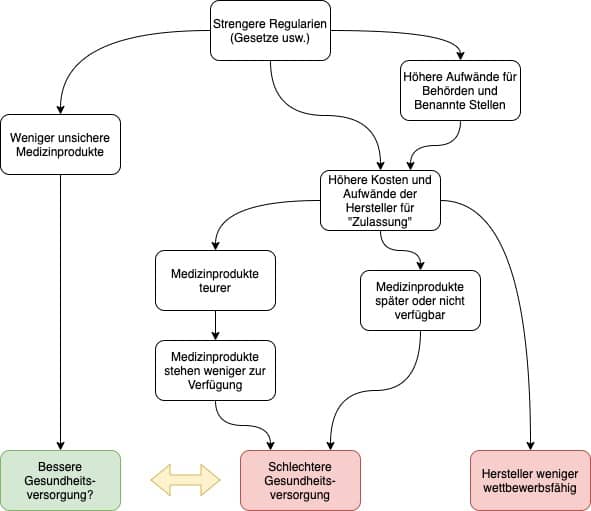
Wer handelt, sollte sich über die Konsequenzen seines Handelns bewusst sein. Doch genau dieser banalen Forderung kommen viele Regulierer (also Gesetzgeber und Behörden) nicht nach. Sie können es auch nicht, denn ihnen fehlt die wissenschaftliche Grundlage.
Die Befragung einiger Experten kann das nicht ersetzen. Einem Hersteller, der sich vornehmlich auf Befragungen von Patienten oder Experten stützen würde, würden die Behörden und Benannten Stellen mangelnde Evidenz vorwerfen. Zu Recht, denn Eminenz ersetzt nicht Evidenz.
Jedes Gesetz, jede Verordnung und jede Richtlinie hat Nebenwirkungen. Um das Nutzen-Risiko-Verhältnis durch die Gestaltung dieser Regularien zu optimieren, bedarf es eines Verständnisses der wirtschaftlichen und technologischen Abhängigkeiten. Diese zu erforschen, ist eine zentrale Aufgabe der Regulatory Science.
Fazit: Ohne ein Verständnis dieser Zusammenhänge fehlen den Gesetzgebern die Entscheidungsgrundlagen. Sie regulieren im kompletten Blindflug.
b) … und gefährdet Märkte und Patienten …
Die Regulierung erfolgt meist in bester Absicht. Zwar dienen Gesetze und andere Vorschriften in einigen Ländern auch zur Marktabschottung. Aber sie verfolgen (hoffentlich) primär das Ziel, dank sicherer und klinisch wirksamer Produkte zu einer optimalen Gesundheitsversorgung beizutragen.
Doch auf zentrale Fragen wissen die Gesetzgeber keine Antwort:
- Werden beispielsweise MDR und IVDR dieses Ziel besser erreichen als die bisherigen EU-Richtlinien (MDD, IVDD und AIMDD)?
- Wie viele Tote und schwer geschädigte Patienten erhofft die EU-Kommission durch die neuen Verordnungen zu vermeiden?
- Und wie viele Tote und schwer geschädigte Patienten nimmt die EU durch Produkte in Kauf, die durch MDR und IVDR verhindert werden?
Auf welcher Datenbasis hat Brüssel seine Entscheidungen getroffen? Es sind Entscheidungen, die direkt den europäischen MedTech-Markt mit einem Volumen von jährlich über 120 Mrd. EUR betreffen und indirekt den Gesundheitsmarkt insgesamt, der alleine in Deutschland eine Milliarde EUR an Wertschöpfung erwirtschaftet – pro Tag! [Quelle]
Die FDA behauptet sogar:
In the U.S., FDA-regulated products account for about 25 cents of every dollar spent by American consumers each year — products that touch the lives of every American every day.
Quelle
c) … oder wir geraten in Abhängigkeiten …
Die FDA engagiert sich aktiv im Bereich der Regulatory Science. Auf der Webseite „Advancing Regulatory Science” bietet sie einen Überblick über ihre Aktivitäten.
Der bequeme europäische Ansatz könnte darin bestehen, diese Ergebnisse für sich zu nutzen, ohne sich zu engagieren und ohne in Regulatory Science zu investieren.
