
Was ist ein Device Master Record (DMR)? Ist er auch für Software gefordert? Falls ja, welche regulatorischen Anforderungen in den USA und Europa sind zu beachten? Und was sollte ein DMR enthalten?
Dieser Beitrag gibt Antworten.
Mit Einführung der neuen Quality Management System Regulation (QMSR) im Februar 2026 schafft die FDA den Device Master Record ab. Ab dann werden die „Akten“ aus der ISO 13485 verlangt, wie das „Medical Device File“.
Was ist ein Device Master Record?
‚Device Master Record‘ (DMR) ist ein durch die FDA geprägter Begriff, der auch außerhalb der USA genutzt wird.
Man versteht unter einem DMR eine „Akte“, die, vereinfacht gesprochen,
- zum einen das Gerät beschreibt und
- zum anderen, wie das Gerät produziert, genutzt und gewartet werden soll.
Daher sollte ein Device Master Record enthalten:
- Zweckbestimmungen
- Spezifikationen (des Geräts und von Komponenten), System-Architektur, Zeichnungen, Berechnungen
- Angaben zur Produktion: Werkzeuge, Methoden, Verfahren, Spezifikationen und Anforderungen an die Produktion
- Verfahren zur Qualitätssicherung: Festlegungen, wie das Gerät (v. a. in der Produktion) zu prüfen ist, z. B. mit welchen Methoden und Werkzeugen. Bei Software, bei der es keine wirkliche Produktion gibt, könnten das Prüfanweisungen nach der Installation oder Konfiguration sein.
- Angaben zum sogannnten „Labeling“. Das geht über die „Labels“ hinaus und umfasst z. B. die Verpackung, Hinweise auf dem Gerät und in Handbüchern, einschließlich der Verwendung von Symbolen
- Hinweise zur Installation, zu Wartung und Service, zur Lagerung und zum Transport, inklusive Methoden und Werkzeuge
Die Ergebnisse des Design Outputs (z. B. Spezifikationen) „landen“ auch im Device Master-Record.
Nicht zu verwechseln ist der DMR mit dem DHR, dem Device History Record, der für jede Instanz eines Produkts zu erstellen ist.
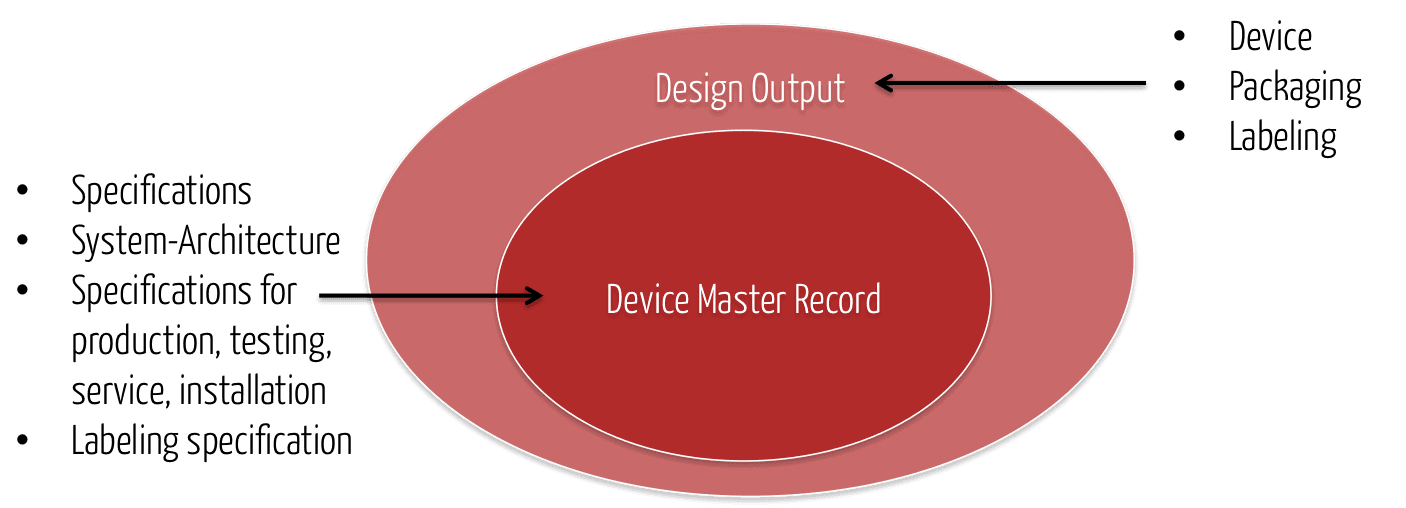
Die FDA schreibt zum Verhältnis von DMR und Design Output: „The total finished design output consists of the device, its packaging and labeling, and the device master record.“ Gleichzeitig enthält laut FDA der Device Master Record die „packaging and labeling specifications“.