Doch es gibt Gründe, die gegen diese Bequemlichkeit sprechen:
- Unnötige Abhängigkeit
Übernahme und Adaption dieser Ergebnisse stellen eine Abhängigkeit dar. Die FDA kann den Zugang zu diesen Daten jederzeit unterbinden. - Limitierte Übertragbarkeit von Daten
Der Fokus der FDA liegt berechtigterweise auf den USA. Das Anliegen der Behörde ist die Sicherheit der US-Patienten. Doch nicht alle Ergebnisse sind auf Europa übertragbar. Weder gibt es in Europa die identischen Produktklassen, noch ist die Bevölkerung in allen Attributen vergleichbar. Die Nutzungskontexte (z.B. Anwender, Arbeitsumgebungen) lassen sich nicht unreflektiert auf Europa übertragen. - Suboptimale Evidenz
Wissenschaft endet nicht an Ländergrenzen. Vielmehr ist eine Wissenschaft wie die Regulatory Science besonders dann erfolgreich, wenn ihr weltweit viele Ressourcen, insbesondere Daten und Wissenschaftler, zur Verfügung stehen. Es geht also nicht um „Europa vs. USA“ oder „Europa vs. den Rest der Welt“. Es geht darum, eine möglichst breite Datenbasis zu schaffen und einen möglichst hohen Erkenntnisgewinn zu erzielen.
d) … und die Hersteller verpassen Chancen und verlieren Zeit und Geld
Die Regulatory Science hat nicht nur die Aufgabe, unsichere Produkte zu verhindern. Sie hat auch die Aufgabe, Innovationen zu fördern und Herstellern neue Möglichkeiten aufzuzeigen, wie sie
- Behörden und Benannten Stellen die Wirksamkeit und Sicherheit ihrer Produkte einfacher, glaubhafter und schneller nachweisen können (z.B. durch Computer-Modelle),
- zuverlässig Risiken durch ihre Produkte während der Entwicklung und im Markt entdecken und beseitigen, bevor es zu Patientenschäden kommt,
- den Stand der Technik verfolgen können,
- von neuen und alternativen Prozessen, Methoden, Materialien und Werkzeugen erfahren, um schnell bessere Produkte zu entwickeln und sich einen Marktvorteil zu verschaffen.
3. Diese Fragen sollte Regulatory Science beantworten
Um die oben genannten Ziele zu erreichen, sollte Regulatory Science Antworten auf die relevanten Fragen aller Stakeholder liefern.
a) Fragen von Regulierern
Fragen zu den Auswirkungen auf Hersteller und den Medizinproduktemarkt
- Was wird es die Hersteller kosten, neue Regularien umzusetzen?
- Wie viele Gesundheitsschäden wird eine neue oder geänderte regulatorische Anforderung zu vermeiden helfen?
- Wie viele Hersteller werden in der Lage sein, diese Anforderung zu erfüllen? Wie lange werden sie dafür benötigen?
- Welcher Prozentsatz an Herstellern wird an dieser Hürde scheitern? Welche Medizinprodukte werden dadurch nicht oder erst später zur Verfügung stehen?
- Was sind die Konsequenzen dieser Verzögerungen oder des Nicht-Inverkehrbringens für das Gesundheitssystem?
- In welcher Weise schaden Regularien kleinen Firmen und/oder der Innovation? Wie kann es gelingen, dass Regularien Innovation sogar befördern?
- Zu welchen Standortnachteilen oder Standortvorteilen führen neue Regulierungen? Wie kann der Standort durch Regularien gestärkt werden?
- Wie können Regularien gestaltet und formuliert werden, damit Medizinprodukte sicherer und leistungsfähiger werden? Führen beispielsweise extra strenge Anforderungen bei einer klinischen Bewertung wirklich dazu, dass Medizinprodukte an Sicherheit, Leistungsfähigkeit und Wirksamkeit gewinnen?
Fragen zu den Auswirkungen auf Behörden und Benannte Stellen
- Wie lange wird es dauern, bis Behörden und Benannte Stellen diese Anforderungen überwachen und einfordern können?
- Zu welchen Engpässen kann es bei diesen Akteuren führen? (Personal, Ausbildung, Verfügbarkeit)
- Sind diese in der Lage, die Anforderungen auch bei Herstellern außerhalb der EU wirksam einzufordern?
- Mit welchen Werkzeugen können/sollen Behörden und Benannte Stellen die Einhaltung der Anforderungen möglichst effizient und effektiv überwachen?
- Welche Zulassungsverfahren bieten sich bei welchen Produkten an?
Fragen zu den Auswirkungen auf Patienten
- Wie sehr dienen Regularien dem Interesse der Patienten? Wo greifen sie unangemessen in die Entscheidungsfreiheit und die Risikoakzeptanz individueller Patienten ein?
Fragen zur künftigen Gesetzgebung
- Auf welche Trends müssen wir uns einstellen? Für welche Produkte, Technologien, Methoden und Materialien werden künftig neue Regularien benötigt?
- Welche derzeitigen regulatorischen Anforderungen entsprechen nicht mehr dem Stand der Technik?
- Wo fehlen Regularien, die notwendig wären, um die Gesundheitsversorgung zu verbessern, z.B. durch sichere Produkte? Wo behindern Regularien und schaden dem Standort oder sogar der Gesundheitsversorgung?
- Wie lässt sich das regulatorische Rahmenwerk so gestalten, dass einerseits dem tatsächlichen Stand der Technik ausreichend schnell Rechnung getragen wird und andererseits die Gesetzgebung dieser Geschwindigkeit gerecht werden kann? Was muss getan werden, dass auch die Hersteller diesem Stand der Technik folgen können?
- Wie können Regularien so spezifisch formuliert werden, dass Hersteller einerseits genau wissen, was verlangt ist, und andererseits nicht unnötig in ihrer Gestaltungsfreiheit eingeschränkt werden?
- Was müssen wir von Herstellern verlangen, damit sie die Sicherheit und Wirksamkeit tatsächlich nachweisen können, aber diese Nachweise (z.B. durch klinische Prüfungen) Patienten nicht unnötig gefährden?
Fragen zur Förderung und Gestaltung
- Wie müssen wir uns als Gesetzgeber personell und inhaltlich aufstellen?
- Welche Empfehlungen sollten wir Behörden und Benannten Stellen geben, um deren Effizienz und Effektivität (z.B. durch Digitalisierung) zu steigern?
- Wie können wir die Aufgaben im Kontext der Regulatory Science auf Behörden, Industrie, Fachgremien, die Wissenschaft, Patienten(vertreter) und andere aufteilen?
- Was können wir den Herstellern als Hilfestellung anbieten? Das betrifft Leitfäden ebenso wie validierte Computermodelle z.B. von Organen.
- In welchen Bereichen fehlen wissenschaftliche Grundlagen? Wo würde sich eine Förderung besonders bezahlt machen?
b) Fragen von Herstellern
- Was ist der Stand der Technik (bezüglich einer Fragestellung)? Wie bestimme ich ihn?
- Welche Optionen habe ich, um die Sicherheit, Leistungsfähigkeit und Wirksamkeit meiner Produkte nachzuweisen? Welche der Optionen sind besonders effizient?
- Werden die Benannten Stellen und Behörden diese Optionen akzeptieren?
- Wie kann ich meine Medizinprodukte mit Hilfe von Computermodellen schneller entwickeln, verifizieren und validieren?
- Was muss ich machen, um diese Computermodelle zu validieren?
- Mit welchem Patientenkollektiv muss ich eine klinische Prüfung durchführen? Welchen Teil dessen kann ich durch in-silico clinical trials ersetzen?
Lesen Sie hier mehr zum Thema Computer-based Modeling and Simulation und erfahren Sie, wie die FDA damit die schnellere Entwicklung und Prüfung von Medizinprodukten fördert.
c) Fragen von Behörden und Benannten Stellen
- Wie bewerte ich die Risiken und die Leistungsfähigkeit neuer
- Produkte und Produktklassen (z.B. Roboter),
- Technologien (z.B. Machine Learning),
- Methoden (z.B. 3D-Druck für die Herstellung) und
- Materialien (z.B. Nanopartikel)?
- Wie entdecke ich bei begrenzten zeitlichen und personellen Ressourcen mit einer möglichst hohen Wahrscheinlichkeit Nicht-Konformitäten bei Produkten und Herstellern?