Welche regulatorischen Anforderungen bestehen an einen DMR?
In den USA ist der Device Master Record verpflichtend. In Europa gibt es mit der „Technical Documentation“ und dem „Medical Device File“ der ISO 13485 vergleichbare Forderungen.
Forderungen der FDA
Die FDA definiert den Device Master Record als „compilation of records containing the procedures and specifications for a finished device“. 21 CFR part 820.181 beschreibt, welche Elemente ein DMR enthalten muss. Eine Zusammenfassung wurde bereits oben gegeben.
Forderungen der ISO 13485:2016
Die ISO 13485:2016 führt im Vergleich zu ihrer Vorgängerversion ein Medical Device File (MDF) ein, was dem Device Master Record ähnelt. Auch hier werden ähnliche Unterlagen verlangt wie oben beschrieben.
Zusätzlich fordert die Norm eine Nachvollziehbarkeit der Änderungen am Produkt über dessen Lebenszeit. Das heißt, dass man im MDF immer die aktuell gültigen Beschreibungen des Produkts (gemäß Liste oben) finden soll.
Will man die einzelnen Versionen und die Änderungen im DMR oder MDF nachvollziehen, muss der Bericht mit jeder Produktversion aktualisiert werden. Um die geforderte Nachvollziehbarkeit zu haben, muss man den Zustand des DMR für jede ältere Version wiederherstellen können.
Forderungen der Medizinprodukteverordnung (MDR) 2017/745
Eine Medizinprodukteverordnung wie die MDR stellt Anforderungen an die technische Dokumentation, die Elemente eines DMR enthalten.
Benötigen wir ein DMR für Software? Falls ja, was muss er enthalten?
Für jedes Medizinprodukt benötigen Sie ein Device Master Record bzw. das europäische Pendant, das Medical Device File. Wenn die Software Teil Ihres Produkts ist, dann sind die Software-Spezifikation oder die Software-Anforderungen Teil des DMR.
Bei Standalone-Software umfasst der DMR einmal den Design Output sowie weitere Dokumente.
Design Output bei Software (als Teil des DMR)
Weitere Elemente eines DMR für eine Standalone-Software
- Zweckbestimmung
- Gebrauchsanweisung
- Installationsanweisung
- Anleitungen zur Produktion, z. B.
- Speichern auf Datenträger
- Hochladen in einen App-Store
- Einschränkungen, die Länder, Betriebssysteme, Hardware betreffen
- Hinweise, ob/wie die Software aktualisiert wird
- Wie lange darf eine Software-Version auf z. B. einer DVD verkauft werden? (DVDs altern)
- Wie werden Updates verteilt?
- Wie wird deren Installation sichergestellt?
- Wie werden alte Versionen zurückgezogen?
- Festlegung der Verpackung
- Trainingsmaterialien
- Lizenzinformationen
Abgrenzung zum Design History File
Das Design History File würde auch folgende Dokumente (einschließlich ihrer Versionen) enthalten.
- Spezifikation und Ergebnisse der Software-Tests wie Unit-Tests, Integrationstests und Software-System-Tests
- Ergebnisse der statischen Code-Analyse und der Code-Reviews
- Spezifikationen, Verifizierung und Validierung der Gebrauchstauglichkeit
- Software-Freigabe
- Risikomanagementakte
- Anleitungen zum Erzeugen der Software einschließlich Build-Scripts und Konfigurationsdateien
Das Ziel des DHF besteht auch darin nachzuweisen, dass die Software gemäß dem Entwicklungsplan entwickelt wurde.