- In welchen Bereichen (Produkte, Hersteller, Märkte, Technologien) werden wir künftig mit Problemen zu rechnen haben? Wir können wir dem gegensteuern?
- Wie kann es uns gelingen, die Zulassungsprozesse und die Marktüberwachung so zu digitalisieren, dass wir
- schwarze Schafe schneller und wirksamer entdecken und
- die eigenen Aufwände dafür begrenzen?
- Wie kann ich abschätzen, wie sehr die Nachweise der Hersteller für möglichst alle Umstände (Nutzungskontexte, Patientenpopulationen) geeignet sind? Wie erkenne ich Lücken?
4. Regulatory Science betreiben
a) Die Akteure der Regulatory Science
Regulatory Science ist nicht nur eine Aufgabe der Universitäten. Das Betreiben von Regulatory Science ist vielmehr eine gesellschaftliche Aufgabe, an der sich beteiligen sollten:
- Nationale und internationale Gesetzgeber, Behörden
- Benannte Stellen
- Fachgremien wie Normengremien, IMDRF usw.
- Hochschulen, Universitäten und andere Forschungseinrichtungen
- Krankenhäuser, Spitäler und sonstige Gesundheitseinrichtungen
- Patienten
- Hersteller
- Dienstleister, Beratungsunternehmen, Testlabore, Weiterbildungsinstitute
Auch das Johner Institut arbeitet seit Jahren aktiv mit:
- Entwicklung von Leitfäden (z.B. zur IT-Sicherheit oder zur künstlichen Intelligenz)
- Beiträge zur Weiterentwicklung von Normen
- Entwicklung von Konzepten, Daten- und Gedankenmodellen
- Ausbildung des wissenschaftlichen Nachwuchses
- Digitalisierung und damit Effizienzsteigerung von Prozessen wie
- der Post-Market Surveillance und
- der kontinuierlichen weltweiten Recherche neuer und geänderter Regularien
- Forschung mit mehreren eigenen Forscher:innen auch in Zusammenarbeit mit Benannten Stellen und Universitäten
Die Akteure in der Forschung konzentrieren sich auf wenige Länder. Eine Analyse der Daten in Pubmed zeigt:
Knapp die Hälfte der Forschenden, die in den letzten 12 Monaten zum Thema „Regulatory Science“ im Kontext von Medizinprodukten publizierten, stammen von Institutionen in den USA. 3 % aus Deutschland.

Wenn man diese Anzahl noch in Korrelation mit dem Umsatz des jeweiligen Medizinproduktemarkts setzt, wird es noch offensichtlicher: Die US-amerikanischen Forschenden publizieren fast dreimal so viel wie die Deutschen. Das zeigt die auf Deutschland normierte Abbildung. Bei China ist es fast ein Faktor 4. Das sind Welten.

Es ist zu beachten, dass die Anzahl der Publikationen und Institutionen kein ultimatives Maß für deren „Impact“ ist. Aber eine genauere Analyse der Relevanz dieser Publikationen lässt Europa nicht unbedingt besser dastehen.
b) Die Methoden der Regulatory Science
Regulatory Science ist eine Wissenschaft.
Das wissenschaftliche Arbeiten zeichnet sich dadurch aus, dass es
- systematisch erfolgt,
- Fragestellungen möglichst vollständig beantwortet,
- objektiv ist,
- im gegebenen Kontext Allgemeingültigkeit hat und damit relevant ist sowie
- überprüfbar ist.
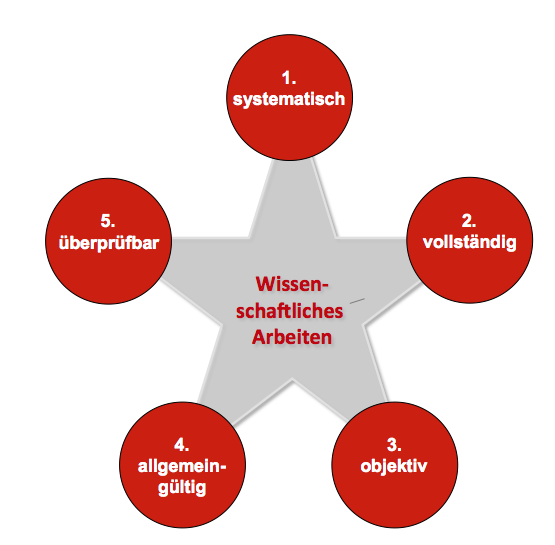
Um diesen Kriterien zu genügen, müssen die Wissenschaftler systematisch vorgehen und geeignete Methoden anwenden.
Zu den Methoden im Bereich der Regulatory Science zählen beispielsweise:
- Berechnungen und Modellierungen, z.B. mit Computer-Modellen
- Experimente; dazu zählen klinische Prüfungen und Usability Tests
- Auswertung vorhandener Daten, das Data-Mining, die Suche nach Abhängigkeiten, der Vergleich von Gesundheitssystemen, Produkten, Methoden, Materialien
- Befragungen und Beobachtungen
- Prototyp-Entwicklung
Professor Haimerl und Professor Johner zeigen in dieser Episode, weshalb diese Wissenschaft ebenso relevant wie lebendig und für uns alle von Bedeutung ist. Sie grenzen die Regulatory Science von der Regulatory Affairs ab und beleuchten den Stand der Wissenschaft in den USA und in Europa.
Diese und weitere Podcast-Episoden finden Sie auch hier.

5. Fazit, Zusammenfassung
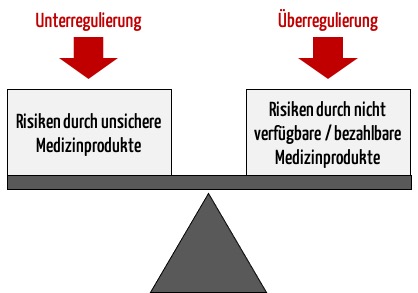
a) Der bisherige Ansatz des Regulierens ist zu naiv
Der Ansatz vieler Regulierer, den Schutz der Patienten vor unsicheren Produkten als einziges Ziel zu verfolgen, greift zu kurz. Denn dann könnte man einfach alle Medizinprodukte verbieten und hätte damit absolute Sicherheit erlangt.
Vielmehr geht es darum, das Nutzen-Risiko-Verhältnis zu optimieren. Das wiederum setzt voraus, dass man den Nutzen und die Risiken kennt und bewertet.
Für die Bewertung benötigt man Daten. Ohne diese Daten verkommt auch eine gut gemeinte Gesetzgebung zu einem wilden „Rumregieren“ im Blindflug. Genau diese Daten, die in anderen Bereichen durch Studien generiert werden, fehlen! Die European Medicine Agency (EMA) plant eine Studie für das Arzneimittelrecht:
Eine Studie zur Zulassung und Überwachung von Humanarzneimitteln wird als Grundlage für die Evaluierung des Regulierungsrahmens mit dem Ziel der Vereinfachung und Straffung der Verfahren und der Kostensenkung dienen
European Medicines Agency’s (EMA) im Dokument COM/2020/761
Danke an Herrn Hentzsch für den Hinweis!
Fazit: Die Gesetzgeber erfüllen nicht die Anforderungen an den Nachweis eines positiven Nutzen-Risiko-Verhältnisses, die sie an die Hersteller stellen.
b) Es geht nicht um das Verhindern, sondern um das Gestalten
Eine professionelle Regulatory Science trägt dazu bei,
- Innovationen zu fördern,
- Märkte zu unterstützen und
- sicherzustellen, dass dem Gesundheitssystem bezahlbare, wirksame und sichere Produkte zur Verfügung stehen.
Die FDA hat das längst erkannt und treibt die Regulatory Science voran. Mit über 15.000 Mitarbeitenden und einem Budget von mehr als 3 Mrd. USD verfügt sie über Ressourcen, von denen europäische Regulierer nur träumen können.
c) Die Regulatory Science braucht uns und wir brauchen die Regulatory Science
Sowohl in Deutschland als auch vielen anderen europäischen Ländern ähnelt die Landschaft der Regulatory Science weitgehend einer Wüste. Dass die Gesetzgeber es besser können, zeigten sie bei der Bekämpfung der Corona-Pandemie: Hier bezogen Politik und Behörden die Wissenschaft(ler) wie z.B. Virologen mit ein.
Würde man bei einer Pandemie so vorgehen wie beim Medizinprodukterecht, würde der Gesetzgeber nicht nur die Virologen ignorieren. Es gäbe nicht einmal eine Virologie.