Unterstützung
Benötigen Sie Unterstützung bei der Zusammenstellung Ihres DMR, MDF oder der technischen Dokumentation? Dann nehmen Sie gleich Kontakt mit uns auf.
Änderungshistorie
- 2024-11-18: Überarbeitung aufgrund der Einführung der neuen QMSR



Wir sind reiner Händler ( keine Eigenprodukte, keine OEMs,…), haben jetzt auf 13485:2016 er umgestellt. Abweichung im Audit lautet ‚keine Meidzinprodukteakte vorhanden‘ Hilfe, was tun? Dachte die ist nur für Hersteller notwendig?
Ich bedanke mich jetzt schon mal für Ihre Antwort / Hilfe,
Mit freundlichen Grüßen,
C. Kurz- Wagner
Danke für Ihre Frage:
Möglicherweise liegt entweder ein Missverständnis des Auditors vor oder es fehlten im QM-Handbuch der entsprechende Ausschluss bzw. der Hinweis auf die Nichtanwendbarkeit. Letzteres könnte man leicht korrigieren.
Hallo Herr Johner,
bei uns in der Firma (IVD Produkte) wird ein DMR und ein DHF erzeugt. Für Changes, welche eine Anpassung dieser beiden Akten nach sich ziehen, sind wir als Qualitätsabteilung oft in der Diskussion, in welcher Reihenfolge diese freigegeben werden sollen:
1.) Erste Ansicht: um eine Validierung vorzunehmen, sollte das zu testende Gerät dem Serienstand entsprechen. Daher muss das DMR freigegeben sein, damit die aktuellen Teile und Qualitätssicherungsmaßnahmen während des Aufbaus sichergestellt werden können. Danach kann der Validierungsreport in den DHF einfließen und dieser freigegeben werden.
2. Zweite Ansicht: die Validierung muss an einem Objekt stattfinden, die dem späteren Serienstand in jeglicher Hinsicht gleicht. Daher kann dies auch eine Nullserie (o.ä.) sein. Somit kann erst der Validierungsreport erstellt und der DHF freigegeben werden. Danach kann dann immer noch der DMR freigegeben werden (die Gefahr, dass dort Änderungen einfließen und somit die Validierungsergebnisse obsolet machen, ignorieren wir einmal).
Gibt es laut FDA bzw. ISO 13485 eine Vorgabe bzgl. der Reihenfolge bzw. Abhängigkeiten von DHF und DMR?
Vielen Dank und schöne Weihnachtsfeiertage,
Lars Fischheiter
Sehr geehrter Herr Fischheiter,
eine von der FDA eingeforderte Reihenfolge ist mir nicht bekannt. Wichtig ist, dass das Gerät dem endgültigen Gerät so gleicht, dass die Prüfungen (es geht möglicherweise nicht nur um die Validierung wie die klinische Prüfung und die summative Bewertung der Usability) die Einhaltung der Anforderungen belegen können. Eine Null-Serie nutzt man i.d.R. dazu. Die Prüfungen, sprich die Nachweise, dass das Gerät sicher entwickelt wurde landn dann einschließlich Planung, Spezifikation und Ergebnissen der Prüfungen im DHF.
Wenn die Prüfungen nachweisen, dass das Produkt richtig produziert wurde, landen sie im DHF.
Ich sehe diese Prüfergebnisse nicht im DMR.
Hilft das? Falls nicht, haken Sie gerne nach.
Viele Grüße, Christian Johner
Hallo, mich würde zum Thema DMR die Aufbewahrungsfrist eines DMRs (für eine Produktart) interessieren! Was würden Sie da empfehlen? Danke und VG, RM
Lieber Herr Morgenstern,
danke für Ihre spannende Frage! Damit ich möglichst gezielt antworten kann: Was wäre der Rechtsbereich? „DMR“ klingt nach FDA, aber wir nutzen den Begriff auch in Europa.
Eine erste Übersicht über die Aufbewahrungsfristen finden Sie hier.
Damit möchte ich mich aber nicht vor einer ausführlicheren Antwort drücken. Haken Sie gerne nach.
Ich danke Ihnen und grüße Sie herzlich, Christian Johner