Um von diesem Niveau auf das der FDA zu gelangen, sind große Kraftanstrengungen notwendig. Diese sind nur gemeinsam zu bewältigen. Das Johner Institut ist dabei.
Änderungshistorie
- 2021-06-28: Hinweis zur EMA im Fazit ergänzt
- 2021-06-07: Abbildungen 3 und 4 und zugehörige Texte eingefügt.



Lieber Kollege Johner,
herzlichen Dank für Ihren sehr wichtigen Beitrag, den ich nur 100%ig unterstützen kann. Solange wir nur im Spannungsfeld zwischen den politischen Akteuren auf der einen Seite und den Industrieunternehmen auf der anderen Seite bleiben, wird wohl die Sichtweise im Vordergrund bleiben, dass die Industrie nur ihre Interessen durchsetzen will und die Politik die Aufgabe hat, das zu verhindern. Eine neutrale und objektive Perspektive durch die Wissenschaft könnten ein ganz zentrales Element sein, um die Diskussion stärker auf einen sachlichen Boden und einen ergebnisorientierten Weg zu bringen. Ich hätte noch ein paar Ergänzungen zu Ihrem Beitrag, die mir persönlich wichtig sind und die ich im Folgenden schildern will.
Die eine betrifft, die Frage warum Regulatory Science bei uns eine so geringe Rolle spielt. Aus meiner Sicht ist es wesentlich, dass gerade ein solches Thema getragen wird von Personen, die konkrete praktische Erfahrungen in der Umsetzung regulatorischer Anforderungen haben. Das ist bei einem Großteil der Kollegen aus dem universitären Bereich nicht der Fall, da sie häufig primär den akademischen Bereich kennen. Sie sind aber nicht unmittelbar mit dem industriellen Umfeld in Kontakt gekommen, wo die regulatorischen Aspekte wirklich zum Tragen kommen. Ich muss selbst sagen, dass ich kein Interesse an regulatorischen Fragestellungen entwickeln konnte, bevor das nicht zentraler Bestandteil meiner eigenen beruflichen Tätigkeit geworden ist.
Insofern ist wohl von den Universitäten nur sehr bedingt ein Beitrag zu diesen Themen zu erwarten – sowohl im Bereich der Lehre als auch in der Forschung. Daher wären die Hochschulen für angewandte Wissenschaften eigentlich der bessere Platz. Auch hier muss man jedoch sagen, dass es bisher nur wenige Hochschulen gibt, die dem Thema einen hohen Stellenwert beimessen und gezielt Schwerpunkte in der Ausbildung setzen – trotz des enormen Fachkräftemangels in diesem Bereich. Hier ist in jedem Fall etwas zu tun und vereinzelt haben das auch schon Hochschulen und Institute aufgegriffen (z.B. bei Ihnen am Johner Institut, in Lübeck oder auch bei uns an der Hochschule Furtwangen / Hochschulcampus Tuttlingen). Durch die Stärkung der Ausbildung in diesem Bereich würde zudem die strategischen Fragestellungen im Bereich Regulatorik stärker zu Tragen kommen, weil die Studierenden das stärker von Grund auf vermittelt bekommen und auf einem ganz anderen Niveau bearbeiten können.
Ein weiterer Nebenaspekt ist, dass Regulatory Science oftmals nicht als Forschungsgegenstand akzeptiert und damit in der Regel nicht in Förderprogrammen berücksichtigt wird. Auch hier mag mitspielen, dass die Frage, was als Forschung betrachtet wird, stärker von Kollegen aus dem universitären Bereich definiert wird. Wie bereits gesagt, wird dort jedoch dieses Thema in der Regel nicht als Schwerpunkt und oftmals eben auch nicht als wissenschaftliches Thema betrachtet.
Eine weitere Ergänzung bezieht sich auf die Fragestellung, in welchen Zeiträumen regulatorische Änderungen greifen. Wenn man sich die Geschichte der MDR anschaut, geht diese bis in das Ende der 2000er Jahre zurück. Das heißt, es hat über 10 Jahre gedauert, bis die MDR verpflichtend in Kraft tritt / treten wird. Bei den heutigen Entwicklungszyklen ist es klar, dass bis zur Umsetzung viele Aspekte neue Betrachtungsformen benötigen, siehe z.B. Fragen der individualisierten Medizintechnik, Nutzung von Real World Data oder „künstlichen Intelligenz“. Es ist auch klar, dass wir manche Überraschung erst feststellen werden (bzw. im Moment gerade feststellen), wenn die enthaltenen Anforderungen wirksam werden.
Das verdeutlicht zwei Ansatzpunkte. Der eine betrifft die Anforderung, dass eine fundierte Kenntnis der Wirkungsweise regulatorischer Anforderungen, Maßnahmen, Prozesse, etc. erlangt werden muss, so wie Sie das in Ihrem Beitrag sehr gut herausgearbeitet haben. Zum anderen werden wir auch dynamischere Ansätze für die Gestaltung regulatorischer Anforderungen benötigen, um mit den Entwicklungen Schritt zu halten. Ein solch „starres Monster“ wie die MDR, das 10 Jahre Entwicklungszeit braucht und dann herausgeschickt wird, ohne dass es vorher getestet ist, erfüllt eine solche Anforderung nur sehr bedingt.
Wenn man sich die FDA als aktuellen Gegenpol anschaut, dann ist dort der Weg etwas anders (auch wenn es sicherlich bei der FDA auch schon andere Zeiten gegeben hat). Hier ist die Gesetzgebung schlanker und die FDA kann dann gezielt über Guidances nachsteuern. Das nutzt die FDA im Moment sehr stark um wichtige inhaltliche wie auch methodische Fragestellungen zu forcieren, die u.a. dynamischere Regulierungsmodelle ermöglichen (siehe z.B. Software Pre-Cert Programm, Diskussionspapier zur künstlichen Intelligenz, Nutzung von Real World Evidence, …). Während wir mit der MDR und Ihren Folgen kämpfen, ist die FDA in der Lage, hier wichtige Zukunftsimpulse zu setzen.
Ich möchte dabei erwähnen, dass ich die grundlegenden Ideen der MDR durchaus positiv betrachte. Allerdings sehe ich so wie Sie, dass die starre Umsetzung in vielen Bereichen hinderlich ist, u.a. weil die Vorgaben nicht praxiserprobt und z.T. auch im Verhältnis zwischen Nutzen und Schaden nicht ausgewogen sind. Im Grunde erfüllt die MDR die Anforderungen nicht, die sie selbst an die Erprobung von Produkten und Validierung ihrer eigenen Anforderungen stellt. Um eine Dynamisierung regulatorische Prozesse aber überhaupt erst möglich und zuverlässig umsetzbar zu machen, wäre gerade eine Etablierung des Bereichs Regulatory Science ein entscheidender Schritt. Nur wenn wir systematisch analysieren, wie regulatorische Prozesse wirken, wo ihre Benefits und wo ihre Risiken sind, sind wir in der Lage, solche Prozesse gut im Sinne aller zu gestalten.
Regulatorische Anforderungen haben in der Tat etwas mit Nachhaltigkeit zu tun und Nachhaltigkeit hat etwas damit zu tun, dass wir Abläufe kontrollieren und immer wieder in die richtigen Bahnen lenken können. Wenn die Zeitskalen zwischen Feststellung von Fehlentwicklungen und den Möglichkeiten darauf zu reagieren, d.h. wirksame Maßnahmen zu ergreifen, nicht zusammenpassen, dann kann ein solches System nicht funktionieren. Auch das gehört aus meiner Sicht zu zentralen Fragestellungen im Bereich Nachhaltigkeit und Regulatory Science. Ich kann mich Ihnen nur anschließen, dass dieser Bereich in Zukunft deutlich gestärkt werden sollte – im Bereich der Ausbildung und im Bereich von Forschung / systematische Erfassung Ihrer Wirkungsweisen. Und das sollte letztendlich im Interesse aller sein – der Gesetzgeber, der Unternehmen und der Gesellschaft.
Mit besten Grüßen
Martin Haimerl
Was für ein wunderbarer Kommentar, lieber Kollege Haimerl!
Danke für diese Gedanken! Lassen Sie uns das bilateral weiter diskutieren, vielleicht in einem Podcast?
Nochmals danke für Ihre großartige Darstellung!
Viele Grüße, Christian Johner